

Abstract
The mechanisms underlying the differential expression of long-term potentiation (LTP) by AMPA and NMDA receptors, are unknown, but could involve G-protein-linked metabotropic glutamate receptors. To investigate this hypothesis we created mutant mice that expressed no metabotropic glutamate receptor 5 (mGluR5), but showed normal development. In an earlier study of these mice we analyzed field-excitatory postsynaptic potential (fEPSPs) in CA1 region of the hippocampus and found a small decrease; possibly arising from changes in the NMDAR-mediated component of synaptic transmission. In the present study we used whole-cell patch clamp recordings of evoked excitatory postsynaptic currents (EPSCs) in CA1 pyramidal neurons to identify the AMPAR- and NMDAR-mediated components of LTP. Recordings from control mice following tetanus, or agonist application (IS, 3R-1-amino-cyclopentane 1,3-dicarboxylic acid) (ACPD), revealed equal enhancement of the AMPA and NMDA receptor-mediated components. In contrast, CA1 neurons from mGluR5-deficient mice showed a complete loss of the NMDA-receptor-mediated component of LTP (LTPNMDA), but normal LTP of the AMPA-receptor-mediated component (LTPAMPA). This selective loss of LTPNMDA was seen in three different genotypic backgrounds and was apparent at all holding potentials (−70 mV to +20 mV). Furthermore, the LTPNMDA deficit in mGluR5 mutant mice could be rescued by stimulating protein kinase C (PKC) with 4β-phorbol-12,13-dibutyrate (PDBu). These results suggest that PKC may couple the postsynaptic mGluR5 to the NMDA-receptor potentiation during LTP, and that this signaling mechanism is distinct from LTPAMPA. Differential enhancement of AMPAR and NMDA receptors by mGluR5 also supports a postsynaptic locus for LTP.
Long-term potentiation (LTP) of glutaminergic synapses may share partly the mechanisms involved in the development of neural circuits, information storage, and neurodegeneration (Bliss and Collingridge 1993). The ionotropic glutamate receptors, of the N-methyl-d-aspartate (NMDA) class, are known to induce LTP via a Ca2+ signal (Nicoll and Malenka 1995). The expression of LTP requires another class of ionotropic glutamate receptor, the α-amino-3-hydroxy-5-methyl-4 isoazolepropionate (AMPA) class, but the contribution of the NMDA receptors in LTP expression has been controversial (for review, see Kullmann and Siegelbaum 1995). In this report we find that both components of NMDA- and AMPA-receptor-mediated synaptic currents (EPSCNMDA and EPSCAMPA) are potentiated equally during LTP and that one of the metabotropic glutamate receptors (mGluR), mGluR5, is required only for the NMDA-receptor-mediated component of LTP (LTPNMDA), with no effect on the AMPA-receptor-mediated component of LTP (LTPAMPA), in the CA1 region of the hippocampus. Our data supports the contributions of both NMDA receptors (NMDAR) and AMPA receptors (AMPAR) to the expression phase of LTP, and shows that the mechanisms underlying these two components are distinct.
Several other lines of evidence invoke mGluRs in LTP. The application of aminocyclopentate-IS,3R-dicarboxylic acid (ACPD), an agonist of broad specificity, or administration of ACPD, combined with subthreshold tetanic stimulation, or coapplication of NMDA and ACPD, induced LTP in the hippocampus (McGuiness et al. 1991; Otani and Ben-Ari 1991; Aniksztejn et al. 1993; Bortolotto and Collingridge 1993; Musgrave et al. 1993; Riedel and Reymann 1993). Consistent with these findings, (+)-α-methyl-4-carboxyphenylglycine (MCPG), a broad-spectrum antagonist for mGluRs, has been shown to inhibit tetanus- or ACPD-induced LTP, but not short-term potentiation in perforant pathway of the dentate gyrus (Richter-Levin et al. 1994; Riedel et al. 1994) and CA1 (Bashir et al. 1993). In contrast, other studies showed a lack of effect of MCPG on LTP (Chinestra et al. 1993; Manzoni et al. 1994). Apparently, MCPG only blocks the initial LTP, but not LTP that arises from later stimulations (Bortolotto et al. 1994).
The class I mGluR, mGluR5, is the main postsynaptic receptor of this type in the CA1 region of the hippocampus (Minakami et al. 1993; Romano et al. 1995), whereas mGluR1a is absent (Shigemoto et al. 1993). mGluR5 is found predominantly in the postsynaptic dendrites and spines of CA1 cells upon EM analysis (Romano et al. 1995). Immunogold staining for mGluR5 shows localization in a perisynaptic annulus on dendritic spines of CA1 pyramidal cells, whereas AMPAR and NMDAR proteins were concentrated in the center of the postsynaptic density (Lujan et al. 1996). Presumably, mGluR5 can only respond to high levels of glutamate, such as may be released during high-frequency tetanic stimulation, or coincident firing at lower frequencies (i.e., theta rhythms). One study using gene-targeting techniques has demonstrated that mGluR1 is not required for LTP in CA1 (Conquet et al. 1994), although a second study did report impaired LTP in this region (Aiba et al. 1994). In fact, LTP could be produced normally in all pathways except in the mossy fiber/CA3 synapses, where activation of the NMDA receptor is not required for LTP induction. These studies indicate that mGluR1 may not be involved in NMDAR-dependent LTP, which is not surprising since mGluR1 is not expressed in CA1 pyramidal neurons (Lujan et al. 1996). The involvement of mGluR5 is unknown because there are no agonists or antagonists specific for mGluR5 currently available (Hayashi et al. 1994; Kingston et al. 1995; Pin and Duvoisin 1995). In the absence of selective agonists or antagonists, we removed mGluR5 by gene targeting and examined its linkage to the induction and expression of LTP. We find that mGluR5 is selectively required to generate LTPNMDA, but not LTPAMPA.
Materials and Methods
VECTOR CONSTRUCTION
A genomic clone containing exon 1 of the mGluR5 gene was isolated from a genomic 129 phage DNA library prepared from embryonic stem (ES) cells of the R1 line after screening with rat mGluR5 cDNA as a probe. The exon 1 region was sequenced and its DNA sequence was 98% identical to the published rat mGluR5 sequence in the corresponding region (Abe et al. 1992). The targeting vector was prepared by inserting a 2.2-kb BamHI–XhoI fragment and a 4.3-kb HindIII fragment 3′ to exon 1 into the pPNT plasmid vector. The 2.2-kb BamHI–XhoI was first subcloned into pBluescript vector (Stratagene) to obtain a NotI restriction site. The 4.3-kb HindIII fragment was cloned into pPNT by BamHI and blunt-end ligation. The construct was designed to delete a 0.4-kb fragment containing part of exon 1 and a part of intron 1, which was replaced by a 1.8-kb fragment containing the PGK–neo cassette. A PGK–tk cassette was inserted downstream of the 4.3-kb HindIII fragment as a negative selection marker.
GENERATION OF MUTANT MICE
The vector DNA was linearized with NotI restriction digestion and transfected into R1 ES cells (Nagy et al. 1993) by electroporation (Bio-Rad, Gene Pulser, set at 240 V and 500 μF). G418 (150–250 μg/ml) and gancyclovir (2 μm) were applied 24 hr after transfection, and double-resistant colonies were isolated during days 7–10 of selection. Genomic DNA of resistant clones was digested with PstI and hybridized with a 1.2-kb HindIII–XhoI DNA fragment 3′ to the 4.3-kb HindIII fragment as an external probe (Fig. 1A). Four clones (from a total of 550 analyzed) were shown to contain desired targeted events, and all of them were used to produce chimeric mice via aggregation with CD1 morulae (Wood et al. 1993). The contribution of ES cells to the germ line of chimerae was determined by mating with CD1 mice and screening for agouti offspring. Germ-line transmission was confirmed by Southern blotting of tail DNA. Three chimerae from two independent ES clones transmitted the mutation through the germ line. Mice heterozygous for the targeted mutation were intercrossed to homozygosity. Homozygous females and males were fertile, but poor breeders. All experimental mice, including wild-type controls, heterozygotes, and homozygotes were generated by heterozygous breeding. Littermate controls were used throughout. Results on mice derived from two separate ES clones were similar.
Figure 1.

Production of mGluR5 null mutants. (A) The 129 mGluR5 genomic clone (exon 1, open box), targeting vector, and mutant mGluR5 locus expected following homologous recombination. (P) Pst1; (B) BamH1; (X) Xho1; (H) HindIII; (Xb) XbaI; (N) NotI. (B). Southern blot analysis of genomic DNA isolated from a representative litter of nine mice derived from a heterozygous (+/−) F1 intercross. DNA was digested with PstI and probed with an external 1.2-kb fragment (above) yielding a 11.5-kb restriction fragment from wild type (+/+), and a 7.5-kb diagnostic band from mutant alleles (−/−). (C). Western blots of crude whole-brain plasma membranes prepared from homozygote mGluR5 mutant (−/−), heterozygote (+/−) and wild-type (+/+) mice, and probed with the antibodies indicated. The amount of mGluR5 detected using antibody to the carboxyl terminus (Romano et al. 1995) relative to wild type was 50% in +/− and 0% in −/− (n = 3 per group). (D) Immunocytochemical staining of +/+ (a,c) and −/− (b,d) mouse hippocampi, using antibodies selective for mGluR5 (a,b) and mGluR1a (c,d). The entire hippocampus (a,b) or the polymorph region of the dentate gyrus (c,d) is shown. Bar, 500 μm (a,b); 200 μm (c,d). Serial sections through the entire CNS (n = 3) revealed normal neuroanatomical organization.
WESTERN ANALYSIS OF mGluR5 MUTANT MICE
Three adult brains from each of +/+, +/−, and −/− mGluR5 mutant mice were pooled and a crude synaptic plasma membrane fraction was prepared. The brains were homogenized in 0.32 m sucrose and centrifuged for 20 min at 2000g. The supernatant was transferred to two volumes of water. The sample was then centrifuged for 15 min at 50,000g. The pellet was resuspended in 50 μm Tris-HCl at pH 7.0 by polytron and the centrifugation and resuspension steps were repeated. The amount of total protein was assayed (Pierce) and the samples were diluted to 5 mg/ml protein. Samples containing 50 μg of total protein were run on 8% Laemmli polyacrylamide gels, transferred to nitrocellulose membranes and probed with anti-GluR1 (UBI); anti-GluR2/3 (UBI), anti-GluR4 (UBI); anti-GluR6/7 (UBI); anti-NR1 (UBI); anti-NR2A/2B [gift of R.J. Wenthold, National Institutes of Health (NIH), Bethesda, MD], anti-mGluR5 (UBI) (Abe et al. 1992) and anti-mGluR1a (A4) (gift of D. Hampson, University of Toronto, Canada), and mGluR4 (gift of R.J. Wenthold). We raised affinity-purified anti-mGluR5 antibody against a carboxy-terminal peptide (Romano et al. 1995). This antibody recognizes both mGluR5a and mGluR5b isoforms (Minakami 1993). The blots were then probed with secondary antibodies linked to alkaline–phosphatase or horseradish peroxidase and the proteins visualized with the appropriate substrates for detection. Some of the blots were developed with the ECL (Amersham) method and the films were scanned for optical density (n = 3) to yield quantative comparisons, with a 1% level of detection.
IMMUNOCYTOCHEMISTRY
Mice were perfusion fixed, as described using 4% paraformaldehyde/0.2% glutaraldehyde in 0.1 m phosphate buffer containing 0.002% CaCl2 at pH 7.35 (Romano et al. 1995). Coronal and parasagittal vibratome sections (50 μm) were incubated in primary antibody in PBS and 0.1% BSA and sodium azide for 24 hr followed by the ABC reagent (Elite, Vector Laboratories) to visualize mGluR5. The antibodies were affinity-purified anti-peptide antibodies against the carboxy-terminal mGluR5 (Ab m5.18) (Romano et al. 1995), or affinity-purified anti-mGluR1a (Ab m1a loop.8) against residues 1116–1130 of mGluR1a. Serial sections through the entire central nervous system (CNS) (n = 3) revealed normal-appearing cell layers, nuclei, and tracts compared to control littermates (n = 3).
ELECTROPHYSIOLOGY
Hippocampal slices ( 400 μm) were prepared from 31- to 42-day-old wild-type or mutant mice and kept for 1 hr in a moist, oxygenated chamber and then transferred to a recording chamber where they were superfused continuously with artificial cerebrospinal fluid (ACSF) (2 ml/min) that had been saturated with 95% O2/5% CO2 at 30 ± 2°C. The composition of ACSF for voltage-clamp recordings, was 124 mm NaCl, 3 mm KCl, 1.25 mm NaH2PO4, 1.3 mm MgCl2, 2 mm CaCl2, 26 mm NaHCO3, 10 mm dextrose, and 100 μm picrotoxin. For field recordings ACSF was the same but without picrotoxin. The whole-cell pipette solution contained 132.5 mmCs-gluconate, 17.5 mm CsCl, 10 mm HEPES, 0.2 mm EGTA, 2 mm Mg-ATP, 0.3 mm GTP, and 5 mm QX-314 (pH 7.4, 292 mOsm). Synaptic currents were evoked at a frequency of 0.1 Hz by bipolar tungsten electrodes. In both field and voltage-clamp recordings, a tetanus consisted of two 100 Hz trains lasting 500 msec and delivered 10 sec apart. In Figures 3, 4, and 6, below, the holding potential during tetanus was 0 mV. In Figures 2 and 5, below, the current-clamp method was used during tetanus. For saturation of LTP (Fig. 4, below) the numbers of trains to induce LTP were indicated by the numbers of arrows. When CNQX was used to isolate NMDAEPSC, a tetanus (two trains, 100 Hz, 500-msec duration, 10 sec apart) was presented at a holding potential 0 mV. Recordings were performed with Axopatch-1D and Axopatch-2B patch-clamp amplifiers (Axon Instruments, Foster City, CA) and data collected with pclamp 6. The change in EPSCs magnitude was calculated by averaging over a 10-min epoch taken 20–30 min after the LTP induction and comparing this value to the 10 min baseline period. No washout was observed up to 1 hr. Each trace shown is the average of six. In Figures 2, a and b, and 5, a–d, below, the NMDAEPSC was measured at a latency of 100 msec, whereas AMPAEPSC was taken 9 msec after stimulation. In figures 1,3,4, and 6 the NMDAEPSC was measured with a latency of 25 msec in the presence of 5 μm CNQX (Fig. 3) or 2 μm CNQX (Figs. 4 and 6, below), as stated. The purity of NMDA current was verified by the addition of 50 μm AP5, which abolished the remaining CNQX resistant current. To reduce the failure rate of LTP induction the following indices were used: Our data indicated that washout was not a major problem for LTP induction. The reason could lie in the following selection criteria. (1) pre-identification of slice quality. Every slice was tested before neuronal patching. The slices were used only if the amplitude of fEPSP recorded in those slices were >1 mV while the stimulating intensity was set to 20% of that inducing the maximum responses. (2) Washout often happened in the young mice (<20 days) in our experiments. The mice used here, however, were >30 days (between 34 to 40 days). (3) The neurons were selected for LTP study only if those neurons showed stability of the resting membrane potentials (not more positive than −60 mV) without oscillation after breakthrough. (4) The pipette solutions were prepared freshly directly before use.
Figure 3.

Complete I/V (current/voltage)curves show LTPNMDA was impaired at all voltages in mGluR5 mutants. (a) Superimposed traces of NMDAEPSCs at three different holding potentials before (0) and 30 min after (1) LTP induction. NMDAEPSCs were measured at 25 msec in CNQX (5 μm). (b) I/V relationship of the peak of NMDAEPSCs before (○) and after (•) LTP induction in the presence of CNQX, (5 μm). At the end of the experiment, the CNQX-resistant EPSC was blocked completely by 50 μm AP5 (not shown). Note the expanded scale (bottom right).
Figure 4.

Saturation of tetanic stimulation failed to induce LTPNMDA in mGluR5 mutant slices. Trains of tetani (100 Hz, 500 msec) (downward arrows) were given to wild-type (•) or mutant (○) neurons in the presence of 2 μm CNQX at 10-min intervals until saturation of LTPNMDA was obtained (eight mice per group). The number of trains is indicated by the arrows. Stimulation intensity to evoke baseline NMDAEPSC was set at 25%–30% of the maximum amplitude. The superimposed traces were obtained as indicated by the letters on the axis.
Figure 6.

PKC stimulation rescues the impaired LTPNMDA in mGluR5 mutants. (a) Bath perfusion of 20 μm 4 β-phorbol-12, 13-dibutyrate (PDBu) for 6 min followed by tetanization (arrow) in wild types (•) and mutants (○) in the presence of 2 μm CNQX. The CNQX-resistant EPSCs was blocked by 50 μm AP5. (b) Initial tetanic stimulation in wild type (○) and mutants (•) was followed by bath-applied PDBu (20 μm) for 6 min.
Figure 2.
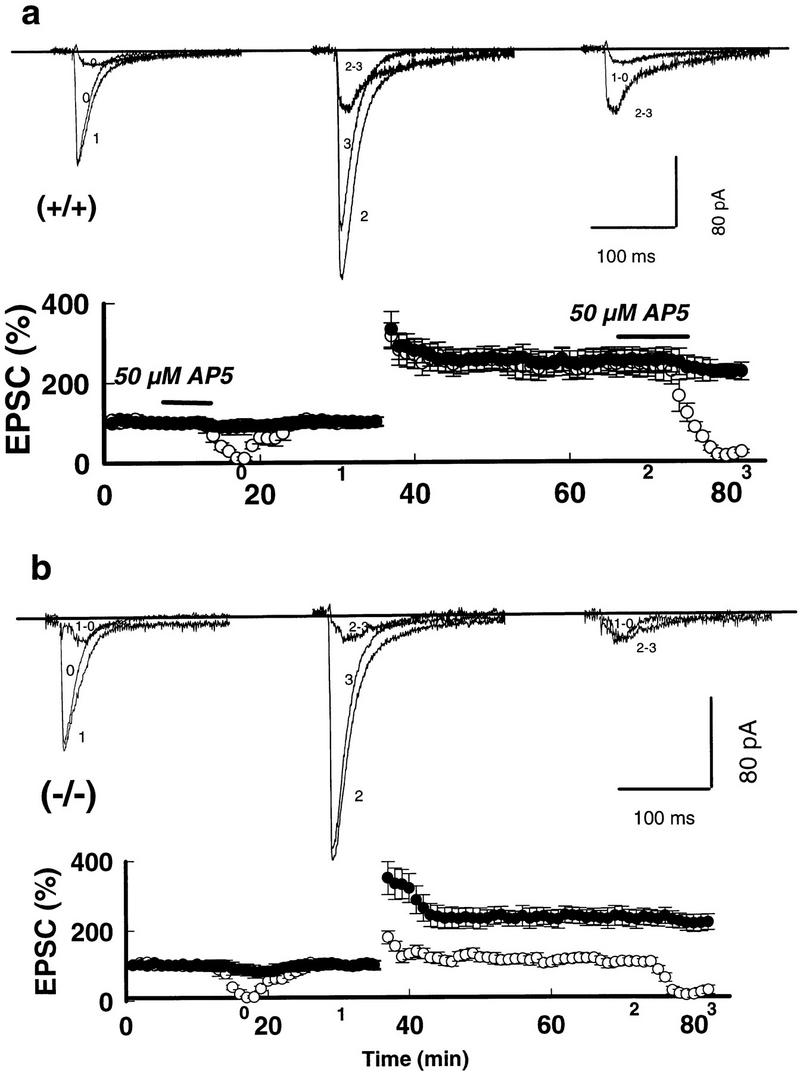
Impaired LTPNMDA with intact LTPAMPA in mGluR5 mutants responding to tetanic stimulation. (a) LTPAMPA (•) and LTPNMDA (○) in area CA1 of the hippocampus of wild type +/+ responding to tetanic stimulation (top) and mutant (−/−) (b, bottom) mice are plotted as a function of time. Horizontal bars indicate that 50 μm D-AP5 was applied for 5 min prior to and following tetanization. Superimposed traces were taken at the indicated times on the axis. (1 − 0) Subtraction of trace 1 from trace 0; (2 − 3) subtraction of trace 2 from trace 3. The AP5-resistant EPSCs was blocked completely by 2 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) at the end of each slice experiment (not shown). The holding potential was −50 mV in this and all other experiments, unless stated otherwise.
Figure 5.
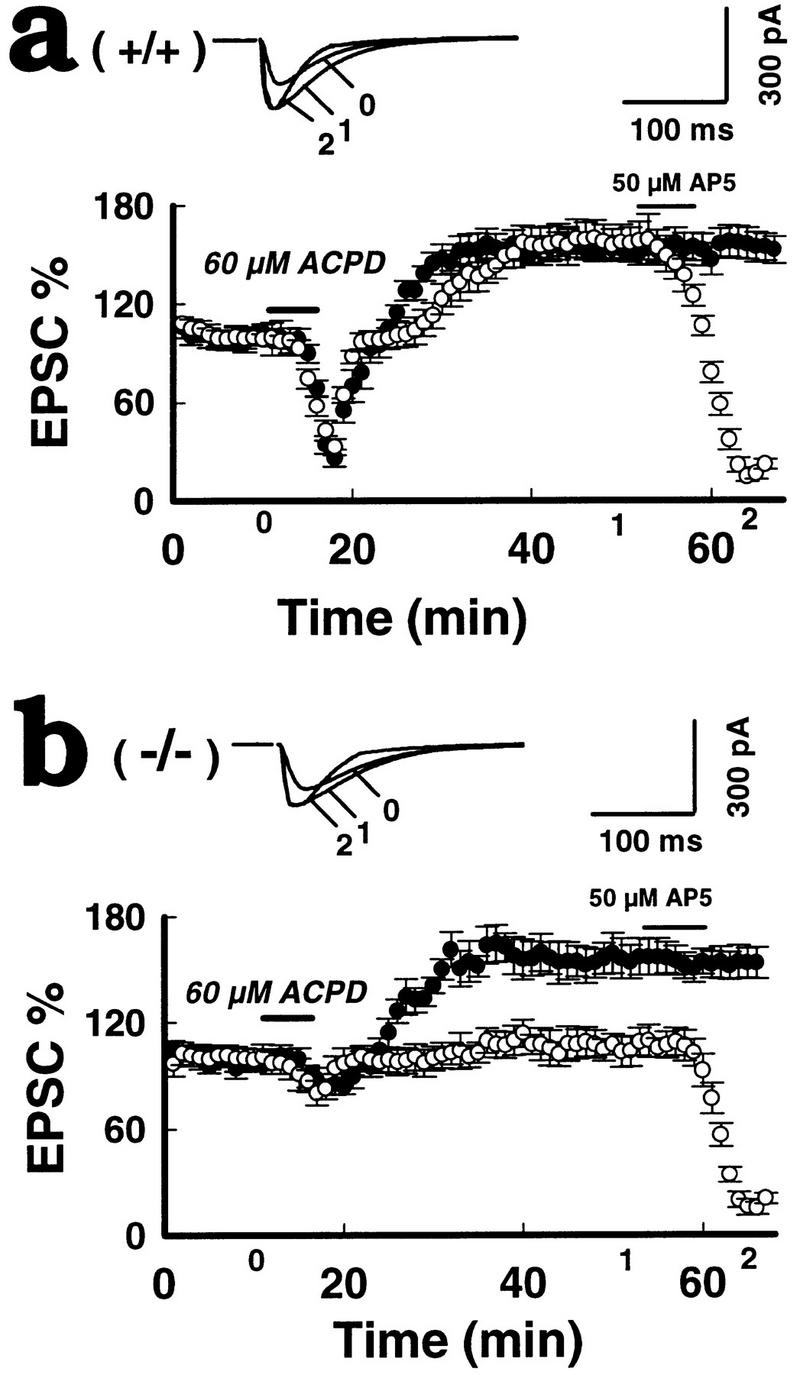
Impaired LTPNMDA in mGluR5 mutants responding to ACPD. (a) Bath perfusions of 60 μm 1S,3R-ACPD for 5 min (horizontal bar) evoked a large initial depression followed by a parallel long-lasting potentiation of the dual components of EPSCs [LTPAMPA (•) and LTPNMDA (○) (at − 50 mV) in the wild-type mice (+/+) (n = 12). (b) In the mutants (−/−) IS-3R-ACPD produced a small initial inhibition of EPSCs followed by a single LTPAMPA (•) without LTPNMDA (○). Thirty minutes after LTP expression, 50 μm AP5 was applied for 5 min to isolate the NMDA component (13 mice per group). Superimposed traces were taken at the indicated points on the x-axis.
The quality of clamping was verified using established criteria (Hestrin et al. 1990; Müller et al. 1992; Spruston et al. 1993; Clark and Collingridge 1995). The input resistance was monitored using voltage steps (−10 mV, 400 msec) at 10-sec intervals throughout the period of the experiment. The mean values of input resistance were 213.12 ± 14.3 MΩ in wild-type controls and 216.6 ± 13.8 MΩ in mGluR5 mutants. Rare neurons were discarded if they showed either a marked change (20%) in holding current or in the amplitude of the capacitance transients, before tetanus or drug application. For each neuron, six successive sweeps to voltage steps were averaged, and if this value changed >20%, relative to a 5-min period prior to tetanus or drug application, the neuron was excluded from the statistical analysis. Series resistance was measured in current clamp and ranged from 34.4 to 46.2 MΩ with a mean in mutants and wild type controls of 36.3 MΩ and 37.2 MΩ, respectively.
All of the electrophysiological recordings were performed by YouMing Lu (Mount Sinai Hospital, Toronto, Canada). Some of the slice recordings were repeated blind, and the experimenter did not know the genotype of the mice before the data was analyzed.
Results
GENERATION OF A NULL MUTATION IN THE mGluR5 GENE OF MICE
A mgluR5-targeting vector was constructed from strain 129 genomic clones that contain exon 1 of the mouse mGluR5 gene and its 3′- and 5′-flanking regions (Fig. 1A). In this vector, a 0.4-kb Xhoi Bindiii fragment, containing the 3′ part of exon I and 5′ part of intron I, was replaced by a 1.8-kb fragment containing the promoter PGK–neo casette, which possessed stop codons in all three frames, and a poly(A) site. The promoter PGK–tk fragment was spliced to the flanking end of the targeting vector, as a negative selection marker. The targeting vector DNA was electroporated into ES cells of the R1 line (Nagy et al. 1993), and the transfectants were selected in G418 and Gancyclovir. Double-resistant colonies were screened for the desired homologous recombination by Southern blotting with a probe 3′ to intron I, and external to the targeting vector (Fig. 1A). Of 550 ES clones analyzed, 4 contained independent targeted events, and these were used separately to make chimerae via aggregation with CD1 morulae (Wood et al. 1993). Chimeric males were mated with CD1, 129, or C57Bl/6 females, to generate three separate lines of offspring. Two independent chimeric mice transmitted the mutation through the germ line, and each showed the same phenotype (see below and Table 1) in all these different genetic backgrounds. Heterozygous offspring were intercrossed to produce homozygous mutants. Wild-type and mutant alleles in the offspring were indicated by the presence of 11.5- and 7.5-kb PstI fragments, respectively, upon Southern blot analysis of tail DNA (Fig. 1b). All mice used in the present experiments were littermates generated by F1 heterozygous matings.
Table 1.
Lack of genetic background effects on LTP
| Strains/ genotypea
|
Pure 129 Sv
|
129 Sv × CD1
|
129 Sv × C57
|
|||
|---|---|---|---|---|---|---|
| (+/+)
|
(−/−)
|
(+/+)
|
(−/−)
|
(+/+)
|
(−/−)
|
|
| LTPAMPA | 161.8 ± 73.3 | 156.6 ± 56.5 | 155.8 ± 68.4 | 157.2 ± 74.7 | ||
| by ACPD | (n = 12) | (n = 13) | (n = 7) | (n = 7) | ||
| LTPNMDA | 160.4 ± 67.1 | 105.1 ± 13.4* | 156.8 ± 71.2 | 101.9 ± 12.8* | ||
| by ACPD | ||||||
| LTPAMPA | 249.6 ± 99.5 | 238.4 ± 86.1 | ||||
| by tetanus | (n = 8) | (n = 8) | ||||
| LTPNMDA | 241.2 ± 96.4 | 111.9 ± 13.2* | 239.9 ± 98.9 | 108.3 ± 11.7 | ||
| by tetanus | (n = 4) | (n = 4)* | ||||
| Saturation of | 323.6 ± 47.6 | 107.9 ± 11.1 | 318.6 ± 126.6 | 103.2 ± 13.6 | ||
| LTPNMDA | (n = 8) | (n = 8)* | (n = 3) | (n = 3)* | ||
Values represent the mean LTP ± s.d. (n) = Number of mice
The values of LTPAMPA from both mutants and wild-type controls show no significant difference between or within the strains (P > 0.2). (*)The values of LTPNMDA in mutants show significant difference from that in wild types between or within strains (P < 0.005). The values of LTPNMDA from mutants show no significant difference between strains (P > 0.3). The values of LTPNMDA from wild types show no significant difference between strains (P > 0.3).
Crude membrane fractions from whole brain were analyzed by Western blots probed with specific antibodies. The mGluR5−/− mice contained no detectable mGluR5 protein (Fig. 1c) even if lanes were overloaded 100-fold over the limit of detection of the antibody (not shown). The antibody used for detection was generated against the carboxyl terminus of mGluR5. Although the antibody did not detect any other proteins in the −/− mice, that were not in the +/+ or +/− mice, we cannot rule out the possibility that some truncated protein, or a fusion with the neomycin-resistance protein, was generated. The level of expression of mGluR5 in heterozygotic mice was ∼50% of wild type. The levels of mGluR1a, mGluR4, NR1, NR2A/2B, GluR1, GluR2/3,GluR4, and GluR6/7 in whole-brain membrane fractions were not detectably different by Western blot in +/+, +/− and −/− mice (Fig. 1C ). Immunocytochemical analysis using antibodies selective for mGluR5 on −/− mice showed no immunoreactivity in any brain region examined, including the molecular layers of the hippocampus (Fig. 1D), whereas antibodies to mGluR1a provided the expected characteristic patterns of staining in the polymorph region of the dentate gyrus, in both +/+ and −/− mice. Serial sections through the entire CNS of several mGluR5 −/− mice showed normal development of neuroanatomical loci and fiber projections (Lu et al. 1997). These data indicate that the mGluR5 mutant mice contain the null alleles of the mGluR5 gene and do not express mGluR5 protein. Also, there is no detectable compensatory alteration in the expression of several other glutamate receptors in the absence of mGluR5.
IMPAIRED LTPNMDA BUT NORMAL LTPAMPA IN mGluR5 MUTANTS IN RESPONSE TO TETANIC STIMULATION
LTP was analyzed using voltage-clamp recordings in the Schaeffer collateral-CA1 synapse in hippocampal slices. The NMDA-receptor-mediated component of the evoked excitatory postsynaptic currents (NMDAEPSCs) was estimated at 100 msec using voltage-clamp recordings. The AMPA-receptor-mediated component was measured at 9 msec (AMPEPSCs) and separated from the NMDA-receptor-mediated component using the NMDA-receptor antagonist D-2-amino-5-phosphonovaleric acid (D-AP5). In 14 wild-type neurons from control littermates (n = 8 mice), the LTP of the NMDA-receptor-mediated component (LTPNMDA) paralleled that of the AMPA-receptor-mediated component (LTPAMPA), at a holding potential of −50 mV, and was stable for at least 1 hr. The peak of LTPNMDA was 241 ± 34%, compared to LTPAMPA at 249 ± 35% 30 min following tetanic stimulation (Fig. 2a). In 12 mutant neurons (n = 8 mice), however, the transient increase in NMDAEPSCs, after the tetanic stimulation, had almost returned to baseline values (112 ± 4.1%) within 10 min, whereas LTPAMPA at 30 min (238 ± 30%) (Fig. 2b) was not different from that in wild type (P > 0.1). The NMDAR-mediated currents were contaminated <15% by AMPA currents. Similar numbers of failures to induce any LTP (i.e., both LTPNMDA and LTPAMPA were absent) were observed (∼20%) in control and mutant neurons.
The amplitude of NMDAEPSCs was also measured in 5 μm CNQX. Lower doses of CNQX (2 μm) were also effective. The remaining EPSCs in 2 μm CNQX were pure NMDAEPSCs as the addition of 50 μm AP5 abolished completely the currents under these experimental conditions. LTPNMDA was also eliminated at all potentials between −70 to +20 mV without observable voltage dependency in the mutants (8 neurons, n = 4 mice) (Fig. 3a,b). This corresponds with previous results showing that NMDAEPSCs prior to and following LTP induction were not altered in the rise and decay times, or the reversal potential, at all voltage levels in control neurons (Bashir et al. 1991). Differences in L-type voltage-gated Ca2+ channels played little role in these experiments since 10 μm nimodipine did not affect wild-type LTPNMDA or LTPAMPA, nor mutant LTPAMPA levels (8 neurons, n = 4 animals; data not shown). Only the amplitude of post-tetanic potentiation was attenuated by nimodipine.
As the basal NMDA-receptor-mediated synaptic responses (expressed as the ratio of NMDAEPSCs to AMPAEPSCs) in mutant mice lacking mGluR5 were reduced slightly at the holding potentials from −40 to +60 mV (Lu et al. 1997), it was possible that NMDA channel function in mutant neurons was set initially at a depressed level prior to the tetanic stimulation and as a consequence of this set value, the threshold may be increased for LTP induction. If so, one would expect to see LTPNMDA, at saturating levels of tetanic stimulation. However, as shown in Fig. 4, LTPNMDA was not seen at all in neurons from mGluR5 mutant mice (16 neurons, n = 8 mice/group), even when the number of trains (100 Hz, 500 msec, 10sec intervals) was increased to 4 (P < 0.005). Like LTP induction in wild-type slices, the induction of LTPAMPA in mutants was blocked when D-AP5 (50 μm) was present during the tetanic stimulation (6 neurons, n = 3, data not shown). These data indicate that the basal NMDA channel was functional in mutant mice, but was uncoupled from mGluR5 activation. In addition, to avoid saturation of responses we stimulated at 25% of the maximum NMDAEPSCs and always observed single-component LTPAMPA in response to tetanic stimulation in the mutants. Presynaptic function as measured by paired pulse facilitation was normal in the mutants (Lu et al. 1997).
IMPAIRED LTPNMDA IN mGluR5 MUTANTS IN RESPONSE TO ACPD
It has been shown that selective activation of mGlu receptors with aminocyclopentane-1S,3R-dicarboxylic acid (ACPD) can evoke a long lasting enhancement of synaptic transmission that was very similar to that following tetanic stimulation in the hippocampus (Bashir et al. 1993; Bortolotto and Collingridge 1993; O’Connor 1994). We find here that ACPD application to wild-type slices induced an initial depression of both AMPAEPSCs and NMDAEPSCs, followed by a slowly rising potentiation of both components, which reached a peak amplitude at ∼15 min following ACPD application and persisted for at least 30 min, without significant decay (six neurons from four animals for each dose). At a concentration of 60 μm, the mean amplitudes of LTPNMDA and LTPAMPA at 30 min following ACPD application were measured at 160.4 ± 19.3% and 161.8 ± 21.2% respectively, in wild-type neurons (24 neurons from 12 mice) at a holding potential of −50 mV (Fig. 5a). In mutants, we found only a slight initial depression of EPSCs and a single LTPAMPA, at 156.7 ± 15.6% with no rebound of NMDA receptor-mediated EPSCs (105 ± 3.3%) following 60 μm ACPD (Fig. 5b; 25 neurons, 13 mice; P < 0.01). The responses of the initial depression and following potentiation to ACPD were never observed at a concentration of <40 μm in the mutants neurons (four neurons from three mice per group).
THE INVOLVEMENT OF PKC IN mGluR5-MEDIATED LTP
The signal downstream of mGluR5 could involve protein kinase C (PKC), since PKC-mediated NMDA-receptor phosphorylation is thought to be involved in LTPNMDA induced by tetanic stimulation, or ACPD (Hu et al. 1987; Malinow et al. 1989; Aniksztejn et al. 1995). We investigated the synaptic response to PKC activation. A protein kinase C stimulator,4β-phorbol-12,13-dibutyrate (PDBu, 20 μm) was bath applied before (Fig. 6a; 16 neurons, eight mice per group) or after tetanic stimulation (Fig. 6b; 16 neurons, eight mice per group). Prior potentiation by PDBu occluded subsequent potentiation of NMDAEPSC by tetanus. Likewise, prior tetanus occluded subsequent PDBu-induced potentiation. Therefore, tetanus and PDBu-induced LTPNMDA may share similar pathways, but other explanations are also possible. This PDBu treatment restored completely the missing LTPNMDA in mutants. Because the effect of PDBu can be pre- or postsynaptic or both, we repeated PDBu application combined with postsynaptic injection of PKC peptide inhibitor (19–36), a potent pseudosubstrate blocker of PKC activity (PKCI) (Malinow 1989; Hvalby et al. 1994). This completely blocked the effect on LTPNMDA in 4/4 CA1 neurons, but had no significant effect on LTPAMPA (data not shown). Therefore, PKC activity is at least required for LTPNMDA induced by PDBu. It is reasonable to propose that differential modulation of LTPAMPA and LTPNMDA occurs. PKC, or one of its isoforms, may be involved in LTPNMDA expression, whereas other enzymes (i.e., αCaMKII) or other isoforms of PKC could be responsible for LTPAMPA. The mutant mice lacking mGluR5 may not activate the PKC pathway required for LTPNMDA.
THE LACK OF GENETIC BACKGROUND EFFECTS ON THE LTP PHENOTYPE
We believe the phenotype ascribed to the loss of mGluR5 cannot be accounted for by the differential inheritance of background genes because (1) we find no difference in LTP in the parental 129 and CD1 strains (Jia et al. 1996); (2) the phenotype of mutants was observed in both 129 × CD1 and 129 × C57Bl/6 offspring (Table 1); (3) the backcrossing of our 129 chimera to the 129 strain showed the same results of selectively impaired LTPNMDA in the absence of mGluR5 (Table 1). Therefore, the phenotype is consistent across different genetic backgrounds.
Discussion
Our finding that LTP of the NMDA-receptor-mediated component of synaptic transmission is eliminated selectively in mGluR5 mutant mice suggests that activation of mGluR5 is required normally for the enhancement of the NMDAR component, but not the AMPAR component. This has several important implications in synaptic plasticity.
Although it is agreed generally that induction of LTP in the CA1 region of the hippocampus requires the activation of postsynaptic NMDA receptors, whether or not the expression phase of LTP is also expressed in the postsynaptic neuron, remains a matter of debate (Bliss and Collingridge 1993; Kullmann and Siegelbaum 1995; Nicoll and Malenka 1995). One approach that has been used to address the nature of LTP was to analyze separately the two components of the glutaminergic signal mediated by NMDA and AMPA receptors. Differential enhancement of these two components would support a postsynaptic locus for LTP, whereas equal potentiation of both signals would argue for a presynaptic view of LTP (Kullmann and Siegelman 1995). Some studies have shown previously equal enhancement of EPSCNMDA and EPSCAMPA following tetanic stimulation (Clark and Collingridge 1995; O’Connor et al. 1995), whereas others showed only a small EPSCNMDA (Tsien and Malinow 1990; Asztely et al. 1992; Müller et al. 1992; Xie et al. 1992; Aniksztejn and Ben-Ari 1995; Kullmann et al. 1996) or no LTPNMDA (Kauer et al. 1988; Perkel and Nicoll 1993; Selig et al. 1995). The reason for these discrepancies are not known, but clearly depend on the experimental conditions used to induce LTP. For example, strong stimulation favored the generation of LTPNMDA, whereas weak stimulation only generated LTPAMPA (Aniksztejn and Ben-Ari 1995). The use of pairing procedures rather than tetanus probably also elicited LTP differentially. In the experiments described in this paper we used a relatively strong stimulation protocol. Therefore, equal enhancement in both components after tetanus was obtained in wild-type mice of several different genetic backgrounds (129, CD1, 129XCD1, 129XC57). A similar result was obtained with agonist (ACPD) application in wild-type mice. However, in the absence of mGluR5, LTPNMDA was not observed in any neurons with either tetanic stimulation or ACPD application, whereas LTPAMPA remained intact. Therefore, activity of mGluR5 is required only for enhancement of NMDA-receptor-mediated signals during LTP. This differential utilization of mGluR5 may explain some of the discrepancies in obtaining LTPNMDA by other investigators, as described above. Since mGluR5 is localized perisynaptically (Lujan et al. 1996), activation of the receptors would be expected only when glutamate in the synaptic cleft reaches a sufficiently high level, either by strong stimulation or by a reduction in glutamate uptake. Therefore, strong LTP-inducing trains would lead to LTPNMDA, as well as LTPAMPA, as a result of the activation of mGluR5, whereas weaker stimuli, or pairing protocols, may not be sufficient to activate mGluR5. Therefore, only LTPAMPA was generated as expression of LTPAMPA does not depend on mGluR5. Our data would also explain why in a brief anoxic–aglycemic episode, LTPNMDA could be obtained much more readily, as excessive neurotransmitter release may have favored the activation of mGluR5 (Hammond et al. 1994).
The selective loss of LTPNMDA in mGluR5 mutants would support strongly the hypothesis that the underlying mechanism for the expression of LTPNMDA and LTPAMPA are distinct. Therefore, postsynaptic neurons would be the predominant locus for LTP. We cannot rule out the possibility that in mutants the properties of NMDA receptors were altered in such a way that they now have a changed sensitivity to glutamate and therefore were not able to respond to increased neurotransmitter release during LTP. This seems unlikely because the NMDA-receptor-mediated current increased as the intensity of stimulation went up, and LTPAMPA, which requires activation of NMDAR, is normal in mutants.
Finally, our data elucidates the identity of a new mGluR with respect to its role in modulating synaptic transmission and plasticity. This issue has also been controversial. Whereas some studies using MCPG, a broadly selective antagonist, showed that coactivation of an unidentified mGluR may be necessary to induce LTP (Bashir et al. 1993, Bortolotto et al. 1994), others were not able to observe any effects of MCPG on LTP (Chinestra et al. 1993; Manzoni et al. 1994). Moreover, as there are no available subtype-specific agonists or antagonists, the question of which one of the eight known mGluRs is important cannnot be answered pharmacologically. Although LTPNMDA was absent in mGluR5 −/− mice stimulated by ACPD application, LTPAMPA could still be elicited, suggesting that it is mediated by a separate mGluR. The nature of this subtype is unknown currently, but analysis of various double or triple knockouts (i.e., mGLuR5XmGluR1, mGluR5XmGluR4/2/7) would help to resolve this issue. It is also possible that the effect of ACPD in generating LTPAMPA is mediated by an unknown mGluR yet to be discovered (for review, see Pin and Duvoisin 1995). Several studies (i.e., Bortolotto and Collingridge 1995) have been reported that elucidate this postsynaptic mechanism.
The signal-transduction pathway underlying potentiation of the NMDA receptor, as a result of the activation of mGluR5 is not defined completely in the present paper, but the activation of PKC may be required. PDBu induced LTPNMDA normally in mGluR5−/−, which suggests that the G-protein coupled signaling involved in the activation of PKC may not be altered in the mutants. In addition, augmentation of NMDAEPSC by PDBu could be blocked by injecting a carefully induced dose of PKCI into the postsynaptic neuron. The blocked PKC was correlated with the lack of LTPNMDA and, therefore, it is reasonable to hypothesize that mGluR5 may be coupled to PKC activity, which in turn, is required for enhancement of the NMDAR component. Several other lines of evidence support a role for PKC in LTP. Postsynaptic injection of the selective 19–31 PKC inhibitory peptide blocked the induction of LTP in the hippocampus (Malenka et al. 1989; Malinow 1989; O’Connor 1995) and a PKCγ knockout also impaired LTP induction (Abeliovitch et al. 1993). Conversely, the injection of PKC into CA1 pyramidal neurons induced a kind of LTP (Hu et al. 1987). PKC may enhance LTP by upregulating the function of the NMDAR (Ben-Ari et al. 1992). However, as the regulation of NMDA receptors can be very complex, potentially involving additional signaling pathways such as protein tyrosine kinases (Wang and Salter 1994; Yu et al. 1997), activation of an independent pathway could well compensate for the loss of the signal caused by lack of mGluR5. Whether or not the IP3 signal generated by the activation of mGluR5 is also important for LTPNMDA is unclear. Dissecting the details of these components and how they are linked to the potentiation of the NMDA receptor will be the focus of future studies.
In summary, we have generated a novel mutant strain deficient in expression of mGluR5 but normal in the expression of other glutamate receptors. Analysis of the mutant mice showed that mGluR5 is not required for the development of the CNS. An earlier study analyzing field potentials in these mice showed a small decrease in the induction of LTP in CA1 and the dentate gyrus but normal induction in the CA3 region of the hippocampus (Lu et al. 1997). The difference between genotypes was abrogated by adding AP5 after the induction of LTP (Lu et al. 1997). We show here using voltage clamp analysis that the expression phase of LTPNMDA is completely lost, whereas LTPAMPA is normal. The selective loss of LTPNMDA in mGluR5−/− mice, suggests that distinct mechanisms must exist in CA1 neurons for LTPNMDA and LTPAMPA components of LTP. Therefore, these mutant mice provide a valuable new tool to dissect the various elements that are used differentially during synaptic plasticity in the mammalian CNS. In addition, the mice show a selective loss in two tasks of spatial learning and memory which supports the role of CA1 in this process as well (Lu et al. 1997).
Acknowledgments
All the electrophysiological recordings presented here were performed by YouMing Lu. This work was supported by the Medical Research Councils, (The Ontario Mental Health Foundation (OMHF), National Institutes of Health, and Research to Prevent Blindness, Inc. H. Mishina for the rat mGluR5 probe; R. Wenthold and D. Hampson for antibodies, and Tatiana Tenkova for the immunocytochemistry. Z. J. was an OMHF fellow.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
References
- Abe T, Sugihara H, Nawa H, Shigemoto R, Mizumo W, Nahanishi J. Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate Ca2+ signal transduction. Biol Chem. 1992;267:13361–13368. [PubMed] [Google Scholar]
- Abeliovitch A, Chen C, Goda Y, Silva A, Stevens C, Tonegawa S. Modified hippocampal long-term potentiation in PKC-K-mutant mice. Cell. 1993;75:1253–1262. doi: 10.1016/0092-8674(93)90613-u. [DOI] [PubMed] [Google Scholar]
- Aiba A, Chen C, Herrup K, Rosenmund C, Stevens CF, Tonegawa S. Reduced hippocampal long-term potentiation and context-specific deficit in associative learning in mGluR1 mutant mice. Cell. 1994;79:365–375. doi: 10.1016/0092-8674(94)90204-6. [DOI] [PubMed] [Google Scholar]
- Aniksztejn L, Ben-Ari Y. Expression of LTP by AMPA and/or NMDA receptors is determined by the extent of NMDA receptor activation during the tetanus. J Neurophysiol. 1995;74:2349–2357. doi: 10.1152/jn.1995.74.6.2349. [DOI] [PubMed] [Google Scholar]
- Asztély F, Wigström H, Gustafsson B. The relative contribution of NMDA receptor channels in the expression of long term potentiation in the hippocampal CA1 region. Eur J Neurosci. 1992;4:681–690. doi: 10.1111/j.1460-9568.1992.tb00177.x. [DOI] [PubMed] [Google Scholar]
- Bashir ZI, Alford S, Davies SN, Randall AD, Collingridge GL. Long-term potentiation of NMDA receptor-mediated synaptic transmission in the hippocampus. Nature. 1991;349:156–158. doi: 10.1038/349156a0. [DOI] [PubMed] [Google Scholar]
- Bashir ZI, Bortolotto ZA, Davies CH, Beretta N, Irving A, Seal AJ, Henley JM, Jane DE, Watkins JC, Collingridge GL. Induction of LTP in the hippocampus needs synaptic activation of glutamate metabotropic receptors. Nature. 1993;363:347–350. doi: 10.1038/363347a0. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Aniksztejn L, Bregestovski P. Protein kinase c modulation of NMDA currents: An important link for LTP induction. Trends Neurosci. 1992;15:333–339. doi: 10.1016/0166-2236(92)90049-e. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL. A synaptic model of memory: Long term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bortolotto ZA, Collingridge GL. Characterization of LTP induced by the activation of glutamate metabotropic receptors in area CA1 of the hippocampus. Neuropharmacology. 1993;32:1–9. doi: 10.1016/0028-3908(93)90123-k. [DOI] [PubMed] [Google Scholar]
- Bortolotto ZA, Collingridge GL. On the mechanism of long-term potentiation induced by (1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid (ACPD) in rat hippocampal slices. Neuropharmacology. 1995;34:1003–1014. doi: 10.1016/0028-3908(95)00054-a. [DOI] [PubMed] [Google Scholar]
- Bortolotto ZA, Bashir ZI, Davies CH, Collingridge GL. A molecular switch activated by metabotropic glutamate receptors regulates induction of long term potentiation. Nature. 1994;368:740–743. doi: 10.1038/368740a0. [DOI] [PubMed] [Google Scholar]
- Chinestra P, Aniksztejn L, Diabira D, Ben-Ari Y. RS-I-methyl-4-carboxyphenylglycine neither prevents induction of LTP nor antagonizes metabotropic glutamate receptors in CA1 hippocampal neurons. J Neurophysiol. 1993;70:2684–2689. doi: 10.1152/jn.1993.70.6.2684. [DOI] [PubMed] [Google Scholar]
- Clark KA, Collingridge GL. Synaptic potentiation of dual-component excitatory post-synaptic currents in the rat hippocampus. J Physiol. 1995;482:39–52. doi: 10.1113/jphysiol.1995.sp020498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, Franz-Bacon K, Reggiani A, Matarese V, Condé F, Collingridge GL, Crépel F. Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature. 1994;372:237–243. doi: 10.1038/372237a0. [DOI] [PubMed] [Google Scholar]
- Hammond C, Crepel V, Gozlan H, Ben-Ari Y. Anoxic LTP sheds light on the multiple facets of NMDA receptors. Trends Neurosci. 1994;17:497–503. doi: 10.1016/0166-2236(94)90140-6. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Sekiyama N, Nakanishi S, Jane DE, Sunter DC, Birse EF, Udvarhelyi PM, Watkins JC. Analysis of agonist and antagonist activities of phenylglycine derivatives for different cloned metabotropic glutamate receptors. J Neurosci. 1994;14:3370–3377. doi: 10.1523/JNEUROSCI.14-05-03370.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hestrin S, Nicoll RA, Perkel DJ, Soh P. Analysis of excitatory synaptic action in pyramidal cells using whole-cell recordings from rat hippocampal slices. J Physiol (London) 1990;422:203–225. doi: 10.1113/jphysiol.1990.sp017980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu GH, Hvalby Ø, Walaas SI, Albert KA, Skjeflo P, Andersen P, Greengard P. Protein kinase C injection into pyramidal cells elicits features of long-term potentiation. Nature. 1987;328:426–429. doi: 10.1038/328426a0. [DOI] [PubMed] [Google Scholar]
- Hvalby Ø, Hemmings HC, Czernik AJ, Nairn AC, Godfraind JM, Jensen V, Raastad M, Storm JF, Andersen P, Greengard P. Specificity of protein kinase inhibitor peptides and induction of long-term potentiation. Proc Natl Acad Sci. 1994;91:4761–4765. doi: 10.1073/pnas.91.11.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia ZP, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna F, Velumian A, MacDonald J, Carlen P, Abramow-Newerly W, Roder J. Enhanced LTP in mice deficient in the AMPA receptor, GluR2. Neuron. 1996;17:945–956. doi: 10.1016/s0896-6273(00)80225-1. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC, Nicoll RA. A persistent post-synaptic modification mediated long term potentiation in the hippocampus. Neuron. 1988;1:911–917. doi: 10.1016/0896-6273(88)90148-1. [DOI] [PubMed] [Google Scholar]
- Kingston AE, Burnett JP, Mayne NG, Lodge D. Pharmacological analysis of 4-carboxyphenylglycine derivatives:comparison of effects on mGluR1I and mGluR5a subtypes. Neuropharmacology. 1995;34:887–894. doi: 10.1016/0028-3908(95)00069-i. [DOI] [PubMed] [Google Scholar]
- Kullmann D, Erdemli G, Asztély F. LTP of AMPA and NMDA receptor-mediated signals: Evidence for pre-synaptic expression and extrasynaptic glutamate spill-over. Neuron. 1996;17:461–474. doi: 10.1016/s0896-6273(00)80178-6. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Siegelbaum SA. The site of expression of NMDA receptor-dependent LTP: New fuel for an old fire. Neuron. 1995;15:997–1002. doi: 10.1016/0896-6273(95)90089-6. [DOI] [PubMed] [Google Scholar]
- Lu YM, Jia Z, Janus C, Henderson J, Gerlai R, Wojtowicz M, Roder J. Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP), but normal CA3-LTP. J Neurosci. 1997;17:5196–5205. doi: 10.1523/JNEUROSCI.17-13-05196.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan R, Nusser H, Shigemoto R, Roberts JD, Ohishi H, Nusser Z, Somogyi P. Peri-synaptic localization of metabotropic glutamate receptors in mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci. 1996;8:1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA, Waxham MN. An essential role for post-synaptic calmodulin and protein kinase activity in LTP. Nature. 1989;340:554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW. Inhibition of post-synaptic PKC or CamKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Manzoni OJ, Weisskopf MG, Nicoll RA. MCPG antagonizes metabotropic glutamate receptors but not long-term potentiation in the hippocampus. Eur J Neurosci. 1994;6:1050–1054. doi: 10.1111/j.1460-9568.1994.tb00599.x. [DOI] [PubMed] [Google Scholar]
- McGuiness W, Anwyl R, Rowan R. Trans-ACPD enhances LTP in hippocampus. Eur J Pharmacol. 1991;197:231–232. doi: 10.1016/0014-2999(91)90529-y. [DOI] [PubMed] [Google Scholar]
- Minakami R, Katsuki F, Sugiyama H. A variant of metabotropic glutamate receptor subtype 5: An evolutionally conserved insertion with no termination codon. Biochem Bioph Res Commun. 1993;194:622–627. doi: 10.1006/bbrc.1993.1866. [DOI] [PubMed] [Google Scholar]
- Müller D, Arai A, Lynch G. Factors governing the potentiation of NMDA receptor-mediated responses in hippocampus. Hippocampus. 1992;2:29–38. doi: 10.1002/hipo.450020105. [DOI] [PubMed] [Google Scholar]
- Musgrave MA, Ballyk BA, Goh JW. Co-activation of metabotropic and NMDA receptors is required for LTP induction. NeuroReport. 1993;4:171–191. doi: 10.1097/00001756-199302000-00014. [DOI] [PubMed] [Google Scholar]
- Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early passage embryonic stem cells. Proc Natl Acad Sci. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC. Contrasting properties of two forms of LTP in the hippocampus. Nature. 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- O’Connor JJ, Rowan MJ, Anwyl R. Long-lasting enhancement of NMDA receptor-mediated synaptic transmission by metabotropic glutamate receptor activation. Nature. 1994;367:557–559. doi: 10.1038/367557a0. [DOI] [PubMed] [Google Scholar]
- O’Connor JJ, Rowan MJ, Anwyl R. Tetanically induced LTP involves a similar increase in the AMPA and NMDA receptor components of the excitatory post-synaptic current: Investigations of the involvement of mGlu receptors. J Neurosci. 1995;15:2013–2020. doi: 10.1523/JNEUROSCI.15-03-02013.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani S, Ben-Ari Y. Metabotropic receptor-mediated LTP in rat hippocampal slices. Eur J Pharmacol. 1991;205:325–326. doi: 10.1016/0014-2999(91)90920-l. [DOI] [PubMed] [Google Scholar]
- Perkel DJ, Nicoll RA. Evidence for all-or-none regulation of neurotransmitter release: Implications for long-term potentiation. J Physiol (Lond.) 1993;471:481–500. doi: 10.1113/jphysiol.1993.sp019911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin J, Duvoisin R. The metabotropic glutamate receptors: Structure and function. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- Richter-Levin G, Errington ML, Megawe H, Bliss TVP. Activation of metabotropic glutamate receptors is necessary for long-term potentiation in the dentate gyrus and for spatial learning. Neuropharmacology. 1994;33:853–857. doi: 10.1016/0028-3908(94)90181-3. [DOI] [PubMed] [Google Scholar]
- Riedel G, Reymann K. An antagonist of the metabotropic glutamate receptor prevents LTP in the dentate gyrus of freely moving rats. Neuropharmacology. 1993;32:929–931. doi: 10.1016/0028-3908(93)90149-w. [DOI] [PubMed] [Google Scholar]
- Riedel G, Witzel W, Reymann KG. MCPG blocks spatial learning in rats and LTP in the dentate gyrus in vivo. Neurosci Lett. 1994;167:141–144. doi: 10.1016/0304-3940(94)91047-2. [DOI] [PubMed] [Google Scholar]
- Romano C, Sesmu M, McDonald CT, O’Malley K, Van der Pol A, Olney J. Distribution of metabotropic glutamate receptor in GluR5 immunoreactivity in rat brain. J Comp Neurol. 1995;355:455–469. doi: 10.1002/cne.903550310. [DOI] [PubMed] [Google Scholar]
- Selig DK, Hjelmstad GO, Herron C, Nicoll RA, Malenka RC. Independent mechanisms for long-term depression of AMPA and NMDA responses. Neuron. 1995;15:417–426. doi: 10.1016/0896-6273(95)90045-4. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Nomura S, Ohishi H, Sugihara H, Nakanishi S, Mizuno N. Immunohistochemical localization of a metabotropic glutamate receptor, mGluR5, in the rat brain. Neurosci Lett. 1993;163:53–57. doi: 10.1016/0304-3940(93)90227-c. [DOI] [PubMed] [Google Scholar]
- Spruston N, Jaffe DB, Williams SH, Johnston D. Voltage-clamp and space-clamp errors associated with the measurement of electronically remote synaptic events. J Neurophysiol. 1993;70:781–802. doi: 10.1152/jn.1993.70.2.781. [DOI] [PubMed] [Google Scholar]
- Tsien RW, Malinow R. LTP: Pre-synaptic enhancement following post-synaptic activation of Ca2+ dependent protein kinase. Cold Spring Harbor Symp Quant Biol. 1990;45:147–1599. doi: 10.1101/sqb.1990.055.01.018. [DOI] [PubMed] [Google Scholar]
- Wang YT, Salter MW. Regulation of NMDA receptors by tyrosine kinases and phosphatase. Nature. 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- Wood SA, Allen ND, Rossant J, Auerbach A, Nagy A. Non-injection methods for the production of embryonic stem cell embryo chimeras. Nature. 1993;365:87–89. doi: 10.1038/365087a0. [DOI] [PubMed] [Google Scholar]
- Xie X, Berger TW, Barionuevo G. Isolated NMDA receptor- mediated synaptic responses express both LTP and LTD. J Neurophysiol. 1992;67:1009–1013. doi: 10.1152/jn.1992.67.4.1009. [DOI] [PubMed] [Google Scholar]
- Yu XM, Askalan R, Keil GJ, Salter MW. NMDA channel regulation by channel-associated protein tyrosine kinase src. Science. 1997;275:674–677. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]