

Non-technical summary
In heart muscle cells, fluctuations of intracellular calcium (Ca2+) concentration ([Ca2+]i) at the frequency defined by the heart rate induce contractions of the cells. Over a longer timescale the same fluctuations define the properties of the cells by regulating expressions of specific genes through the activation of a variety of cellular enzymes. In this study, we have characterized a specific cell signalling pathway, explaining how [Ca2+]i regulates the expression of the L-type calcium channel (LTCC). We show that [Ca2+]i-activated calmodulin kinase II (CaMKII) activates downstream regulatory element (DRE) binding transcription factor DREAM, which consequently suppresses the expression of LTCCs. By experiments and mathematical modelling we demonstrate that the LTCC downregulation through the Ca2+–CaMKII–DREAM cascade constitutes a physiological feedback mechanism enabling cardiomyocytes to adjust the calcium intrusion through LTCCs to the amount of intracellular calcium detected by CaMKII.
Abstract
Abstract
Recent studies have demonstrated that changes in the activity of calcium–calmodulin-dependent protein kinase II (CaMKII) induce a unique cardiomyocyte phenotype through the regulation of specific genes involved in excitation–contraction (E–C)-coupling. To explain the transcriptional effects of CaMKII we identified a novel CaMKII-dependent pathway for controlling the expression of the pore-forming α-subunit (Cav1.2) of the L-type calcium channel (LTCC) in cardiac myocytes. We show that overexpression of either cytosolic (δC) or nuclear (δB) CaMKII isoforms selectively downregulate the expression of the Cav1.2. Pharmacological inhibition of CaMKII activity induced measurable changes in LTCC current density and subsequent changes in cardiomyocyte calcium signalling in less than 24 h. The effect of CaMKII on the α1C-subunit gene (Cacna1c) promoter was abolished by deletion of the downstream regulatory element (DRE), which binds transcriptional repressor DREAM/calsenilin/KChIP3. Imaging DREAM–GFP (green fluorescent protein)-expressing cardiomyocytes showed that CaMKII potentiates the calcium-induced nuclear translocation of DREAM. Thereby CaMKII increases DREAM binding to the DRE consensus sequence of the endogenous Cacna1c gene. By mathematical modelling we demonstrate that the LTCC downregulation through the Ca2+–CaMKII–DREAM cascade constitutes a physiological feedback mechanism enabling cardiomyocytes to adjust the calcium intrusion through LTCCs to the amount of intracellular calcium detected by CaMKII.
Introduction
Heart muscle cells are capable of adapting to both acute and chronic changes in the circulatory demands of the body. Short-term adaptation is mediated by phosphorylation and de-phosphorylation of cytosolic proteins involved in contraction, excitability and signalling. Over the long term, these same kinases and phosphatases shape the cardiac phenotype through activation of a myriad of transcription factors. One of the central pathways is mediated by calcium–calmodulin-dependent protein kinase II (CaMKII) which decodes changes in [Ca2+]i into corresponding levels of kinase activity. In cardiac muscle cells CaMKII phosphorylates cytosolic targets such as L-type calcium channels (LTCCs), ryanodine receptors (RyRs) and the sarcoplasmic reticulum (SR) Ca2+-ATPase (SERCA)-regulating protein phospholamban to establish positive feedback between cardiomyocyte beating rate and calcium signalling through calcium-dependent facilitation of plasmalemmal calcium flux and enhanced SR calcium dynamics (Frey et al. 2000; Maier, 2009). However, in the case of the cytosolic CaMKII activity becoming continuously exaggerated, this physiological feedback becomes maladaptive. This was demonstrated by acute overexpression of cytosolic CaMKII, which through excessive phosphorylation of CaMKII target proteins caused changes in the cardiomyocyte calcium signalling resembling those seen in cardiac failure models (Kohlhaas et al. 2006). Furthermore, long-term potentiation of CaMKII activity by transgenic overexpression of the cytosolic isoform of CaMKII (CaMKIIδC) leads to cardiac hypertrophy and failure in mice through reprogramming of cardiomyocyte gene expression (Zhang et al. 2003).
Increases in the expression and activity of the endogenous CaMKII are seen in a wide variety of animal models of cardiac hypertrophy and failure (Zhang & Brown, 2004), and consequently ablation of CaMKIIδ has been shown to prevent load-induced cardiac hypertrophy and remodelling in mice (Backs et al. 2009). Several lines of evidence indicate that transgenic overexpression of CaMKII in mouse heart can recruit the expression of hypertrophic and fetal cardiac genes. Furthermore, overexpression of the nuclear CaMKIIδB isoform alone is able to induce a pattern of genes that leads to development of cardiac hypertrophy with signs of dilated cardiomyopathy and failure in mice (Zhang et al. 2002). These effects may be due to CaMKII interaction with the family of class II histone deacetylases (HDACs), especially HDAC4, acting as a holistic transcriptional repressor of cardiac hypertrophy and fetal gene expression (Backs et al. 2006; Little et al. 2007; McKinsey, 2007). CaM-kinases have also been shown to regulate a plethora of transcription factors including CREB, CBP and SRF (Ikura et al. 2002) and CaMK-dependent myocyte-enhancing-factors 2 (MEF2a, -b, -c and -d isoforms), which are transcription factors controlling myogenesis, muscle hypertrophy and mitochondrial function (Frey et al. 2000). In addition, CaMKII seems to have specific targets among cardiac genes, such as cardiomyocyte hypertrophy markers A- and B-type natriuretic factors (Ramirez et al. 1997; Ronkainen et al. 2007), and chronic CaMKII overexpression induces a unique cardiomyocyte phenotype related to changes in the expression of genes involved in excitation–contraction coupling (E–C-coupling) (Maier et al. 2003). This suggests a mechanism that mediates subtle regulation of defined genes among the genes involved in E–C-coupling. Interestingly, it was recently demonstrated that ventricular myocytes of CaMKIIδ knockout mice have increased L-type Ca2+ channel current density due to increased expression of the gene (Cav1.2) coding the pore-forming subunit of the channel (Xu et al. 2010), but the mechanism behind this phenomenon has remained unknown.
The aim of the present study was to find out the mechanisms behind the CaMKII-induced transcriptional modifications of the cardiomyocyte E–C-coupling. We studied CaMKII-induced changes in gene expression, calcium currents and calcium signals of cardiomyocytes. Gene expression mechanisms were studied by identifying CaMKII-regulated transcription factors by analysing native and mutated target gene promoter fragments, followed by measurement of transcription factor binding on the native gene. Physiological implications of the described mechanisms were demonstrated by implementing the experimental data into a mathematical ventricular cell model. The new mathematical model with a transcriptional feedback loop allowed us to simulate the impact of the CaMKII-induced transcriptional changes on the cardiomyocyte E–C-coupling.
Methods
Ethical approval
All procedures were carried out in compliance with the policies and regulations set out in Drummond (2009) and the experimental designs were approved by the Animal Use and Care Committee of the University of Oulu.
Myocyte isolation and cell culture
Rat neonatal ventricular cardiomyocytes were isolated 1–2 days after birth. The animals (n = 196) were killed by cervical dislocation and the ventricles were excised and cut into small pieces and thereafter incubated 1 h in a solution containing 100 mm NaCl, 10 mm KCl, 1.2 mm KH2PO4, 4.0 mm MgSO4, 50 mm taurine, 20 mm glucose, 10 mm Hepes, 2 mg ml−1 collagenase type 2 (Worthington, Lakewood, NJ, USA), 2 mg ml−1 pancreatin (Sigma-Aldrich, St Louis, MO, USA) and 1% penicillin–streptomycin (PS). After incubation the detached cells were collected in 15 ml Falcon tubes and centrifuged for 5 min at 160 g. The supernatant and the top layer of the pellet containing damaged cells were discarded and the isolated cardiomyocytes were plated on 35 mm fibronectin-coated dishes at a density of 780 cells mm−2. The cells were cultured to reach confluence in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% PS for 48–72 h after which the plasmid transfections and pharmacological interventions took place.
The HL-1 cardiomyocytes line was a kind gift from Dr William Claycomb (Louisiana State University, New Orleans, USA). The cell culture was maintained as described earlier (Claycomb et al. 1998). Briefly, the cells were cultured in Claycomb medium (Sigma-Aldrich), supplemented with 10% FBS, 2 mm l-glutamine, 100 μm noradrenaline and 1% PS. The medium was changed daily; passages were performed every 4 days. The cells were plated on fibronectin-coated dishes at a density of 210 cells mm−2. The temperature in the culture chamber was 37°C with 5% CO2 and 95% air atmosphere. The transfections and pharmacological interventions with KN-93 (Calbiochem, San Diego, CA, USA) or KN-92 (Calbiochem) took place 48–72 h after passage.
H9c2(2-1) cells, derived from embryonic rat ventricle (American Type Cell Culture Collection (ATCC), Manassas, VA, USA), were grown according to ATCC's instructions in DMEM (ATCC) supplemented with 10% FBS (HyClone, Logan, UT, USA) and 1% PS (100 U ml−1 penicillin and 100 μg ml−1 streptomycin) (Sigma-Aldrich). The cells were plated on culture dishes at a density of 260 cells mm−2. The transfections and pharmacological interventions with Ca2+ ionophore A23187 (Teflabs, Austin, TX, USA) took place 48–72 h after passage. The temperature in the culture chamber was 37°C with 5% CO2 and 95% air atmosphere.
Overexpression plasmids
To specifically increase either cytosolic or nuclear CaMKII activity in the cultured cells, an expression vector coding the nuclear (pCaMKIIδB) or cytosolic (pCaMKIIδC) CaMKIIδ splicing variant was used (Ramirez et al. 1997). The expression plasmids were kind gifts from Dr Joan Brown (University of California, San Diego, USA). To increase CaMKII activity independent of [Ca2+]i, an expression vector coding the constitutively active T286D mutant CaMKIIα (pCaMKII-T286D) (Brickey et al. 1994) was used. The plasmid was a kind gift from Dr K. Ulrich Bayer (University of Colorado, Aurora, USA). To study the subcellular localization of DREAM we used a plasmid coding a GFP-DREAM fusion protein (Matsuda et al. 2006) kindly donated by Dr Miho Matsuda (Kyushu University, Fukuoka, Japan). To produce a DREAM overexpression plasmid without GFP, the DREAM insert was digested out of the GFP-DREAM fusion vector with HindIII and KpnI and subcloned into pBS-SK (Stratagene, La Jolla, CA, USA). The DREAM insert was then digested out of pBS-SK with EcoRI and KpnI and ligated to the pSRα vector (Brickey et al. 1994). The integrity of the resulting plasmid was confirmed by sequencing.
Transfection of cultured cells
Cultured H9c2 and HL-1 cells and neonatal cardiomyocytes were transiently transfected 48 h after passage/isolation with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Briefly, the cells cultured on 35 mm dishes were kept in 750 μl of Opti-MEM (Invitrogen) containing 10 μl of Lipofectamine and 8 μg of plasmid DNA for 5 h, after which the transfection medium was replaced with normal culture medium. For 12-well plates the amounts were 300 μl, 4 μl and 3.2 μg per well, respectively. The transfection efficiency evaluated by transfecting pEGFP-N1 (Clontech, Mountain View, CA, USA) was routinely approximately 60% in HL-1 cells, 30% in neonatal cardiomyocytes and 80% in H9c2 cells after 48 h. COS-1 cells were plated on 6-well plates 350 cells mm−2 and maintained for 24 h in DMEM containing 10% FBS and 1% PS before transfection with 1 μg of DREAM overexpression plasmid and 3 μl FuGENE 6 (Roche, Indianapolis, IN, USA) reagent per well.
Preparation of the Cacna1c promoter activity reporter constructs
The original full-length (−2014–(+31)) and D2 (−764–(+31)) Cacna1c promoter constructs were kind gifts from Dr Marsh (Wayne State University, Detroit, USA) (Liu et al. 2000). Because the D2 construct displayed CaMKII-mediated repression and the majority of this construct's activity lies in its distal region (Fan et al. 2002), a series of proximal deletion mutants was made by PCR using primers within the D2 construct sequence with HindIII and SacI linkers, yielding transcripts of 171, 228, 357, 484 and 594 bp. The shortened Cacna1c promoter regions were ligated to a minimal tyrosine kinase promoter containing a luciferase reporter gene (pT81-Luc) (Nordeen, 1988). The forward primer with a HindIII linker underlined was: 5′-AAGCTTGACTAACGCATTCCGGGGGGC-3′. The reverse primers with SacI linkers underlined, numbered according to their position within the Cacna1c construct, were: −594, 5′-CGAGCTCGGTTTTCTGTTGTTTTTCTGC-3′; −537, 5′-CGAGCTCGGTTCTAAGCAATGACAAACTG-3′; −408, 5′-CGAGCTCGTTTTATCTTTCCCTCTCCC-3′; −281, 5′-CGAGCTCGGAATGTGTGCTTCCATGGAG-3′; −171, 5′-CGAGCTCGGTCTTTGTATATCAGCACAG-3′. Mutations to the original constructs were carried out with the QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions. All plasmids were sequenced to confirm the presence and integrity of the promoter segments and mutations.
Luciferase reporter assays
The Cacna1c promoter constructs expressing the luciferase gene were co-transfected with the pRL-TK-luciferase control plasmid (Promega, Madison, WI, USA) and pCaMKIIδB to study the mechanism of CaMKII-mediated Cacna1c gene expression regulation. Luciferase signals were measured and normalized with the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. For the luciferase construct transfections, cells were transfected on 12-well plates with 1.6 μg of Cacna1c-luc constructs; 1.6, 0.4 or 0.1 μg of pCaMKIIδB or empty control plasmid pUC18 and 0.2 μg of pRL-TK per well.
Intracellular calcium measurements
The intracellular Ca2+ signals of the HL-1 cells and neonatal rat cardiomyocytes were recorded in normal Tyrode solution containing 130 mm NaCl, 4 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 10 mm Hepes and 10 mm glucose (pH adjusted to 7.35 with NaOH) using Fluo-3 Ca2+-indicator. Cells were incubated with 5 μm Fluo-3-AM (Molecular Probes) at room temperature for 20 min or with 2 μm Fluo-4-AM (Invitrogen) for 20 min at 37°C. A pre-incubation period of 30 min was used to ensure de-esterification of Fluo-3-AM or Fluo-4-AM and to exclude direct effects of KN-93. [Ca2+]i elevations were produced by application of 10 mm caffeine (Sigma-Aldrich) or by application of 5 μm of the L-type Ca2+ channel opener Bay K 8644 (Sigma-Aldrich). The fluorescence signals were detected with an Olympus FV1000 confocal microscope (excitation: 488 nm, detection: 510–610 nm). Laser intensity was minimized to reduce photobleaching and phototoxicity. Line-scanning was performed to obtain a sufficient sampling rate of 100 Hz for detection of fast changes in the intracellular Ca2+ concentration. Fluo fluorescence intensity is expressed as an F/F0-ratio, where F is the background subtracted fluorescence intensity and F0 is the background subtracted minimum fluorescence value measured from each cell at rest.
For intracellular Ca2+ measurements H9c2 cells were loaded with 2 μm Fluo-4 acetoxy methyl ester (Fluo-4-AM; Invitrogen) at 37°C for 20 min, rinsed twice and pre-incubated for another 30 min at 37°C before measurements. During measurements H9c2 cells were perfused with 5% CO2–95% O2 gas mix-bubbled 35°C DMEM containing different concentrations of A23187. Full-frame images of the cells were taken with an Olympus FV1000 laser-scanning confocal microscope every minute after application of A23187. The cells were excited at 488 nm wavelength and fluorescence was detected at 510–610 nm. The intracellular Ca2+ concentration of the control cells was calculated using min–max calibration with solutions containing 110 mm NaCl, 5.4 mm KCl, 0.9 mm MgCl2, 44 mm NaHCO3, 25 mm glucose and either 5 mm EGTA (min calibration) or 15 mm CaCl2 (max calibration). In experiments involving manipulation of [Ca2+]i with A23187, only max calibration was conducted after measurements. In these experiments the [Ca2+]i was calculated based on the known baseline [Ca2+]i of the H9c2 cells.
Measurement of the L-type calcium channel current
For whole-cell patch-clamp Ca2+ current recordings, single HL-1 cells were enzymatically isolated from the KN-93-treated and control dishes. Cultured HL-1 cells were rinsed with solution containing 100 mm NaCl, 10 mm KCl, 1.2 mm KH2PO4, 4 mm MgSO4, 50 mm taurine, 20 mm glucose and 10 mm Hepes (pH adjusted to 6.9 with KOH) and gently shaken in the same solution with 2 mg ml−1 collagenase type 2 (Worthington) for 20–30 min in 37°C. The isolated cells were centrifuged for 4 min at 750 g and plated on dishes treated with laminin. The cells were stored in HL-1 cell growth medium at 37°C and used within 9 h after the isolation. Whole-cell Ca2+ currents were recorded at 25°C. The normal Tyrode solution containing 146 mm NaCl, 4.5 mm KCl, 1.1 mm CaCl2, 1 mm MgCl2, 10 mm glucose and 10 mm Hepes (pH adjusted to 7.4 with NaOH) was used as the initial bath solution. Patch-clamp pipettes with a resistance of 2–5 MΩ were filled with solution containing 120 mm caesium aspartate, 10 mm TEA-Cl, 10 mm EGTA, 1 mm MgATP, 5 mm sodium phosphocreatine, 1 mm NaGTP and 10 mm Hepes (pH adjusted to 7.2 with CsOH). After gaining intracellular access, the normal Tyrode solution was changed to a solution containing 140 mm TEA-Cl, 5 mm CaCl2, 2 mm MgCl2, 10 mm Hepes and 10 mm glucose (pH adjusted to 7.4 with TEA-OH) (Yang et al. 2005). The cell membrane capacitance was measured by applying a 5 mV pulse to the holding potential. The L-type Ca2+ current was measured by applying a command potential from the −40 mV holding potential. Cells also displaying the T-type Ca2+ current were not used in the analysis. A cell was considered to contain the T-type Ca2+ current if the Ca2+ current was activated with a –80 mV to –40 mV clamp. Whole-cell currents were filtered at 2 kHz and acquired at 10 kHz. Clampex 9.2 software, an Axopatch-1D amplifier and Digidata 1322A A/D-D/A (Axon Instruments, Union City, CA, USA) were used for controlling the command potentials and data acquisition. Data analysis was made using Clampfit 9.2 (Axon Instruments) and Origin 7.5 (OriginLab Corporation, Northampton, MA, USA). The membrane currents were scaled by dividing them with the cell membrane capacitance.
Immunofluorescence labelling and microscopy of HL-1 cells
For the immunolabelling of haemagglutinin-tagged CaMKIIδ and L-type calcium channel α1c-subunits, commercial anti-HA FITC-conjugate (Sigma-Aldrich, Saint Louis, MO, USA) and anti-α1c (Santa Cruz Biotechnology, Santa Cruz, CA, USA) antibodies were used. For the secondary labelling of the α1c-antibodies, chicken anti-rabbit-IgG Alexa 488 conjugate was used (Molecular Probes). After 24 h treatment with 3 μm KN-93 or transfection with haemagglutinin-tagged CaMKIIδ-isoforms, the HL-1 cells were rinsed with 0.1 m Tris-HCl, pH 7.3, fixed with 3% paraformaldehyde for 2 min, and permeabilized for 10 min with 0.5% Triton X-100 (Sigma-Aldrich). After washing with 0.1 m Tris-HCl, pH 7.3 twice for 5 min, the primary antibody (anti-HA-FITC or anti-α1c) was incubated for 1 h in 0.1 m Tris-HCl, pH 7.3 containing 10% FBS and 0.05% Triton X-100. For the secondary labelling of the α1c antibodies, the specimens were again washed twice and the secondary antibody (Alexa Fluor 488 chicken anti-rabbit IgG) was incubated (0.1 m Tris-HCl, pH 7.3) for 1 h. After the antibody incubations the specimens were rinsed twice for 10 min before the microscopic studies. The dilution for all the antibodies used was 1:750 in all groups. After labelling, images were taken freshly with an Olympus FV1000 confocal microscope with a 488 nm excitation wavelength and detection at 510–610 nm. For the quantification of the α1c protein levels, a background signal from samples incubated without the primary antibody was subtracted and the fluorescence levels were scaled to the control.
DREAM immunoblotting
The nuclear proteins were extracted as described earlier (Schreiber et al. 1989). Western blots of the nuclear extracts were performed using an anti-DREAM antibody (Upstate, Lake Placid, NY, USA). Samples (10 μg) were loaded onto an SDS-PAGE gel and transferred to nitrocellulose filters. The membranes were blocked in 5% non-fat milk and incubated with the indicated primary antibody overnight. The protein levels were detected by enhanced chemiluminescence.
RNA preparation and quantitative RT-PCR
Total RNA was isolated from HL-1 cells using the QuickPrep Total RNA Extraction Kit (Amersham Pharmacia Biotech). RNA was used as the template for cDNA synthesis in reverse transcriptase reactions (First Strand cDNA Synthesis Kit, MBI Fermentas). The quantitative PCR reactions were performed with ABI 7700 Sequence Detection System (Applied Biosystems, USA) using the TaqMan chemistry. The forward and reverse primer and bifunctional fluorogenic probe sequences used were: L-type calcium channel α1c-subunit (rat): 5′-TTGACAATGTTCTGGCAGCC-3′, 5′-TCTGGCCACCCCTCGA-3′ and 5′-Fam-TGATGGCCCTCTTTACCGTCTCCACC-Tamra-3′ (GenBank accession No. NM_009781); L-type calcium channel α1c-subunit (mouse): 5′-TTGACAATGTTTTGGCAGCC-3′, 5′-TCTGGCCACCCTTCGA-3′ and 5′-FamTGATGGCTCTCTTCACCGTCTCCACC-Tamra-3′ (NM_009781); L-type calcium channel β2-subunit (mouse): 5′-CCCGTCAAAAAATCCCAACA-3′, 5′-TCCCGCTGCGGTGGT-3′ and 5′-Fam-CGTTCCTCCTCAGCCACACACCAA-Tamra-3′ (NM_023116); sarcoplasmic reticulum Ca2+-ATPase isoform 2a (mouse): 5′-CAGCCATGGAGAACGCTCA-3′, 5′-TCGTTGACCCCGAAGTGG-3′ and 5′-Fam-ACAAAGACCGTGGAGGAGGTGCTGG-Tamra-3′ (NM_009722); ryanodine receptor isoform 2 (mouse): 5′-CAGCAGCCCCCACAGG-3′, 5′-TTCCATGTAGCCGCTGCTC-3′ and 5′-Fam-TCATTGCGGTTCACTATGTCCTGGAGG-Tamra-3′ (NM_023868); Na+/Ca2+-exchanger isoform 1 (mouse): 5′-TTGTTTTCCCATGTTGACCATATAA-3′, 5′-GAGCCAGTACATTCAGTGGTTTCA-3′ and 5′-Fam-TGCAGATACAGAGGCAGAAACAGGAGGAA-Tamra-3′ (NM_011406) and B-type natriuretic peptide (mouse): 5′-AGGCGAGACAAGGGAGAACA-3′, 5′-GGAGATCCATGCCGCAGA-3′ and 5′-Fam-CATCATTGCCTGGCCCATCGC-Tamra-3′ (NM_008726). The results were normalized to 18S rRNA quantified from the same samples using the forward and reverse primers: 5′-TGGTTGCAAAGCTGAAACTTAAAG-3′ and 5′-AGTCAAATTAAGCCGCAGGC-3′. The probe for the 18S amplicon was 5′-Vic-CCTGGTGGTGCCCTTCCGTCA-Tamra-3′.
Electrophoretic mobility shift assay
A double-stranded oligonucleotide probe corresponding to the putative DREAM binding region of the rat Cacna1c promoter with the sense sequence: 5′-TGTGAATACTCAGTTTGTCATTGCTTAGAACC-3′ and antisense sequence: 5′-GGTTCTAAGCAATGACAAACTGAGTATT-3′ as well as another double-stranded oligonucleotide from the same region containing double point mutations in the putative DREAM binding region with the sense sequence: 5′-TGTGAATACTCAGTTAGTCTTTGCTTAGAACC-3′ and antisense sequence 5′-GGTTCTAAGCAAAGACTAACTGAGTATT-3′ were designed (Oligomer, Helsinki, Finland). The probes were sticky-end-labelled with [α-32P]dCTP by Klenow enzyme. For each reaction mixture (20 μl) 6 μg of nuclear protein and 2 μg of poly(dI-dC)2 was used in a buffer containing 24 mm Hepes (pH 7.9), 5 mm MgCl2, 135 mm NaCl, 40 mm KCl, 2.5 mm DTT, 0.6 mm EDTA, 0.3 mm EGTA, 8% glycerol, 0.02% Nonidet P-40, 0.65 mm phenylmethylsulfonyl fluoride, 25 μg ml−1 leupeptin, 2.5 μg ml−1 pepstatin and 25 μg ml−1 aprotinin. Reaction mixtures were incubated each with 2 μg of the labelled probe for 20 min followed by non-denaturating gel electrophoresis on a 5% polyacrylamide gel. Subsequently, gels were dried and exposed in a PhosphorImager screen (Molecular Dynamics, Sunnyvale, CA, USA) and analysed with the Molecular Imager FX imaging system and Quantity One 4.2.1 software (Bio-Rad). To confirm the DNA sequence specificity of the protein–DNA complex formation, competition experiments with 10-, 100- and 1000-fold molar excess of non-radiolabelled oligonucleotides were performed. Oligodeoxynucleotides or the anti-DREAM antibody (Upstate) were added to the reaction mixture 20 min before addition of the labelled probe.
Mathematical modelling of the Ca2+–CaMKII–DREAM interaction
As previously described (Tavi et al. 2003; Aydin et al. 2007), a model of the CaMKII activation reaction scheme (Bhalla & Iyengar, 1999) was employed to simulate CaMKII activity with different Ca2+ concentrations (Fig. 6B). The CaMKII-dependent regulation of excitation–contraction coupling was modelled as described earlier (Koivumaki et al. 2009). The original scheme was modified with the following additions (Fig. 6C). The DREAM-dependent modulation of the LTCC expression, XCaL, was formulated in the dynamic and steady states (ss) as follows:
Figure 6. The Ca2+–CaMKII–DREAM–LTCC cascade establishes feedback between [Ca2+]i and Ca2+ influx.
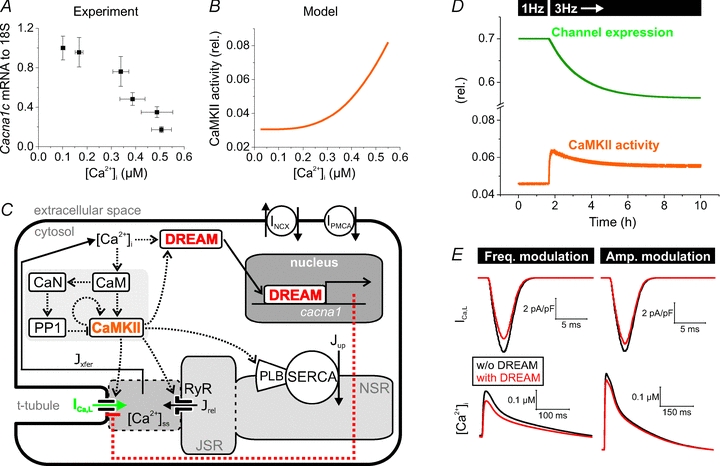
A, elevation of [Ca2+]i resulted in a dose-dependent repression of Cacna1c in H9c2 cells. The level of calcium accumulation caused by 0.1–10 μm A23187 Ca2+ ionophore and the corresponding levels of Cacna1c mRNA were determined by Fluo-4 calcium imaging (n = 6–16) and quantitative real-time PCR analysis (n = 6). B, relative CaMKII activity shown as a function of [Ca2+]i in a mathematical model of the cardiomyocyte. The CaMKII reaction chain was stimulated with a constant calcium input. Steady-state activity has been scaled to the total amount of CaMKII. C, schematic presentation of the myocyte model. For simplicity, only calcium-related sarcolemmal ion transporters and channels are shown. DREAM is activated by [Ca2+]i and CaMKII, and it causes negative feedback to the LTCC by decreasing the channel conductance. Colour coding matches B, D and E. D, representative simulation shows the effect of increased pacing frequency (1 → 3 Hz) on CaMKII activity and the consequent decrease in the expression of the LTCC. The time constant for the channel expression change was 125 min. E, the DREAM feedback decreases the LTCC current (ICa,L) and the calcium transient amplitude. Results from pacing steady states indicate that a rather similar end-effect can be obtained with both frequency and amplitude modulation of CaMKII and DREAM activation. Simulations were run at 3 Hz pacing, and at 2 Hz pacing combined with enhanced intracellular calcium transient amplitude (stimulation of SR Ca2+-ATPase), with and without DREAM–LTCC feedback, respectively. PMCA, plasma membrane Ca2+ ATPase; CaN, Calcineurin; PP1, protein phosphatase 1; PLB, phospholamban; JSR, junctional sarcolpasmic reticulum; NSR, network sarcoplasmic reticulum; J, calcium flux.
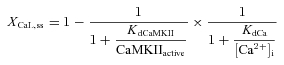 |
(1) |
| (2) |
where KdCa = 0.1 μm, KdCaMKII = 0.3 μm, CaMKIIactive = total amount of active CaMKII and [Ca2+]i = cytosolic free calcium concentration. The parameter values were set so that (1) CaMKII inhibition increases the ICa,L 1.5-fold and (2) increasing [Ca2+]i to 0.5 μm decreases the relative channel expression to 0.2 (see Fig. 6A). The time constant of the changing channel expression, τgCaL, was varied between 1 and 125 min. The range was limited by the computational load, because a simulation with τgCaL = 125 min needed to be run for 500 min to reach steady state. With a pacing frequency of 3 Hz this results in a computation time of ∼8 days. However, the value of the time constant does not affect the actual phenomenon; it just dictates the rate of the events. The conductance of LTCC was made dependent on the relative channel expression level:
| (3) |
where gCaL,0 = 0.1729 ms μF−1. All the simulations were run in the Matlab (The Mathworks, Inc., Natick, MA, USA) environment of technical programming as described previously (Koivumaki et al. 2009).
Statistical testing
Statistical testing was done with one-way or two-way ANOVA followed by the Bonferroni test using Origin 7.5 software (OriginLab Corp., Northampton, MA, USA). P < 0.05 was considered statistically significant (***, P < 0.001; **, P < 0.01; *, P < 0.05). All values are expressed as mean ± SEM.
Results
Nuclear (δB) and cytosolic (δC) CaMKII selectively downregulate LTCC α1C-subunit expression
First we tested if direct CaMKII targets would be found among the proteins with a direct impact on calcium handling. We screened putative CaMKII-regulated genes after overexpressing either nuclear (δB) or cytosolic (δC) CaMKII in HL-1 cardiomyocytes (Fig. 1A). Expression of the majority of the genes was not sensitive to CaMKII; transcripts for the Na+/Ca2+-exchanger (NCX1), ryanodine receptor (RyR2), SR Ca2+-ATPase (Serca2a) and LTCC β-subunit (Cacnb2a) were all unaltered with CaMKII overexpression (Fig. 1B). In contrast, both nuclear (δB) and cytosolic (δC) CaMKII isoforms decreased the expression of LTCC α1C-subunit gene (Cacna1c) by ∼49.6 and ∼73.4%, respectively. At the same time, expression of B-type natriuretic peptide (BNP), a commonly used cardiac hypertrophy marker and known transcriptional target of CaMKII (Ronkainen et al. 2007), was significantly increased with both CaMKII isoforms.
Figure 1. The Cacna1c gene is selectively repressed by CaMKII in cardiomyocytes.
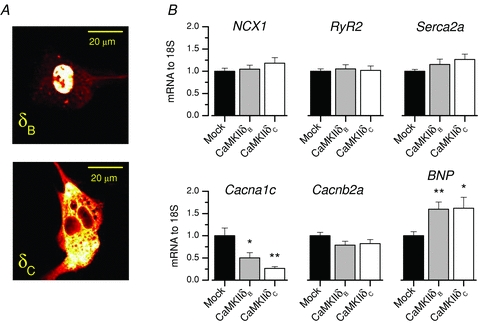
A, FITC-conjugated haemagglutinin antibody labelling confirmed specific nuclear and cytosolic expression of haemagglutinin-tagged CaMKIIδB and CaMKIIδC in HL-1 cells. B, overexpression of CaMKIIδB and CaMKIIδC isoforms in HL-1 cells resulted in 49.6% and 73.4% repression of Cacna1c, respectively. The B-type natriuretic peptide gene (BNP) was induced 59.8% and 62.0%. Other measured calcium activity-related genes did not change. Expression levels were measured by quantitative real-time PCR (n = 6).
Inhibition of CaMKII augments the expression of LTCCs and induces specific changes in the E–C-coupling
Accepting that CaMKII downregulates LTCC expression, inhibition of CaMKII in active cardiomyocytes should have an opposite effect. KN-93 (3 μm, n = 10), an inhibitor of CaMKII, or its inactive analogue KN-92 (3 μm, n = 9) had no acute effect (up to 5 min exposure) on the amplitudes of spontaneous [Ca2+]i transients of HL-1 cardiomyocytes (see Supplemental Fig. S1, available online only). To test if chronic CaMKII inhibition has an effect in LTCC expression, we exposed spontaneously active HL-1 cardiomyocytes to KN-93 (3 μm), for 24 h. After this, the cells were incubated several hours without KN-93 to exclude direct effects of KN-93. When the LTCC current was measured, we observed that KN-93-treated cells had larger LTCC densities compared to control cells (Fig. 2A). The current–voltage relationship was similar in both groups, but the absolute current values were larger in the KN-93 group with ∼1.5-fold increase in the peak value at +10 mV (−4.7 ± 0.8 pA pF−1vs. −7.2 ± 0.9 pA pF−1, n = 11 and 18, respectively, P < 0.05) (Fig. 2A). These changes were accompanied by ∼90.5% increased expression of the α1C-subunit gene (Cacna1c) in the KN-93-treated group (n = 12, P < 0.05), as well as an ∼3-fold increase in the α1C-subunit immunoreactivity in KN-93-treated cells (Fig. 2B). Similarly KN-93 (24 h) increased the Cacna1c expression in rat neonatal cardiomyocytes (P < 0.001, n = 5), whereas KN-92 did not (n = 5) (Supplemental Fig. S2).
Figure 2. Twenty-four hour treatment with the CaMKII inhibitor KN-93 increased LTCC current, protein and mRNA as well as Ca2+ transient amplitude and SR Ca2+ load in HL-1 cells.
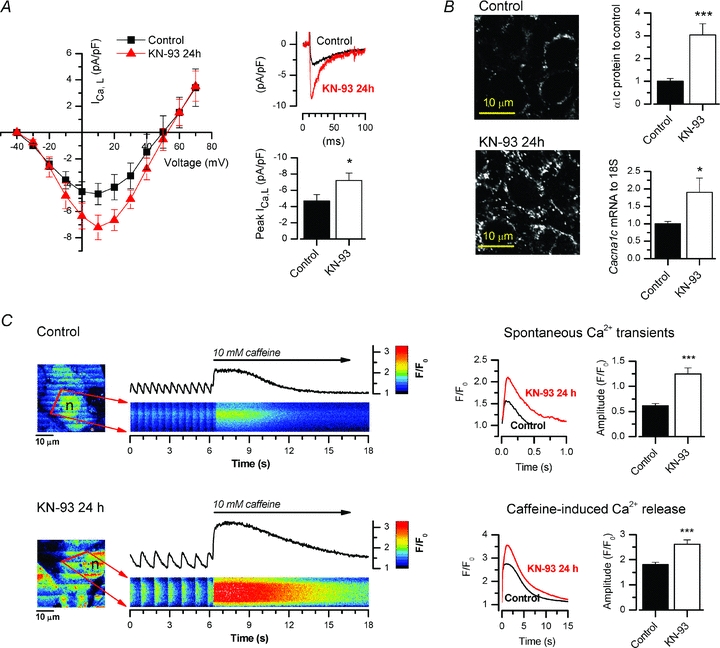
A, the LTCC current–voltage relationship was measured from control (n = 11) and KN-93-treated (n = 18) HL-1 cells with patch-clamp. Representative current recordings from control and KN-93-treated HL-1 cells are shown with voltage-clamp from a −40 mV holding potential to +10 mV. The mean peak LTCC current showed a 54.3% increase in cells treated 24 h with 3 μm KN-93. B, confocal microscopy of specimens treated 24 h with 3 μm KN-93 and immunofluorescence-labelled against LTCC α1c-subunit revealed a 3.0-fold increase in the protein level (n = 4). A corresponding 90.5% increase in Cacna1c expression was measured by quantitative real-time PCR (n = 12). C, laser scanning confocal x–y images (left) from control and KN-93-treated, spontaneously active HL-1 cells loaded with Fluo-3 calcium indicator with corresponding line scans (red line) through the cytosol surrounding the brighter nuclear area (n). Caffeine (10 mm) was applied during recordings to evaluate SR calcium content. Average traces (right) showing 2.0- and 1.8-fold increases in calcium transient and caffeine-induced SR calcium release amplitudes in 24 h KN-93-treated (n = 12) HL-1 cells in comparison to control cells (n = 14).
If the effect of CaMKII on the expression of the genes involved in cardiac calcium signalling is selective and targeted to LTCCs, CaMKII inhibition should have a specific effect on the cardiomyocyte phenotype and especially on calcium signals. To test this, HL-1 cardiomyocytes exposed to KN-93 for 24 h were loaded with Fluo-3-AM and intracellular calcium transients were measured with a laser scanning confocal microscope. KN-93-treated cells had prominent changes in the calcium signals (Fig. 2C), the amplitudes of the calcium transients were significantly increased, which should naturally result from increased density of LTCCs. Increased calcium influx through LTCCs could also augment the SR calcium stores. As an indicator of the increased SR calcium stores in KN-93-treated cells, the amplitudes of the caffeine-induced calcium releases (Tavi et al. 2005) were increased ∼45% (P < 0.001). The calcium extrusion rate after the caffeine pulse (time constant of one-exponential fit) which is mediated by the Na+/Ca2+-exchanger, was not changed (n = 12). When the trigger of calcium-induced calcium release, i.e. the LTCC current, is increased, one would expect a greater fraction of SR calcium to be released upon each excitation. Supporting this, we observed that the ratio between calcium transient and caffeine pulse amplitudes, i.e. the fractional release, was increased from 0.35 ± 0.03 to 0.48 ± 0.04 (P < 0.01) with chronic CaMKII inhibition. Altogether these changes in the calcium handling of the KN-93-treated cardiomyocytes indicate that CaMKII inhibition selectively affects LTCC expression with a functionally significant impact on the cardiomyocyte E–C-coupling.
A specific site in the Cacna1c promoter mediates the CaMKII effects
To study the mechanism behind the CaMKII-mediated α1C-subunit gene (Cacna1c) repression, we co-expressed Cacna1c promoter constructs of two different lengths together with CaMKII. Both the full-length (−2014 to +31) and the shorter D2 (−764 to +31) constructs were repressed by CaMKIIδB overexpression in both HL-1 cells and in primary cultures of rat neonatal ventricular cardiomyocytes (Fig. 3A). It was shown earlier that c-Ets (CAGGATGC) and AP-1 (ATGCTGACACAC) sites located at −690 and −629 in the Cacna1c promoter, are responsible for the majority of the promoter activity (Fan et al. 2002). Therefore, to preserve this endogenous basal activity of the promoter we prepared new promoter constructs from the original D2 construct sequence. The new promoter inserts were cloned into the pT81-luc plasmid which coupled them to a thymidine kinase minimal promoter (TK). The inserts were designed to contain both the c-Ets and the AP-1 sites with varying lengths of proximal deletions of the original D2 construct. Proximal deletions of the promoter insert between −408 and −171 showed decreased activity of the construct, suggesting that segments in this area are required for the maximal activation of the Cacna1c promoter (Fig. 3B). Furthermore, deleting a more distal segment between −594 and −537 increased the construct activity, indicating a repressor site within this sequence. Overexpression of CaMKIIδB decreased the activity of all of the constructs except the shortest Cacna1c (−764 to −594) (Fig. 3C), suggesting that the CaMKII-mediated repressor activity was dependent on the sequence between −594 and −537.
Figure 3. Promoter analysis for the transcription factor binding site responsible for the Cacna1c repression by CaMKIIδB.
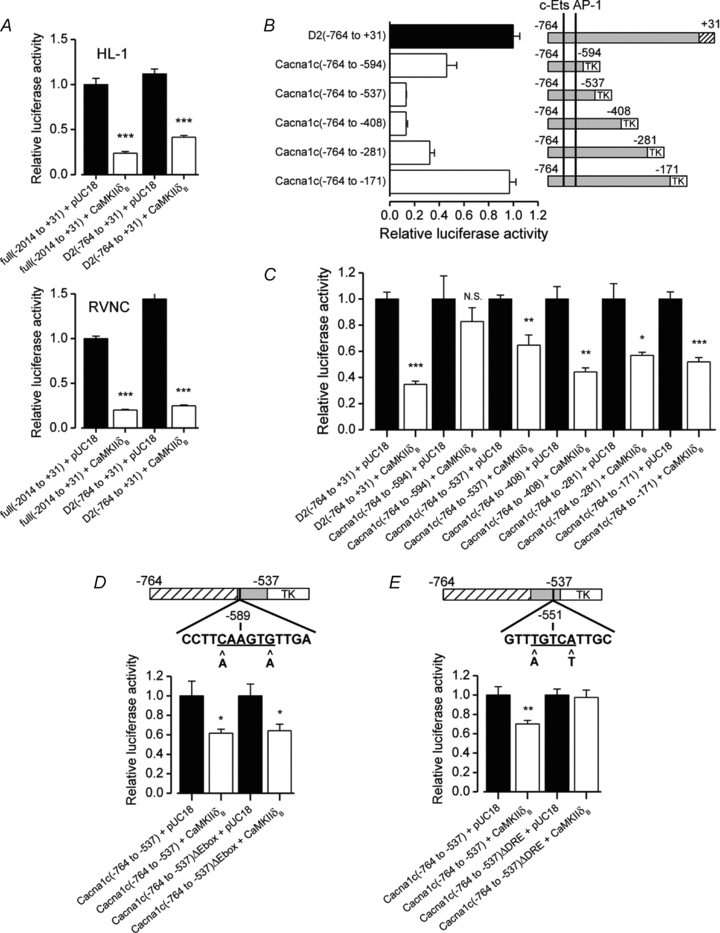
A, co-expressions of Cacna1c promoter–luciferase constructs with CaMKIIδB showing that both a full-length (−2014 to +31) and a shorter promoter segment (−764 to +31) containing construct were repressed by CaMKIIδB in HL-1 cells and rat neonatal ventricular cardiomyocytes (n = 4–5). B, activities of different length thymidine kinase (TK) minimal promoter-coupled promoter constructs, containing the c-Ets and AP-1 sites. The basal activity of these promoter constructs (n = 4) showing that the proximal part of the Cacna1c promoter between −408 and −171 is required for the maximal activity of the promoter. Deletion of the more distal segment between −594 and −537 increases the activity of the promoter, suggesting a repressor segment in this area. C, co-transfections of CaMKIIδB with the promoter constructs in B showing that all others except the shortest promoter construct Cacna1c(−764 to −594) were repressed by CaMKIIδB (n = 4). Genomatix database analysis revealed at least two potentially repressive binding sites in the area between −594 and −537. These candidate sites were a putative E-Box at −589 and a putative DRE (downstream regulatory element) at −551. D, double point mutations in the E-Box (−591 C→A and −586 G→A) did not have any effect on the promoter construct repression by CaMKIIδB (n = 6). E, double point mutations in the putative DRE (−553 T→A and −549 A→T) abolished the CaMKIIδB repression (n = 6).
On the basis of a database search (Genomatix software GmbH, Munich, Germany) the −594 to −537 promoter sequence contains at least two putative transcription factor binding sites reported to mediate calcium-dependent repression; an E-Box at −589 and a DRE (downstream regulatory element) at −551. CaMKII phosphorylation of the E-Box-binding factor myogenin has been reported earlier to repress nicotinic acetylcholine receptor gene expression in skeletal muscle (Tang et al. 2004). DRE antagonist modulator (DREAM) on the other hand has been reported to act as a transcriptional repressor that directly binds calcium (Carrion et al. 1999). To study the involvement of these sites in the CaMKII-mediated Cacna1c repression, we introduced two point mutations (–591 C→A and −586 G→A) into the putative E-Box in the Cacna1c (−764 to −537) promoter construct. Co-expressions of CaMKIIδB together with the original and the modified promoter construct revealed that the mutations had no effect on the CaMKIIδB-induced repression (Fig. 3D). Next we mutated the putative DRE in a similar manner (−553 T→A and −549 A→T) and found out that corrupting this putative binding site abolished the CaMKII-induced repression of the promoter construct (Fig. 3E). On these grounds we concluded that CaMKII is able to repress the Cacna1c gene promoter and that this effect is likely to be carried out by a DRE at −551 in the Cacna1c promoter.
CaMKII promotes nuclear translocation of DREAM
It was shown previously that intracellular calcium modulates the nuclear translocation of DREAM/calsenilin/KChIP3 (Zaidi et al. 2004). Therefore, we next wanted to study if calcium-activated CaMKII has a role in this process. When DREAM-overexpressing neonatal cardiomyocytes were exposed to CaMKII inhibitor KN-93, the amount of DREAM protein in the nuclear fraction of the cell lysates was significantly decreased (Fig. 4D), suggesting that CaMKII modulated the DREAM translocation. In line with this, when GFP-DREAM-fusion protein-expressing rat neonatal cardiomyocytes were exposed to caffeine to induce robust calcium increase in the cytosol, GFP-DREAM was translocated to the nucleus (τ = 4.2 ± 0.4 min, n = 10) (Fig. 4A and B). Similarly, when the cells were exposed to the LTCC opener Bay K 8644 (5 μm), which produced an ∼2-fold increase in the calcium transient amplitude and lengthening of the duration (Supplemental Fig. 3A), the nuclear-to-cytosolic ratio of GFP-DREAM was increased from 0.96 ± 0.02 to 1.44 ± 0.11 (P < 0.05, n = 4, Supplemental Fig. 3B). The time constant of caffeine-induced GFP-DREAM translocation was not dependent on CaMKII activity, but remained similar in cells co-expressing constitutively active CaMKII (CaMKIIT286D) (τ = 4.6 ± 1.1 min, n = 13) as well as in cells acutely treated with CaMKII inhibitor KN-93 (τ = 3.0 ± 0.5 min, n = 10) (Fig. 4B). The extent of GFP-DREAM translocation, however, was altered by modulation of CaMKII activity (Fig. 4B and C). In control cells the maximal translocation as indicated by the ratio between the nuclear and cytosolic fluorescence levels was 1.43 ± 0.08 and it was significantly increased in CaMKIIT286D cells (1.70 ± 0.05, n = 13, P < 0.01) and reduced in KN-93-treated cells (1.08 ± 0.05, n = 10, P < 0.01) (Fig. 4B and C). In all cells, the subcellular localization of DREAM was dependent on CaMKII but also on [Ca2+]i because without calcium stimulation DREAM was located mostly in the cytosol, even in myocytes expressing constitutively active CaMKIIT286D (Fig. 4C). In line with a previous report (Zaidi et al. 2004), we noticed that DREAM translocation requires Ca2+, but in addition we show that this effect was augmented by CaMKII overexpression and suppressed with CaMKII inhibition. These results suggest that [Ca2+]i and CaMKII co-operatively regulate the subcellular localization of DREAM.
Figure 4. Nuclear translocation of DREAM is dependent on CaMKII activity.
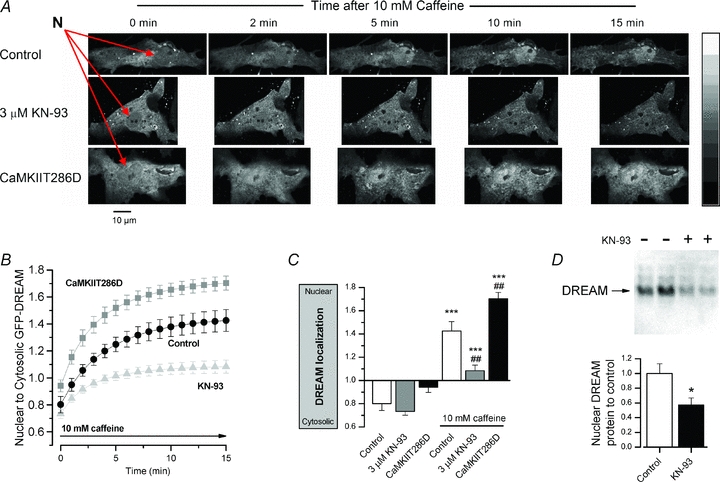
A, representative measurements showing laser scanning confocal microscope images at selected time points in a control cell, cell acutely treated with 3 μm KN-93 and a cell co-expressing constitutively active CaMKII (CaMKIIT286D) after introduction of 10 mm caffeine (nuclei marked with red arrows). B, confocal images were taken each minute after caffeine stimulation and the rate of DREAM translocation was determined based on the calculated ratio between nuclear and cytosolic fluorescence in control (n = 10), CaMKIIT286D overexpressing (n = 13) and CaMKII inhibitor KN-93 (n = 10) groups. C, 15 min after caffeine stimulation DREAM was translocated from the cytosol to nucleus in each experimental group (statistical significance marked with *). The translocation was significantly attenuated with 3 μm KN-93 and enhanced in CaMKIIT286D-expressing cells in comparison to the control group (statistical significance marked with #). D, representative bands of a Western blot utilizing DREAM-specific antibody and the corresponding quantitative analysis of the nuclear protein fragments of the DREAM-overexpressing neonatal cardiomyocytes showed that the CaMKII inhibitor KN-93 decreases the amount of the nuclear DREAM (n = 6).
Repression of endogenous Cacna1c by [Ca2+]i and DREAM is mediated by a DREAM-binding DRE sequence at −551 in the Cacna1c promoter
If the expression of the endogenous Cacna1c is suppressed by a mechanism involving CaMKII and DREAM, suppression should depend on [Ca2+]i and on DREAM expression. Overexpression of DREAM alone also suppressed Cacna1c expression, but did not potentiate the calcium-induced suppression significantly (Fig. 5A). This suggests that although DREAM is involved in Ca2+-dependent repression of Cacna1c, its amount is not the limiting factor in the pathway. To confirm that the putative DRE sequence in the Cacna1c gene promoter is functional and binds DREAM, we performed electrophoretic mobility shift assays. The nuclear extracts from cultured DREAM-overexpressing and control COS cells were incubated with a Cacna1c promoter 28 bp oligonucleotide probe containing the sequence surrounding the putative DRE at −551. Nuclear extracts with overexpressed DREAM protein increased binding with the Cacna1c 28 bp probe and the binding was attenuated by the addition of DREAM-specific IgG (Fig. 5B), resulting in a super-shifted protein–DNA complex. Furthermore, introduction of the same point mutations as used before in the Cacna1c promoter luciferase construct resulted in attenuated binding. Altogether these results indicate that the endogenous Cacna1c gene is [Ca2+]i-dependently repressed by DREAM, and in the rat Cacna1c promoter this effect is mediated by a DREAM-binding DRE sequence at position −551.
Figure 5. Cacna1c gene repression by [Ca2+]i is mediated by the DREAM DNA binding to the−551 site in the Cacna1c promoter.
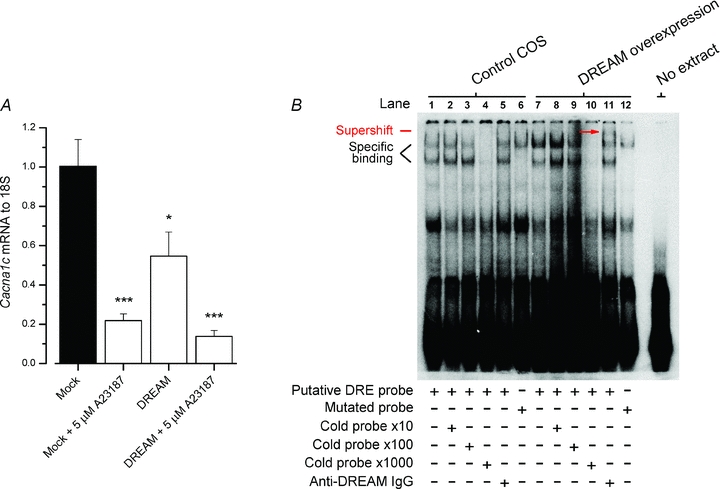
A, quantitative real-time PCR analysis in H9c2 cells (n = 6) showing that Cacna1c is repressed in addition to [Ca2+]i by DREAM overexpression alone. A23187 (5 μm) or DREAM overexpression repressed Cacna1c 78.1% and 45.3%, respectively. B, electrophoretic mobility shift assay showing DREAM binding to the putative DRE in the Cacna1c promoter. The nuclear extracts from cultured DREAM overexpressing and control COS cells were incubated with a 28 bp oligonucleotide probe containing the sequence surrounding the putative DRE at the position –551 in the Cacna1c promoter. The presence of overexpressed DREAM in the binding reaction increased binding in nuclear extracts with the Cacna1c 28 bp probe (lanes 1 and 7). Specific binding was determined using a 10-, 100- or 1000-fold excess of competing unlabelled probe (lanes 2–4 and 8–10). The presence of DREAM in the protein–DNA complexes was further verified using an antibody specific for DREAM (lanes 5 and 11) which induced a supershift band (red arrow) in the binding reaction containing overexpressed DREAM protein (lane 11). Introduction of double point mutations in the putative DRE sequence markedly reduced but did not completely abolish specific protein–DNA interactions in the Cacna1c 28 bp probe.
Does the Ca2+–CaMKII–DREAM–LTCC pathway establish a transcriptional feedback between cytosolic Ca2+ and Ca2+ influx?
It was shown previously that genetic deletion of CaMKIIδ results in alterations of LTCC in mice (Xu et al. 2010). Complementing this our findings suggest a mechanism where [Ca2+]i and subsequent CaMKII activation together act to repress Cacna1c expression by promoting DREAM nuclear translocation. If this was true, in a timescale of hours to tens of hours Cacna1c expression should be proportional to [Ca2+]i. To assess this experimentally we maintained different levels of steady [Ca2+]i in Cacna1c-expressing H9c2 cardiomyoblasts for 24 h by a calcium ionophore in order to construct a [Ca2+]i–Cacna1c repression curve. As expected, Cacna1c was progressively downregulated by [Ca2+]i between 0.1 and 0.5 μm up to ∼83% at 0.5 μm[Ca2+]i (Fig. 6A). This relationship between [Ca2+]i and Cacna1c expression defines the minimal and maximal repression of Cacna1c induced by [Ca2+]i and calcium-activated CaMKII. To further estimate the impact of this repression on the dynamic function of cardiomyocytes, we implemented these data into a mathematical model of the cardiomyocyte E–C-coupling (Koivumaki et al. 2009) including the CaMKII activation (Fig. 6B) and interaction with phosphorylation targets (Fig. 6C). The updated model was designed to include the effect of the transcriptional output on the LTCC density (Fig. 6C). Simulations with the model demonstrate that in conditions with initially elevated [Ca2+]i, CaMKII activation gradually leads to decreased LTCC expression, thus providing feedback through lower [Ca2+]i and reduced CaMKII activity (Fig. 6D). Whether the CaMKII activation is induced by either increased frequency or amplitude of the calcium transients, the change is subsequently counterbalanced by reduced LTCC current and calcium transient amplitude (Fig. 6E). Simulations suggest that with these border values the feedback mechanism is sufficient to regulate the calcium balance of the cardiomyocyte.
Discussion
This study establishes a new cascade in calcium-dependent transcription where CaMKII potentiates the calcium-induced translocation of the transcriptional repressor DREAM into the nucleus and thereby promotes DREAM-induced transcriptional repression. A prime example of this is the LTCC α1C-subunit, which is selectively downregulated upon [Ca2+]i rise and CaMKII activation. As such the Ca2+–CaMKII–DREAM–LTCC cascade constitutes a physiological feedback mechanism which enables cardiomyocytes to adjust the capacity of calcium intrusion through LTCC to the amount of intracellular calcium-activated CaMKII.
Regulation of LTCC function and expression in cardiomyocytes
The LTCC has a central role in cardiomyocyte calcium signalling and it constitutes an indispensable element in the physiological regulation of cardiomyocyte calcium balance. Being directly regulated by protein kinase A (Lory & Nargeot, 1992; Ferrier et al. 1998), CaMKII (Dzhura et al. 2000; Lee et al. 2006) and calcium–calmodulin (Peterson et al. 1999), the LTCC is an effective endpoint of a variety of intracellular signalling cascades. A number of reports indicate that LTCC regulation through phosphorylation is important for the normal adaptation of cardiomyocytes facing an increase in the heart rate or increased β-adrenergic activity. On the other hand, failure to appropriately regulate the activity of the LTCC can cause contractile dysfunction and trigger cardiac arrhythmias (Anderson, 2004). Compromised calcium handling of the cardiomyocytes is one of the reasons for impaired contractile function of the heart involved in cardiac hypertrophy and heart failure (Jiang et al. 2002; Piacentino et al. 2003; Saito et al. 2003; Engelhardt et al. 2004; Reuter et al. 2004). Impaired calcium signalling is caused by altered expression of calcium-handling proteins, among these being the reduced LTCC expression and density (Takahashi et al. 1992; Chen et al. 2002).
In contrast to the acute regulation of the LTCCs, the cellular signalling pathways controlling LTCCs at the transcriptional level are not known in detail, although several transcription factors such as CRE-binding protein CREB (Tsai et al. 2007), GATA-4 and Csx/Nkx2.5 (Wang et al. 2007) have been implicated in the expressional regulation of the Cav1.2 gene Cacna1c. Specific transcriptional repression of Cav1.2 by TNFα-induced activation of p50 and p65 has been reported in human colonic smooth muscle (Shi et al. 2005). Here we report that the activity of calcium–calmodulin-dependent kinase (CaMKII) regulates the expression of the LTCC pore-forming α1c-subunit in cardiac myocytes. This suggests a novel feedback mechanism that controls the calcium flux through the LTCC depending on the activity of CaMKII, which is in turn activated by cytosolic (Anderson, 2004) and nuclear (Wu et al. 2006) calcium signals. This feedback was convincingly demonstrated in vivo recently when it was shown that LTCC density is increased in CaMKIIδ knockout mice (Xu et al. 2010). From the physiological point of view, the feedback loop established by Ca2+–CaMKII–DREAM–LTCC acts to dynamically adjust the [Ca2+]i and Ca2+ influx together. As demonstrated by the model simulations, if either the frequency or the amplitude of the intracellular Ca2+ transients is increased, this feedback downregulates the LTCC current to suppress enlarged or frequent Ca2+ signals. It is interesting that while CaMKII-induced phosphorylation induces acute facilitation of the LTCCs (Dzhura et al. 2000; Wu et al. 2001), the CaMKII–DREAM cascade promotes the opposite effect by repressing LTCC expression. Upon Ca2+ activation of CaMKII, this two-phasic mechanism first acts to increase the Ca2+ influx, but if Ca2+ remains elevated, transcriptional repression of the LTCCs will gradually limit the Ca2+ influx. Subsequently, in pathological situations where intracellular calcium is constantly elevated this mechanism should downregulate LTCC expression, promoting distinct changes in calcium balance and action potential configuration. For example, in atrial fibrillation high frequency electrical activity leads to accumulation of [Ca2+]i and LTCC downregulation (Brundel et al. 1999; Lai et al. 1999; Klein et al. 2003; Christ et al. 2004; Gaborit et al. 2005). In addition, when this regulation is incapacitated by artificially manipulating CaMKII activity, the impact of this feedback on the phenotype of the cell is revealed. Demonstrating this, 24 h pharmacological inhibition of CaMKII resulted in upregulation of the LTCC transcripts and current, subsequently altering the calcium balance of the cardiomyocytes (Fig. 2). In a wider context, it could be suggested that when this feedback mechanism is uncoupled either genetically (Xu et al. 2010) or pharmacologically, acute changes in the cell phenotype ensue.
Transcriptional regulation of LTCCs by a Ca2+–CaMKII–DREAM pathway
Calcium signalling and calcium-dependent transduction cascades are essential in the physiological regulation of cardiac contraction (Bers, 2002) as well as important determinants of cardiomyocyte adaptation, growth and differentiation (Molkentin et al. 1995; McKinsey et al. 2000; Ikura et al. 2002; Olson & Schneider, 2003; Wu et al. 2006). Here we report a novel cascade in the calcium-dependent remodelling of cardiac myocytes. Compared to other CaMKII-mediated mechanisms regulating cardiomyocyte transcription, such as CaMKII-regulated histone deacetylase activity (Backs et al. 2006; Little et al. 2007; McKinsey, 2007), the CaMKII–DREAM–LTCC cascade established in this study provides more specific and subtle regulation of cardiomyocyte gene expression and phenotype. To what extent this mechanism contributes to Ca2+- and CaMKII-dependent cardiac remodelling in vivo remains to be studied, but it could be suggested that it might be one of the first adaptive mechanisms activated in cardiomyocytes facing enhanced [Ca2+]i activity and might explain the effects of CaMKII on LTCC expression (Xu et al. 2010). According to our results Ca2+–CaMKII induces a rapid nuclear translocation of DREAM with a time constant of ∼4 min (Fig. 4) enabling relatively prompt repression of DREAM target genes upon activation of the cascade. In the nucleus DREAM is known to bind to the DRE-core sequence GTCA in the DNA of the gene promoter areas (Carrion et al. 1999; Ledo et al. 2000) and we found that this type of sequence also mediates expressional repression of the Cacna1c gene (Fig. 5). Interestingly, this type of element has been earlier reported to be subject not only to regulation by calcium-dependent signalling but also PKA-mediated cascades (Carrion et al. 1999; Ledo et al. 2000). DRE-mediated repression has earlier been reported for the NCX3 gene in neurons (Gomez-Villafuertes et al. 2005) as well as several other genes (Carrion et al. 1999; Sanz et al. 2001; Rivas et al. 2004; Savignac et al. 2005). While DREAM has been reported to specifically bind DRE to suppress gene expression (Carrion et al. 1999), a product of the same gene named KChIP3 has been reported to interact with and modulate the Kv4.2-type subunits of potassium channels (An et al. 2000). However, the fact that knockout of the DREAM/calsenilin/KChIP3 locus in mouse showed no obvious cardiac phenotype (Cheng et al. 2002) suggests that the effects of DREAM/calsenilin/KChIP3 on transcription or K+ channel modulation are not indispensable in the heart. On the other hand, as suggested earlier (Mellstrom & Naranjo, 2001), it is also possible that other proteins closely related to DREAM/calsenilin/KChIP3, namely KChIP 1 and 2, might be functionally alternative to DREAM/calsenilin/KChIP3 in both transcriptional regulation and K+ channel modulation.
Limitations and applicability of the results
Our results indicate that CaMKII activity in cardiac myocytes controls LTCC density by regulating the transcription of Cav1.2 through activation of the transcriptional repressor DREAM. The experiments in this study were done in cardiomyocyte cultures and we did not characterize to what extent and under which conditions the described mechanisms are utilized in the adult heart. Comparable studies with primary isolated adult cardiomyocytes would be challenging as these are much less viable in culturing conditions than less mature cardiomyocytes or cell lines. However, while immature cultured cardiomyocytes and cardiomyocyte cell lines have structural and functional differences compared to adult myocytes, their transcriptional regulation shares many common features. For example, CaMKII ablation in adult mouse myocardium results in an increase in the expression of LTCCs (Xu et al. 2010), which is in line with our results showing a similar response with pharmacological inhibition of CaMKII in HL-1 cardiomyocytes. Whether the DREAM pathway is involved in in vivo regulation of LTCC expression remains to be studied, but it could be speculated that any mechanism establishing a link between [Ca2+]i and LTCC density would be a central part of the cardiomyocyte physiology and as such, malfunction of this feedback may precede the development of cardiomyocyte phenotypes with impaired function. Our findings may help to explain why compromised CaMKII-dependent regulation initiates a spectrum of different cardiac pathologies and sheds light on the role of CaMKII-dependent transcriptional regulation of cardiomyocyte E–C-coupling in cardiac pathogenesis, development and physiology.
Acknowledgments
The authors wish to thank Eero Kouvalainen for help with statistical analysis and Anneli Rautio for technical assistance. This study was supported by the Aarne Koskelo Foundation, Academy of Finland, Emil Aaltonen Foundation, Finnish Foundation of Cardiovascular Research, Jenny and Antti Wihuri Foundation, Maud Kuistila Foundation, Paavo Ilmari Ahvenainen Foundation, Research and Science Foundation of Farmos, Sigrid Juselius Foundation and Finnish Medical Foundation.
Glossary
Abbreviations
- CaMKII
Ca2+–calmodulin-dependent protein kinase II
- DRE
downstream regulatory element
- DREAM
DRE agonist modulator
- E–C-coupling
excitation–contraction coupling
- GFP
green fluorescent protein
- LTCC
L-type calcium channel
- NCX
Na+/Ca2+ exchanger
- PS
penicillin–streptomycin
- RyR
ryanodine receptor
- SERCA
SR Ca2+-ATPase
- SR
sarcoplasmic reticulum
Author contributions
The experimental work was performed at the Institute of Biomedicine, University of Oulu and the modelling was done at the Department of Biotechnology and Molecular Medicine, A.I. Virtanen Institute for Molecular Sciences, University of Eastern Finland, Kuopio. Conception and design of this study was conducted by P.T. and J.J.R. Experiments were performed by J.J.R., S.L.H., T.K., J.T.K., R.S., S.R., V.-P.R. and P.T. Data were interpreted and analysed, and article drafted, by J.J.R. and P.T. All authors approved the final version to be published.
Supplementary material
Supplementary Fig. S1
Supplementary Fig. S2
Supplementary Fig. S3
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, Rhodes KJ. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403:553–556. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- Anderson ME. Calmodulin kinase and L-type calcium channels: A recipe for arrhythmias? Trends Cardiovasc Med. 2004;14:152–161. doi: 10.1016/j.tcm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Aydin J, Korhonen T, Tavi P, Allen DG, Westerblad H, Bruton JD. Activation of Ca2+-dependent protein kinase II during repeated contractions in single muscle fibres from mouse is dependent on the frequency of sarcoplasmic reticulum Ca2+ release. Acta Physiol (Oxf) 2007;191:131–137. doi: 10.1111/j.1748-1716.2007.01725.x. [DOI] [PubMed] [Google Scholar]
- Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, Bassel-Duby R, Maier LS, Olson EN. The δ isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci U S A. 2009;106:2342–2347. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest. 2006;116:1853–1864. doi: 10.1172/JCI27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Cardiac excitation–contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Bhalla US, Iyengar R. Emergent properties of networks of biological signaling pathways. Science. 1999;283:381–387. doi: 10.1126/science.283.5400.381. [DOI] [PubMed] [Google Scholar]
- Brickey DA, Bann JG, Fong YL, Perrino L, Brennan RG, Soderling TR. Mutational analysis of the autoinhibitory domain of calmodulin kinase II. J Biol Chem. 1994;269:29047–29054. [PubMed] [Google Scholar]
- Brundel BJJM, Van Gelder IC, Henning RH, Tuinenburg AE, Deelman LE, Tieleman RG, Grandjean JG, Van Gilst WH, Crijns HJGM. Gene expression of proteins influencing the calcium homeostasis in patients with persistent and paroxysmal atrial fibrillation. Cardiovasc Res. 1999;42:443–454. doi: 10.1016/s0008-6363(99)00045-0. [DOI] [PubMed] [Google Scholar]
- Carrion AM, Link WA, Ledo F, Mellstrom B, Naranjo JR. DREAM is a Ca2+-regulated transcriptional repressor. Nature. 1999;398:80–84. doi: 10.1038/18044. [DOI] [PubMed] [Google Scholar]
- Chen XW, Piancentino V, Furukawa S, Goldman B, Margulies KB, Houser SR. L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ Res. 2002;91:517–524. doi: 10.1161/01.res.0000033988.13062.7c. [DOI] [PubMed] [Google Scholar]
- Cheng HY, Pitcher GM, Laviolette SR, Whishaw IQ, Tong KI, Kockeritz LK, Wada T, Joza NA, Crackower M, Goncalves J, Sarosi I, Woodgett JR, Oliveira-dos-Santos AJ, Ikura M, van der Kooy D, Salter MW, Penninger JM. DREAM is a critical transcriptional repressor for pain modulation. Cell. 2002;108:31–43. doi: 10.1016/s0092-8674(01)00629-8. [DOI] [PubMed] [Google Scholar]
- Christ T, Boknik P, Wohrl S, Wettwer E, Graf EM, Bosch RF, Knaut M, Schmitz W, Ravens U, Dobrev D. L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation. 2004;110:2651–2657. doi: 10.1161/01.CIR.0000145659.80212.6A. [DOI] [PubMed] [Google Scholar]
- Claycomb WC, Lanson NA, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME. Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol. 2000;2:173–177. doi: 10.1038/35004052. [DOI] [PubMed] [Google Scholar]
- Engelhardt S, Hein L, Dyachenkow V, Kranias EG, Isenberg G, Lohse MJ. Altered calcium handling is critically involved in the cardiotoxic effects of chronic β-adrenergic stimulation. Circulation. 2004;109:1154–1160. doi: 10.1161/01.CIR.0000117254.68497.39. [DOI] [PubMed] [Google Scholar]
- Fan QI, Vanderpool K, Marsh JD. A 27 bp cis-acting sequence is essential for L-type calcium channel α1C subunit expression in vascular smooth muscle cells. Biochim Biophys Acta. 2002;1577:401–411. doi: 10.1016/s0167-4781(02)00441-4. [DOI] [PubMed] [Google Scholar]
- Ferrier GR, Zhu JQ, Redondo IM, Howlett SE. Role of cAMP-dependent protein kinase A in activation of a voltage-sensitive release mechanism for cardiac contraction in guinea-pig myocytes. J Physiol. 1998;513:185–201. doi: 10.1111/j.1469-7793.1998.185by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey N, McKinsey TA, Olson EN. Decoding calcium signals involved in cardiac growth and function. Nat Med. 2000;6:1221–1227. doi: 10.1038/81321. [DOI] [PubMed] [Google Scholar]
- Gaborit N, Steenman M, Lamirault G, Le Meur N, Le Bouter S, Lande G, Leger J, Charpentier F, Christ T, Dobrev D, Escande D, Nattel S, Demolombe S. Human atrial ion channel and transporter subunit gene-expression remodeling associated with valvular heart disease and atrial fibrillation. Circulation. 2005;112:471–481. doi: 10.1161/CIRCULATIONAHA.104.506857. [DOI] [PubMed] [Google Scholar]
- Gomez-Villafuertes R, Torres B, Barrio J, Savignac M, Gabellini N, Rizzato F, Pintado B, Gutierrez-Adan A, Mellstrom B, Carafoli E, Naranjo JR. Downstream regulatory element antagonist modulator regulates Ca2+ homeostasis and viability in cerebellar neurons. J Neurosci. 2005;25:10822–10830. doi: 10.1523/JNEUROSCI.3912-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura M, Osawa M, Ames JB. The role of calcium-binding proteins in the control of transcription: structure to function. Bioessays. 2002;24:625–636. doi: 10.1002/bies.10105. [DOI] [PubMed] [Google Scholar]
- Jiang MT, Lokuta AJ, Farrell EF, Wolff MR, Haworth RA, Valdivia HH. Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ Res. 2002;91:1015–1022. doi: 10.1161/01.res.0000043663.08689.05. [DOI] [PubMed] [Google Scholar]
- Klein G, Schroder F, Vogler D, Schaefer A, Haverich A, Schieffer B, Korte T, Drexler H. Increased open probability of single cardiac L-type calcium channels in patients with chronic atrial fibrillation: Role of phosphatase 2A. Cardiovasc Res. 2003;59:37–45. doi: 10.1016/s0008-6363(03)00357-2. [DOI] [PubMed] [Google Scholar]
- Kohlhaas M, Zhang T, Seidler T, Zibrova D, Dybkova N, Steen A, Wagner S, Chen L, Brown JH, Bers DM, Maier LS. Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ Res. 2006;98:235–244. doi: 10.1161/01.RES.0000200739.90811.9f. [DOI] [PubMed] [Google Scholar]
- Koivumaki JT, Korhonen T, Takalo J, Weckstrom M, Tavi P. Regulation of excitation–contraction coupling in mouse cardiac myocytes: integrative analysis with mathematical modelling. BMC Physiol. 2009;9:16. doi: 10.1186/1472-6793-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai LP, Su MJ, Lin JL, Lin FY, Tsai CH, Chen YS, Huang SKS, Tseng YZ, Lien WP. Down-regulation of L-type calcium channel and sarcoplasmic reticular Ca2+-ATPase mRNA in human atrial fibrillation without significant change in the mRNA of ryanodine receptor, calsequestrin and phospholamban: An insight into the mechanism of atrial electrical remodeling. J Am Coll Cardiol. 1999;33:1231–1237. doi: 10.1016/s0735-1097(99)00008-x. [DOI] [PubMed] [Google Scholar]
- Ledo F, Carrion AM, Link WA, Mellstrom B, Naranjo JR. DREAM-αCREM interaction via leucine-charged domains derepresses downstream regulatory element-dependent transcription. Mol Cell Biol. 2000;20:9120–9126. doi: 10.1128/mcb.20.24.9120-9126.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TS, Karl R, Moosmang S, Lenhardt P, Klugbauer N, Hofmann F, Kleppisch T, Welling A. Calmodulin kinase II is involved in voltage-dependent facilitation of the L-type Cav1.2 calcium channel: Identification of the phosphorylation sites. J Biol Chem. 2006;281:25560–25567. doi: 10.1074/jbc.M508661200. [DOI] [PubMed] [Google Scholar]
- Little GH, Bai Y, Williams T, Poizat C. Nuclear calcium/calmodulin-dependent protein kinase IIδ preferentially transmits signals to histone deacetylase 4 in cardiac cells. J Biol Chem. 2007;282:7219–7231. doi: 10.1074/jbc.M604281200. [DOI] [PubMed] [Google Scholar]
- Liu L, Fan QI, El-Zaru MR, Vanderpool K, Hines RN, Marsh JD. Regulation of DHP receptor expression by elements in the 5′-flanking sequence. Am J Physiol Heart Circ Physiol. 2000;278:H1153–H1162. doi: 10.1152/ajpheart.2000.278.4.H1153. [DOI] [PubMed] [Google Scholar]
- Lory P, Nargeot J. Cyclic AMP-dependent modulation of cardiac Ca channels expressed in Xenopus laevis oocytes. Biochem Biophys Res Commun. 1992;182:1059–1065. doi: 10.1016/0006-291x(92)91839-i. [DOI] [PubMed] [Google Scholar]
- McKinsey TA. Derepression of pathological cardiac genes by members of the CaM kinase superfamily. Cardiovasc Res. 2007;73:667–677. doi: 10.1016/j.cardiores.2006.11.036. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Lu JR, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier LS. Role of CaMKII for signaling and regulation in the heart. Front Biosci. 2009;14:486–496. doi: 10.2741/3257. [DOI] [PubMed] [Google Scholar]
- Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic CaMKIIδC overexpression uniquely alters cardiac myocyte Ca2+ handling: Reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. 2003;92:904–911. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- Matsuda M, Yamamoto TA, Hirata M. Ca2+-dependent regulation of calcitonin gene expression by the transcriptional repressor DREAM. Endocrinology. 2006;147:4608–4617. doi: 10.1210/en.2006-0254. [DOI] [PubMed] [Google Scholar]
- Mellstrom B, Naranjo JR. Ca2+-dependent transcriptional repression and derepression: DREAM, a direct effector. Semin Cell Dev Biol. 2001;12:59–63. doi: 10.1006/scdb.2000.0218. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Black BL, Martin JF, Olson EN. Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell. 1995;83:1125–1136. doi: 10.1016/0092-8674(95)90139-6. [DOI] [PubMed] [Google Scholar]
- Nordeen SK. Luciferase reporter gene vectors for analysis of promoters and enhancers. Biotechniques. 1988;6:454–458. [PubMed] [Google Scholar]
- Olson EN, Schneider MD. Sizing up the heart: development redux in disease. Genes Dev. 2003;17:1937–1956. doi: 10.1101/gad.1110103. [DOI] [PubMed] [Google Scholar]
- Peterson BZ, DeMaria CD, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- Piacentino V, Weber CR, Chen XW, Weisser-Thomas J, Margulies KB, Bers DM, Houser SR. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. 2003;92:651–658. doi: 10.1161/01.RES.0000062469.83985.9B. [DOI] [PubMed] [Google Scholar]
- Ramirez MT, Zhao XL, Schulman H, Brown JH. The nuclear δB isoform of Ca2+/calmodulin-dependent protein kinase II regulates atrial natriuretic factor gene expression ventricular myocytes. J Biol Chem. 1997;272:31203–31208. doi: 10.1074/jbc.272.49.31203. [DOI] [PubMed] [Google Scholar]
- Reuter H, Han TY, Motter C, Philipson KD, Goldhaber JI. Mice overexpressing the cardiac sodium–calcium exchanger: defects in excitation-contraction coupling. J Physiol. 2004;554:779–789. doi: 10.1113/jphysiol.2003.055046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas M, Mellstrom B, Naranjo JR, Santisteban P. Transcriptional repressor DREAM interacts with thyroid transcription factor-1 and regulates thyroglobulin gene expression. J Biol Chem. 2004;279:33114–33122. doi: 10.1074/jbc.M403526200. [DOI] [PubMed] [Google Scholar]
- Ronkainen JJ, Vuolteenaho O, Tavi P. Calcium-calmodulin kinase II is the common factor in calcium-dependent cardiac expression and secretion of A- and B-type natriuretic peptides. Endocrinology. 2007;148:2815–2820. doi: 10.1210/en.2006-1676. [DOI] [PubMed] [Google Scholar]
- Saito T, Fukuzawa J, Osaki J, Sakuragi H, Yao N, Haneda T, Fujino T, Wakamiya N, Kikuchi K, Hasebe N. Roles of calcineurin and calcium/calmodulin-dependent protein kinase II in pressure overload-induced cardiac hypertrophy. J Mol Cell Cardiol. 2003;35:1153–1160. doi: 10.1016/s0022-2828(03)00234-7. [DOI] [PubMed] [Google Scholar]
- Sanz C, Mellstrom B, Link WA, Naranjo JR, Fernandez-Luna JL. Interleukin 3-dependent activation of DREAM is involved in transcriptional silencing of the apoptotic hrk gene in hematopoietic progenitor cells. EMBO J. 2001;20:2286–2292. doi: 10.1093/emboj/20.9.2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savignac M, Pintado B, Gutierrez-Adan A, Palczewska M, Mellstrom B, Naranjo JR. Transcriptional repressor DREAM regulates T-lymphocyte proliferation and cytokine gene expression. EMBO J. 2005;24:3555–3564. doi: 10.1038/sj.emboj.7600810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding-proteins with mini-extracts, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419–6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi XZ, Pazdrak K, Saada N, Dai B, Palade P, Sarna SK. Negative transcriptional regulation of human colonic smooth muscle Cav1.2 channels by p50 and p65 subunits of nuclear factor-κB. Gastroenterology. 2005;129:1518–1532. doi: 10.1053/j.gastro.2005.07.058. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Allen PD, Lacro RV, Marks AR, Dennis AR, Schoen FJ, Grossman W, Marsh JD, Izumo S. Expression of dihydropyridine receptor (Ca2+ channel) and calsequestrin genes in the myocardium of patients with end-stage heart failure. J Clin Invest. 1992;90:927–935. doi: 10.1172/JCI115969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang HB, Macpherson P, Argetsinger LS, Cieslak D, Suhr ST, Carter-Su C, Goldman D. CaM kinase II-dependent phosphorylation of myogenin contributes to activity-dependent suppression of nAChR gene expression in developing rat myotubes. Cell Signal. 2004;16:551–563. doi: 10.1016/j.cellsig.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Tavi P, Allen DG, Niemela P, Vuolteenaho O, Weckstrom M, Westerblad H. Calmodulin kinase modulates Ca2+ release in mouse skeletal muscle. J Physiol. 2003;551:5–12. doi: 10.1113/jphysiol.2003.042002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavi P, Hansson A, Zhang SJ, Larsson NG, Westerblad H. Abnormal Ca2+ release and catecholamine-induced arrhythmias in mitochondrial cardiomyopathy. Hum Mol Gen. 2005;14:1069–1076. doi: 10.1093/hmg/ddi119. [DOI] [PubMed] [Google Scholar]
- Tsai CT, Wang DL, Chen WP, Hwang JJ, Hsieh CS, Hsu KL, Tseng CD, Lai LP, Tseng YZ, Chiang FT, Lin JL. Angiotensin II increases expression of α1C subunit of L-type calcium channel through a reactive oxygen species and cAMP response element-binding protein-dependent pathway in HL-1 myocytes. Circ Res. 2007;100:1476–1485. doi: 10.1161/01.RES.0000268497.93085.e1. [DOI] [PubMed] [Google Scholar]
- Wang Y, Morishima M, Zheng M, Uchino T, Mannen K, Takahashi A, Nakaya Y, Komuro I, Ono K. Transcription factors Csx/Nkx2.5 and GATA4 distinctly regulate expression of Ca2+ channels in neonatal rat heart. J Mol Cell Cardiol. 2007;42:1045–1053. doi: 10.1016/j.yjmcc.2007.03.905. [DOI] [PubMed] [Google Scholar]
- Wu X, Zhang T, Bossuyt J, Li XD, McKinsey TA, Dedman JR, Olson EN, Chen J, Brown JH, Bers DM. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest. 2006;116:675–682. doi: 10.1172/JCI27374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YJ, Dzhura I, Colbran RJ, Anderson ME. Calmodulin kinase and a calmodulin-binding ‘IQ’ domain facilitate L-type Ca2+ current in rabbit ventricular myocytes by a common mechanism. J Physiol. 2001;535:679–687. doi: 10.1111/j.1469-7793.2001.t01-1-00679.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Xu L, Lai D, Cheng J, Lim HJ, Keskanokwong T, Backs J, Olson EN, Wang Y. Alterations of L-type calcium current and cardiac function in CaMKIIδ knockout mice. Circ Res. 2010;107:398–407. doi: 10.1161/CIRCRESAHA.110.222562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Shen W, Rottman JN, Wikswo JP, Murray KT. Rapid stimulation causes electrical remodeling in cultured atrial myocytes. J Mol Cell Cardiol. 2005;38:299–308. doi: 10.1016/j.yjmcc.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Zaidi NF, Thomson EE, Choi EK, Buxbaum JD, Wasco W. Intracellular calcium modulates the nuclear translocation of calsenilin. J Neurochem. 2004;89:593–601. doi: 10.1046/j.1471-4159.2004.02362.x. [DOI] [PubMed] [Google Scholar]
- Zhang T, Brown JH. Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc Res. 2004;63:476–486. doi: 10.1016/j.cardiores.2004.04.026. [DOI] [PubMed] [Google Scholar]
- Zhang T, Johnson EN, Gu YS, Morissette MR, Sah VP, Gigena MS, Belke DD, Dillmann WH, Rogers TB, Schulman H, Ross J, Brown JH. The cardiac-specific nuclear δB isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J Biol Chem. 2002;277:1261–1267. doi: 10.1074/jbc.M108525200. [DOI] [PubMed] [Google Scholar]
- Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Bers DM, Brown JH. The δC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92:912–919. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.