

Abstract
Earlier work has suggested that Fyn tyrosine kinase plays an important role in synaptic plasticity. To understand the downstream targets of Fyn signaling cascade in neurons, we generated transgenic mice expressing either a constitutively activated form of Fyn or native Fyn in neurons of the forebrain. Transgenic mice expressing mutant Fyn exhibited higher seizure activity and were prone to sudden death. Mice overexpressing native Fyn did not show such an obvious epileptic phenotype, but they exhibited accelerated kindling in response to once-daily stimulation of the amygdala. Tyrosine phosphorylation of at least three proteins was enhanced in the forebrains of both native and mutant fyn transgenic mice; tyrosine phosphorylation of these three proteins was reduced in fyn knockout mice, suggesting that they are substrates of Fyn. One of these proteins was identified as the subunit 2B (NR2B) of the N-methyl-d-aspartate (NMDA) receptor. Administration of MK-801, a noncompetitive NMDA receptor antagonist, retarded kindling in mice overexpressing native Fyn, as well as wild-type mice, suggests that the accelerated kindling in mice overexpressing Fyn is also mediated by the NMDA receptor activity. Our results thus suggest that tyrosine phosphorylation by Fyn might be involved in regulation of the susceptibility of kindling, one form of the NMDA receptor-mediated neuronal plasticity.
The brain exhibits a high level of protein–tyrosine kinase (PTK) activity. Two recent lines of evidence suggest that protein tyrosine phosphorylation is not only important for various developmental events, but also modulates synaptic transmission in the adult (for review, see Boxall and Lancaster 1998). First, inhibitors of tyrosine kinases block long-term potentiation (LTP) and depression (LTD), two types of synaptic plasticity that are thought to be important for learning and memory (O’Dell et al. 1991; Abe and Saito 1993; Boxall et al. 1996). Second, tyrosine phosphorylation modulates several ion channels and neurotransmitter receptors (Wang and Salter 1994; Moss et al. 1995; Swope et al. 1995; Valenzuela et al. 1995; Jonas and Kaczmarek 1996; Yu et al. 1997). Specifically, enhanced tyrosine phosphorylation of the N-methyl-d-aspartate (NMDA)-receptor subunit 2B (NR2B) is reported after the induction of LTP (Rosenblum et al. 1996; Rostas et al. 1996) and in the context of certain kinds of learning tasks (Rosenblum et al. 1995, 1997). However, the signaling pathways leading to these phosphorylation events have not been well characterized yet.
Earlier work has shown that the targeted disruption of Fyn, a member of Src family of nonreceptor PTKs, leads to altered hippocampal anatomy, a deficit in LTP and spatial learning (Grant et al. 1992), and a retardation of kindling (Cain et al. 1995). To determine whether the impairment of LTP in fyn knockout mice is caused directly by the lack of Fyn in adult hippocampal neurons or indirectly by a disruption in neuronal development, we recently generated fyn rescue mice by reintroducing native Fyn into the fyn knockout mouse as a transgene under the control of the calcium/calmodulin-dependent protein kinase IIα (CaMKIIα) promoter (Kojima et al. 1997). This promoter drives expression restricted to forebrain neurons late in neuronal development; therefore it is unlikely to rescue a developmental deficit. In fyn rescue mice, LTP was restored to normal, even though the morphological abnormalities characteristic of fyn knockout mice were still present. These results suggest that the Fyn protein modulates the threshold for the induction of LTP in the adult mouse directly.
The rescue experiment raises the next question: How is Fyn involved in synaptic plasticity? To answer this question, the cascade of signaling in which Fyn participates needs to be delineated. To identify the downstream components of this cascade, we generated transgenic mice expressing either native Fyn or the constitutively activated mutant under the CaMKIIα promoter. We then examined the physiological and biochemical consequences of overexpressing Fyn or the constitutively activated mutant in the adult brain.
Materials and Methods
GENERATION OF FYN TRANSGENIC MICE
Construction of transgenic mice expressing native Fyn has been described previously (Kojima et al. 1997). To construct the activated version of fyn transgene, we subcloned the EcoRI–DraI fragment of the fynB cDNA (pmBF, Cooke and Perlmutter 1989) into the vector M13mp18. Single-stranded phage DNA was prepared, and codon 531 was mutated from TAT to TTT by site-directed mutagenesis (Kunkel 1985) using the oligonucleotide 5′-CGGGCTGAAACTGGGGCTCTG-3′. The resulting point mutation was confirmed by DNA sequencing. The mutated cDNA was cloned into the vector pNN265 and then into pNN279 (provided by N. Nakanishi, Harvard Medical School, Boston, MA), as described previously (Kojima et al. 1997). The transgene was excised from the vector by SalI digestion and gel purified. The transgene was microinjected into the pronuclei of fertilized eggs collected from C57BL6/CBA/F1 females as described by Hogan et al. (1986). The transgene-injected eggs were transferred into the oviducts of pseudopregnant females. Founder mice were screened by PCR of tail DNA using the transgene-specific primer pair. Mouse lines were established by crossing founders with the hybrid strain (C57BL/6J × 129Sv) of heterozygous fyn knockout mice (Stein et al. 1992). The F1 offspring were used for detecting transgenic fyn mRNA by in situ hybridization and for surveying the viability of each transgenic line. To obtain the fyn transgenic mice, which were on wild-type and fyn knockout genetic backgrounds for the fyn gene, the F1 offspring of heterozygous fyn knockout mice were crossed with nontransgenic littermates of heterozygous fyn knockout mice. The F2 offspring on wild-type (fyn+/+) and fyn knockout (fyn−/−) genetic backgrounds were used for detecting the Fyn protein and its kinase activity. To examine the seizure threshold, kindling, and changes in protein tyrosine phosphorylation, we analyzed the F2 offspring that generated on wild-type genetic background for fyn gene (Tg+/fyn+/+) with a comparison of nontransgenic wild-type mice (Tg−/fyn+/+) , and in some experiments with a comparison of fyn knockout mice (Tg−/fyn−/−).
IN SITU HYBRIDIZATION
Transgenic fyn mRNA was detected in brain sections prepared from F1 offspring of each line as described (Kojima et al. 1997). Briefly, brains were removed quickly and frozen on dry ice. Cryostat sections (12 μm thick) were dried at 42°C and kept at −80°C until use. Antisense digoxigenin-labeled RNA homologous to the transgene 3′ noncoding sequence was prepared and used as a probe for detecting the transgene transcript specifically. Procedures for pretreatment of sections, hybridization, and detection were done as described (Boerhinger Mannheim, Germany).
WESTERN BLOTTING AND IN VITRO IMMUNE COMPLEX KINASE ASSAY
Tissue was homogenized in 10 volumes of RIPA buffer [10 mm Tris-HCl (pH 7.5), 1% NP-40, 0.1% sodium deoxycholate, 0.1% SDS, 0.15 m NaCl, 1 mm EDTA, 1 mm sodium orthovanadate, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mm phenylmethylsulfonyl fluoride (PMSF)] and centrifuged at 10,000g for 10 min. Supernatants were frozen rapidly in liquid nitrogen and stored at −80°C until use. Protein concentration was determined with the BCA protein assay kit (Pierce, Rockford, IL). For immunoblotting, 20–40 μg of protein was separated on 12% gel (for the Fyn protein) or 7.5% SDS–polyacrylamide gel [for phosphotyrosine (PY)-containing proteins], transferred to nitrocellulose membrane (Schleicher & Schuell, Germany), and probed with anti-Fyn antibody (Upstate Biotechnology, Lake Placid, NY) or anti-PY antibody PT-66 (Sigma, St. Louis, MO). After incubation with HRP-conjugated anti-mouse IgG, signals were detected by ECL Western blotting detection reagents (Amersham, England). The intensity of the signals detected on an autoradiography film (Hyperfilm-ECL, Amersham) was quantified by an imaging analyzer (Quantity One; PDI, Huntington Station, NY).
For kinase assay, RIPA extract (200 μg protein) was reacted with 4 μg of either anti-Fyn antibody or anti-Src antibody (Ab-1; Oncogene Science, Cambridge, MA), then precipitated with Protein G Sepharose (Pharmacia Biotech, Sweden). The immunoprecipitate was incubated with 125 μm [γ-32P]ATP (specific activity: 37 kBq/nmole) and 10 μg of the Src-substrate peptide (KVEKIGEGTYGVVYK, Upstate Biotechnology) at 30°C for 10 min. The reaction solution was spotted onto P81 phosphocellulose paper. After washing the paper with 0.75% of phosphoric acid, insoluble counts were measured with a liquid scintillation counter. Samples precipitated without antibody were used as a baseline for the kinase activity. Kinase activity was represented as fmoles phosphate incorporated into the substrate peptide/(min)(μg protein).
PTZ INFUSION
Adult male mice 2–6 months of age weighing 23–40 grams were held loosely in a plastic bag and administered a continuous infusion of 1.0% pentylenetetrazole (PTZ, Sigma) in saline into the tail vein at a constant flow rate (0.1 ml/min). The time required for first twitch and tonic extension was monitored. Data were corrected for body weight and converted to the corresponding PTZ dose (mg/kg of body weight).
ELECTRODE IMPLANTATION AND KINDLING PROCEDURE
Adult male mice 2.5–4 months of age weighing 23–36 grams were anesthetized with 15–17 μl/gram of 2.5% Avertin [0.25% (wt/vol) tribromoethyl alcohol, and 0.25% (vol/vol) tertiary amyl alcohol]. A bipolar electrode was implanted stereotaxically into the left basolateral amygdala (2.4 mm anterior to interaural zero, 2.8 mm lateral to the midline, 4.8 mm ventral to the skull surface). The electrode was consisted of twisted Teflon-insulated stainless steel wires (each wire 100 μm in diameter; A-M Systems, Everett, WA). After 10 days recovery, mice received the electrical stimulus through the electrode once per day. The stimulus consisted of a 1-sec train of 60-Hz sine-wave pulses. The intensity of the stimulus for each animal was determined by delivering a series of stimulations beginning at 20 μA and increasing gradually at 90-sec intervals until afterdischarges (ADs) were observed. ADs were recorded from the left amygdala through the same electrode, digitized at 10 kHz and analyzed by MacLab (ADInstruments, Australia). Behavioral seizures were classified according to a modified version of Racine’s criteria (Racine 1972) as follows: (1) rhythmic mouth and facial movement; (2) head nodding; (3) unilateral forelimb clonus; (4) rearing and bilateral forelimb clonus; (5) falling and/or hindlimb clonus; (6) running or bouncing seizure; (7) tonic and clonic seizure; and (8) tonic extension culminating in death. Mice were defined as fully kindled when greater than stage 4 convulsions were elicited on three consecutive days. To examine the effect of MK-801 on the development of kindling, MK-801 (1 mg/kg, Research Biochemicals International, Natick, MA) was administered intraperitoneally 1–2 hr before the daily stimulation on days 2–11. On completion of the experiment, animals were anesthetized deeply with pentobarbital and decapitated. The brains were removed and frozen quickly on dry ice. Serial coronal sections (20 μm thick) were stained with cresyl violet, and examined on a light microscope to confirm the positioning of the electrode tips within the amygdala.
IMMUNOPRECIPITATION AND WESTERN BLOTTING OF THE NR2 subunits
Immunoprecipitation of the NR2 subunits was carried out as described by Rosenblum et al. (1996). The forebrains were homogenized in 9 volumes of ice-cold 0.32 m sucrose in phosphate-buffered saline containing 1 mm sodium vanadate, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mm PMSF using a glass–Teflon homogenizer. The homogenate was centrifuged at 1000g for 10 min at 4°C. The supernatant was recentrifuged at 15,000g for 20 min at 4°C yielding the crude synaptosomal (P2) fraction. The P2 pellet was lysed in 10 mm Tris-Cl (pH 8.0) containing 1 mm sodium vanadate, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mm PMSF, then centrifuged at 15,000g for 20 min. The resulting pellet was resuspended in 10 mm Tris-Cl (pH 8.0) containing 1 mm sodium vanadate, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mm PMSF, diluted with an equal volume of 2% SDS, boiled for 5 min, then centrifuged at 15,000g for 10 min at 4°C. The supernatant (500 or 1000 μg protein) was diluted 1:10 in 10 mm Tris-Cl (pH 7.5), 1% NP-40, 0.1% sodium deoxycholate, 0.15 m NaCl, 1 mm EDTA, incubated with 2 μg of either anti-NR2A antibody (Chemicon, Temecula, CA) or anti-NR2B antibody (Transduction Laboratories, Lexington, KY) at 4°C overnight, then precipitated with protein G–Sepharose. The precipitate was washed with RIPA buffer five times, then separated on 6 or 7.5% of SDS–polyacrylamide gel and blotted with anti-PY, NR2A, or NR2B antibody.
Results
FYN TRANSGENES ARE EXPRESSED AT HIGH LEVELS IN THE FOREBRAIN NEURONS
We obtained four independent transgenic mouse lines overexpressing the native form of Fyn (N8, 39, 85, and 92) and six lines expressing a mutant form of Fyn (M3, 27, 47, 58, 78, and 84). Transgenic fyn mRNA was detected at a high level in the brain by Northern blotting, by ribonuclease protection assay, or by RT–PCR, whereas it was hardly detected in nonneural tissues with the exception of weak expression in the testis (data not shown). The spatial and temporal pattern of expression of the fyn transgene in the brain of line N8 has been described previously (Kojima et al. 1997). To assess the expression of transgenic fyn mRNA in the brain of each transgenic line, we performed in situ hybridization of the brain sections from the F1 offspring older than 4 weeks using a transgene-specific cRNA probe. As shown in Figure 1A and B, transgenic fyn mRNA was detected mainly in the neurons of the forebrain, including the neocortex, the hippocampus, and the amygdala. In the neocortex, the transgene was expressed most strongly in layers II and III and moderately in other layers except for layer I. In the hippocampus, the pyramidal cells in all subregions were stained uniformly, as were the granule cells of the dentate gyrus. Neurons in the striatum were stained moderately. By contrast, signals were weak in the cerebellum and brain stem, with an exception of some neurons, such as cerebellar Purkinje cells, which were clearly positive (Fig. 1C). Although the intensity of signal was different, the distribution of transgenic fyn mRNA was similar in all the lines.
Figure 1.

The distribution of transgenic fyn mRNA in the brain. (A) Transgenic fyn mRNA was detected in horizontal brain sections prepared from F1 offspring of fyn transgenic mice using a digoxigenin-labeled transgene-specific cRNA probe. No signal was detected in nontransgenic littermates (non-Tg). The regional pattern restricting to the forebrain was essentially similar among the individual lines. (B) In the forebrain of line N8, the transgenic fyn mRNA was expressed highly in neurons of the neocortex (Cx), hippocampal pyramidal layer (Py), dentate granular layer (DG), and lateral amygdala nucleus (LA). Scale bar, 500 μm. (C) In the cerebellum of line N8, the transgenic fyn mRNA was also detected in Purkinje cells (Pu). (ML) Cerebellar molecular layer; (GL) cerebellar granular layer; (WM) white matter. Scale bar, 100 μm.
We next examined the level of Fyn protein and Fyn kinase activity in RIPA extracts prepared from the forebrain of mice expressing native (N8, 39, 85, and 92) and mutant Fyn (M27, 58, and 78). To distinguish between the transgenic Fyn protein and the endogenous one, we performed these biochemical experiments in the F2 offspring, which were generated on either wild-type (fyn+/+) or fyn knockout (fyn−/−) genetic background. Animals from the three lines of mutant fyn transgenic mice, M3, 47, and 84, showed higher mortality (described below) and bred poorly; these lines were excluded from biochemical analysis because of lack of material. The transgenic Fyn protein was detected in the forebrain of each transgenic line generated on fyn knockout (Tg+/fyn−/−) genetic background (Fig. 2). The protein level was different among lines, in parallel with the mRNA level detected by in situ hybridization. The amount of transgenic Fyn protein, normalized to the level of endogenous Fyn detected in nontransgenic wild-type mice, was as follows; N8, 5.93 ± 0.49; N39, 2.08 ± 0.60; N85, 3.74 ± 0.44; N92, 4.81 ± 0.45; M27, 1.10 ± 0.15; M58, 0.41 ± 0.31; M78, 1.93 ± 0.28 (mean ± s.e.m., n = 7). To determine whether the transgenic Fyn protein has a tyrosine kinase activity, in vitro immune complex kinase assays were performed on RIPA extracts prepared from the forebrain of mice generated on either wild-type (fyn+/+) or fyn knockout (fyn−/−) genetic background. Fyn kinase activity measured in the wild-type background (Tg+/fyn+/+) was consistently higher than that in the fyn knockout background (Tg+/fyn−/−), because the former presented a summation of both the transgenic Fyn and endogenous Fyn and the latter was derived only from the transgenic Fyn (Table 1). The kinase activity of transgenic Fyn was consistent with the relative amount of the transgenic Fyn protein estimated by immunoblotting. When the Fyn kinase activity measured on fyn knockout background was subtracted from that on wild-type background, the difference was equal to the endogenous Fyn kinase activity in nontransgenic wild-type animals. Thus, overexpression of the fyn transgene does not appear to affect endogenous Fyn kinase activity. In this in vitro kinase assay using the Src-substrate peptide, we did not see enhanced kinase activity with the constitutively activated Fyn in which negatively regulated tyrosine residue Y531 was replaced by phenylalanine: The specific activity of mutant Fyn, calculated as kinase activity versus amount of Fyn protein, was equivalent to that of the native Fyn. Although we could not interpret this unexpected result, it might be possible that in this in vitro kinase assay using the synthetic peptide we fail to distinguish the inactivated form of Fyn kinase from the activated one, despite the presence of an excess amount of the phosphatase inhibitor, sodium vanadate. The small substrate peptide, but not the endogenous substrates, may be accessible to the inactivated Fyn kinase, as well as to the activated kinase. In contrast to the data from in vitro kinase assay, we observed that in vivo the enhancement of tyrosine phosphorylation of several endogenous proteins was higher in mutant fyn transgenic mice than that in native fyn transgenic mice as described below.
Figure 2.
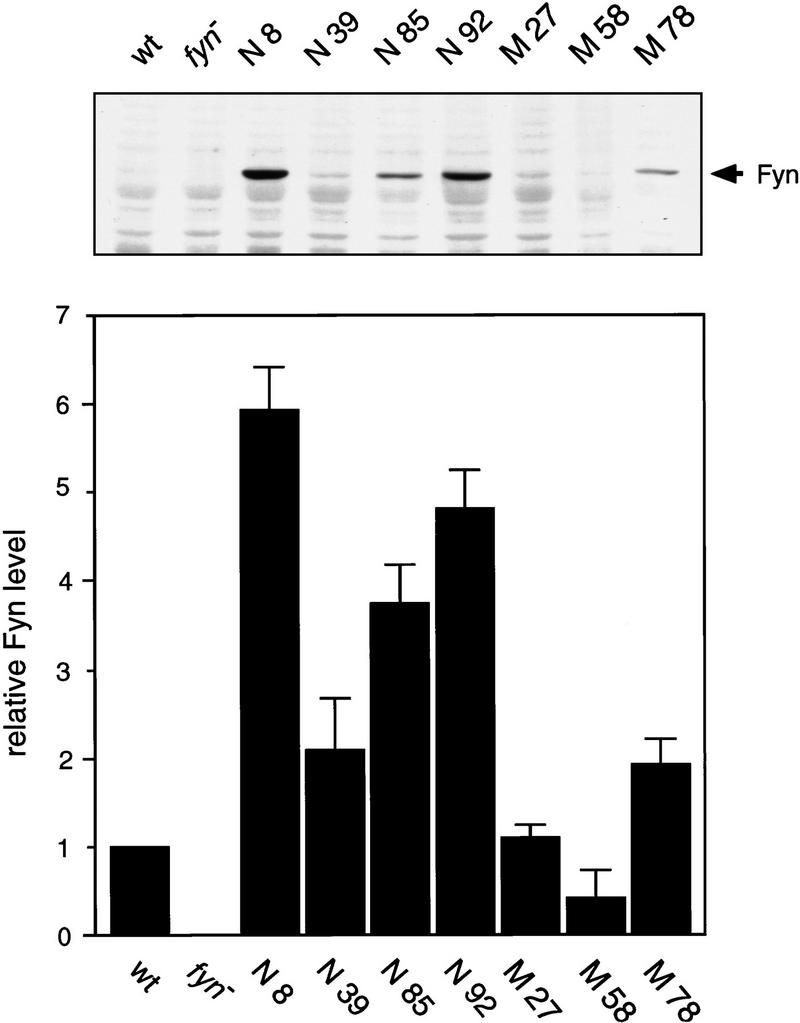
The levels of Fyn protein in fyn transgenic mice. Transgenic Fyn protein was detected by Western blotting of RIPA extract (20 μg of protein) prepared from the forebrain of each transgenic mouse line generated on fyn knockout genetic background (Tg+/fyn−/−). Relative amount of Fyn protein was determined by densitometry of the fluorographic image and compared to that in wild-type mice (wt). Each bar represents the mean ± s.e.m. from seven independent experiments.
Table 1.
Fyn and Src kinase activities in the forebrain of fyn transgenic mice
| Mouse line
|
Fyn kinase activity (fmoles/min/μg− ± s.e.m.)
|
Src kinase activity (fmoles/min/μg ± s.e.m.)
|
||
|---|---|---|---|---|
|
fyn+/+ background
|
fyn−/− background
|
sp. act.a
|
||
| Nontransgenic | ||||
| wild-type | 1.91 ± 0.29 (7) | — | 1.86 ± 0.29 | 3.54 ± 0.18 (6) |
| fyn− | — | 0.01 ± 0.03 (4) | — | 6.33 ± 0.27 (4) |
| Native fyn transgenic | ||||
| N 8 | 16.47 ± 1.40 (7) | 13.44 ± 1.37 (6) | 2.27 ± 0.21 | 3.21 ± 0.29 (6) |
| N 39 | 5.63 ± 0.96 (7) | 3.76 ± 0.54 (6) | 1.81 ± 0.24 | 3.60 ± 0.60 (3) |
| N 85 | 14.20 ± 0.95 (7) | 9.41 ± 1.29 (4) | 2.52 ± 0.30 | 3.52 ± 0.28 (6) |
| N 92 | 13.21 ± 0.91 (7) | 9.75 ± 0.74 (6) | 2.03 ± 0.14 | 3.43 ± 0.48 (6) |
| Mutant fyn transgenic | ||||
| M 27 | 4.08 ± 0.50 (7) | 1.72 ± 0.22 (6) | 1.56 ± 0.18 | 3.79 ± 0.06 (3) |
| M 58 | 3.77 ± 0.25 (7) | 1.00 ± 0.14 (6) | 2.44 ± 0.35 | 3.78 ± 0.31 (3) |
| M 78 | 5.50 ± 0.56 (7) | 3.30 ± 0.32 (6) | 1.71 ± 0.12 | 3.61 ± 0.38 (3) |
Fyn kinase activity of RIPA extract of forebrain was measured in wild-type mice, fyn knockout mice (fyn−) and several lines of native and mutant fyn transgenic mice generated on either wild-type (fyn+/+) or fyn knockout (fyn−/−) genetic background. Src kinase activity was also measured in each group of mice, wild-type mice, fyn knockout mice (fyn−), and several lines of native and mutant fyn transgenic mice generated on wild-type (fyn+/+) genetic background. Value represents the mean ± s.e.m.. Total number of animals examined is represented in a parenthesis.
The specific activity was determined by dividing the kinase activity, which was measured in wild-type mice and in fyn transgenic mice on fyn knockout genetic background, from the relative amount of Fyn protein estimated by immunoblotting (Fig. 2).
OVEREXPRESSION OF FYN TRANSGENE DOES NOT AFFECT SRC KINASE ACTIVITY
Previous reports show that Src kinase activity was up-regulated by the disruption of Fyn (Grant et al. 1995). To determine whether overexpression of Fyn affects Src kinase activity, we measured Src kinase activity in forebrain extracts prepared from wild-type mice, fyn knockout mice, and fyn transgenic mice on wild-type genetic background (Tg+/fyn+/+). Although Src kinase activity was elevated twofold by the disruption of Fyn, we found that Src kinase activity was not changed by overexpression of either native or mutant Fyn (Table 1). Similarly, whereas the level of Src protein was also elevated twofold in fyn knockout mice, we did not see any changes in the level of Src protein in the transgenic animals, as estimated by immunoblotting (data not shown).
MICE EXPRESSING MUTANT FYN ARE PRONE TO SPONTANEOUS DEATH
Because we used the heterozygous fyn knockout mice for establishing each transgenic line, the genotype of F1 offspring was a mixture of wild-type (fyn+/+) and heterozygote (fyn+/−). In overall appearance, transgenic mice expressing native and mutant Fyn were indistinguishable from their nontransgenic littermates. The fyn transgenic mice also show normal reproductive behavior and no obvious mortality at the prenatal stages. Histological examination of the brain of the fyn transgenic mice did not reveal any overt abnormalities at the light microscopic level (. Nevertheless, we found that mice expressing mutant Fyn were prone to spontaneous death, generally after weaning. We surveyed the viability of postweaning F1 offspring for up to 20 weeks (Table 2). We observed no difference in the viability between wild-type mice and heterozygotes in the absence of the transgene. In three out of the six lines that expressed the mutant Fyn (M3, 47, and 84), animals died within 8, 5, and 15 weeks, respectively. We detected high levels of transgenic fyn mRNA in line M84 by ribonuclease protection assay, comparable to the level in line N92 (data not shown). In the remaining three lines (M27, 58, and 78), the survival rate was lower than that in native fyn transgenic lines. In these three lines, the survival rate correlated inversely with the level of mutant Fyn protein (Fig. 2); survival was higher in line M58 (which shows the lowest level of mutant Fyn protein) than in lines M27 and 78. The sudden death was observed generally in mice expressing mutant Fyn when the mice were >4 weeks of age. In these lines, we have often observed spontaneous running and bouncing fits as well as tonic convulsion followed by death. By contrast, >88% of transgenic mice expressing native Fyn survived >20 weeks, although on rare occasion we also observed seizure in lines N8 and 92.
Table 2.
Survival rate of fyn transgenic mice
| Mouse line | Percent survival (n) |
|---|---|
| Nontransgenic | |
| fyn+/+ | 100 (49) |
| fyn+/− | 94 (45) |
| Native fyn transgenic | |
| N 8 | 96 (46) |
| N 39 | 100 (9) |
| N 85 | 100 (29) |
| N 92 | 88 (34) |
| Mutant fyn transgenic | |
| M 3 | 0 (3) |
| M 27 | 54 (26) |
| M 47 | 0 (10) |
| M 58 | 71 (17) |
| M 78 | 57 (21) |
| M 84 | 0 (7) |
The survival rate of postweaning transgenic F1 offspring was surveyed up to 20 weeks and represented as a percentage of mice survived. The total number surveyed is represented in a parenthesis.
For further physiological and biochemical studies we analyzed fyn transgenic mice generated on wild-type genetic background (Tg+/fyn+/+) with a comparison of nontransgenic wild-type mice (Tg−/fyn−/−), and in some experiments with a comparison of fyn knockout mice (Tg−/fyn−/−) .
THRESHOLD FOR PTZ-INDUCED SEIZURE IS REDUCED IN MICE EXPRESSING MUTANT FYN
To determine whether the threshold for seizure is altered in the transgenic mice expressing mutant Fyn, we administered penthylenetetrazole (PTZ) to wild-type mice, to fyn knockout mice, and to several lines of fyn transgenic mice (N8, N85, M27, and M78). An intravenous infusion of PTZ with a constant flow rate elicits a series of stereotyped seizure responses, beginning with head-twitching, proceeding to clonic seizure, followed by tonic extension, and death. The drug doses required for first twitch and tonic convulsion were compared among the groups of animals (Fig. 3A). In contrast to the report that another line of fyn knockout mice shows higher seizure susceptibility (Lu et al. Miyakawa et al. 1996), our fyn knockout mice did not show any significant differences in PTZ sensitivity from wild-type mice: The dose required for first twitch and tonic extension was 42.0 ± 2.2 and 84.8 ± 6.8 in wild-type mice (n = 10), respectively, and 46.2 ± 3.9 and 83.2 ± 11.1 in fyn knockout mice (n = 4). None of the transgenic lines showed significant change in the PTZ dose for first twitch [36.1 ± 2.4 in N8 (n = 10), 37.1 ± 2.2 in N85 (n = 6), 38.9 ± 3.4 in M27 (n = 8), 34.9 ± 3.1 in M78 (n = 5)]. However, in mice expressing mutant Fyn, we observed a reduction in the drug dose required for tonic extension. Consistent with their low survival rate, in the lines expressing mutant Fyn, M27 and M78, the lethal dose of PTZ was reduced significantly (by 31% and 38%, respectively), as compared with wild-type mice [62.6 ± 7.7 in M27 (n = 8), 56.2 ± 6.8 in M78 (n = 5); wild-type vs. M27, P = 0.0469; wild-type vs. M78, P = 0.021, two-tailed, unpaired t-test]. A small reduction of the lethal dose also was observed in mice expressing native Fyn, lines N8 and N85, but the difference was not significant statistically [70.9 ± 8.6 in N8 (n = 10), 75.2 ± 11.3 in N85 (n = 6); wild-type vs. N8, P = 0.2201; wild-type vs. N85, P = 0.4583].
Figure 3.



Seizure susceptibility in fyn transgenic mice induced by penthylenetetrazole and electrical stimulation of the amygdala. (A) Threshold for penthylenetetrazole (PTZ)-induced seizure in the group of mice was determined by a continuous infusion of 1.0% of PTZ (0.1 ml min−1) intravenously. The time required for first twitch and tonic extension was monitored. Data were corrected for body weight and converted into the corresponding PTZ dose (mg/kg body weight). Each bar represents the mean ± s.e.m. (Solid area) Twitch; (open area) extension. The PTZ dose required for tonic extension was reduced significantly in mice expressing mutant Fyn (M27 and 78). (*) P < 0.05 (two-tailed, unpaired t-test). (B) Representative electroencephalograms recorded from the amygdala of a wild-type (wt, upper trace) and a mutant fyn transgenic mice (M78, lower trace) after the electrical stimulation. Thick bar represents the duration of the stimulation. Prolonged electrical response associated by behavioral seizure was often elicited in mice expressing mutant Fyn. (C) The AD duration and convulsion stage of the individual animals after the initial stimulation were plotted. Convulsion stage after the stimulation was determined according to the modified Racine’s classification (see Materials and Methods).
ELECTRICAL STIMULATION OF AMYGDALA ELICITS HIGHER SEIZURE ACTIVITY IN MICE EXPRESSING MUTANT FYN
A brief electrical stimulation to the amygdala induces an elctrographic seizure, an AD. To examine whether overexpression of Fyn or the constitutively active mutant, which is also expressed in the amygdala (Figs. 1 and 5A, below), affects AD generation, we stimulated the left amygdala of several lines of fyn transgenic mice and recorded the behavioral response and stimulus-induced electroencephalogram through the same electrode. Although we did not determine the precise threshold for AD, the intensity of the stimulus required to induce AD did not differ among the groups [42.2 ± 4.1 μA in wild-type mice (n = 26), 39.1 ± 11.7 μA in N8 (n = 18), 39.5 ± 9.4 μA in N39 (n = 8), 46.0 ± 7.7 μA in M27 (n = 8), 35.8 ± 5.3 μA in M78 (n = 7)]. Nonetheless, we observed that higher seizure activity was frequently elicited in mice expressing mutant Fyn. Whereas the stimulus induced a brief AD in wild-type mice (<20 sec without any convulsive responses), a prolonged AD lasting longer than 30 sec was often elicited in mice expressing mutant Fyn (Fig. 3B). In three out of eight mice in line M27 and three out of seven in line M78, this prolonged AD was accompanied by a convulsive response more severe than convulsion stage 1 (Fig. 3C). By contrast, the majority of two lines expressing native Fyn, N8, and N39, showed normal AD and no obvious convulsive responses, although in three out of 18 mice of line N8 there was a prolonged AD lasting >20 sec accompanying stage 3 or 4 convulsion.
Figure 5.




Change in tyrosine phosphorylation of the brain proteins in fyn transgenic mice. (A) PY-containing proteins and Fyn were detected by immunoblotting of RIPA extract (25 μg protein) prepared from wild-type mice (wt) and line N8 (tg) using anti-PY antibody (αPY) and anti-Fyn antibody (αFyn), respectively. Tyrosine phosphorylation of PY180, 120, 100, and Fyn (arrowheads) was enhanced in some brain regions where Fyn was overexpressed. The highest level of the enhancement was observed in the telencephalon, including the cerebral cortex, hippocampus, and amygdala/pyriform cortex (amygdala), where Fyn was overexpressed. Weak but significant enhancement of tyrosine phosphorylation was observed in the olfactory bulb, diencephalon and midbrain. By contrast, the enhancement of tyrosine phosphorylation was not observed in the medulla oblongata and pons (medulla/pons) and cerebellum where transgenic Fyn was expressed at a low level. Molecular weight markers (in kilodaltons) are indicated at left. (B) PY-containing proteins were detected by immunoblotting of the forebrain extracts (25 μg protein) prepared from wild-type (wt), fyn knockout (fyn−) and several lines of fyn transgenic mice. Tyrosine phosphorylation of PY180, 120, and 100 (arrowheads) was enhanced in most of fyn transgenic lines and conversely reduced in fyn knockout mice. Molecular mass markers (in kilodaltons) are indicated at left. (C) The level of PY180 (open bars) and 100 (hatched bars) was determined by densitometry of the fluorographic image. Each bar represents the mean value normalized with the level in wild-type mice ± s.e.m. from seven independent experiments. Significant change from the value in wild-type mice is indicated by asterisks; (*) P < 0.05, (**) P < 0.01, (***) P < 0.005 (two-tailed, one group t-test). (D) The relationship between relative level of PY proteins and relative amount of Fyn normalized with that in wild-type mice is plotted. The values shown are obtained from the (□) endogenous or native Fyn and (█) mutant Fyn, respectively. The phosphorylation of each PY protein was enhanced in parallel to the relative level of Fyn protein, and the level of enhancement was higher in mutant Fyn than that in native Fyn.
KINDLING IS ACCELERATED IN FYN TRANSGENIC MICE
The majority of mice expressing native Fyn and half of mice expressing mutant Fyn (M27 and 78) showed no overt convulsive responses after the initial stimulation. To determine whether overexpression or constitutive activation of Fyn alters kindling, we kindled those mice that failed to show convulsions more severe than stage 1 at the initial stimulation. Kindling was induced by stimulating the amygdala once per day at the intensity equal to that of the initial stimulation. As shown in Figure 4A, both the AD duration and convulsion stage progressed more rapidly in line N8, which shows the highest level of native fyn transgene expression, and in mice expressing mutant Fyn (data from M27 and 78 were combined). The number of ADs required for reaching fully generalized seizure was significantly different among the groups of mice [ANOVA F(3,33) = 5.50, P = 0.0035, Fig. 4B]. A significant decrease in the number of ADs was observed in N8 and in mice expressing mutant Fyn [11.5 ± 0.5 ADs in wild-type mice (n = 17), 7.9 ± 0.4 ADs in N8 (n = 7), 8.2 ± 0.7 ADs in M27 and 78 (n = 9); wild-type vs. N8, P = 0.0056, wild-type vs. M27 and 78, P = 0.0066, post hoc Fisher’s PLSD test]. On the other hand, in line N39, which expresses native fyn transgene at a low level, kindling developed normally and the AD number required for generalized seizure was similar to that in wild-type mice [12.5 ± 3.2 ADs (n = 4), wild-type vs. N39, P = 0.50]. Thus, the acceleration of kindling is likely to depend on the expression level of native fyn transgene.
Figure 4.
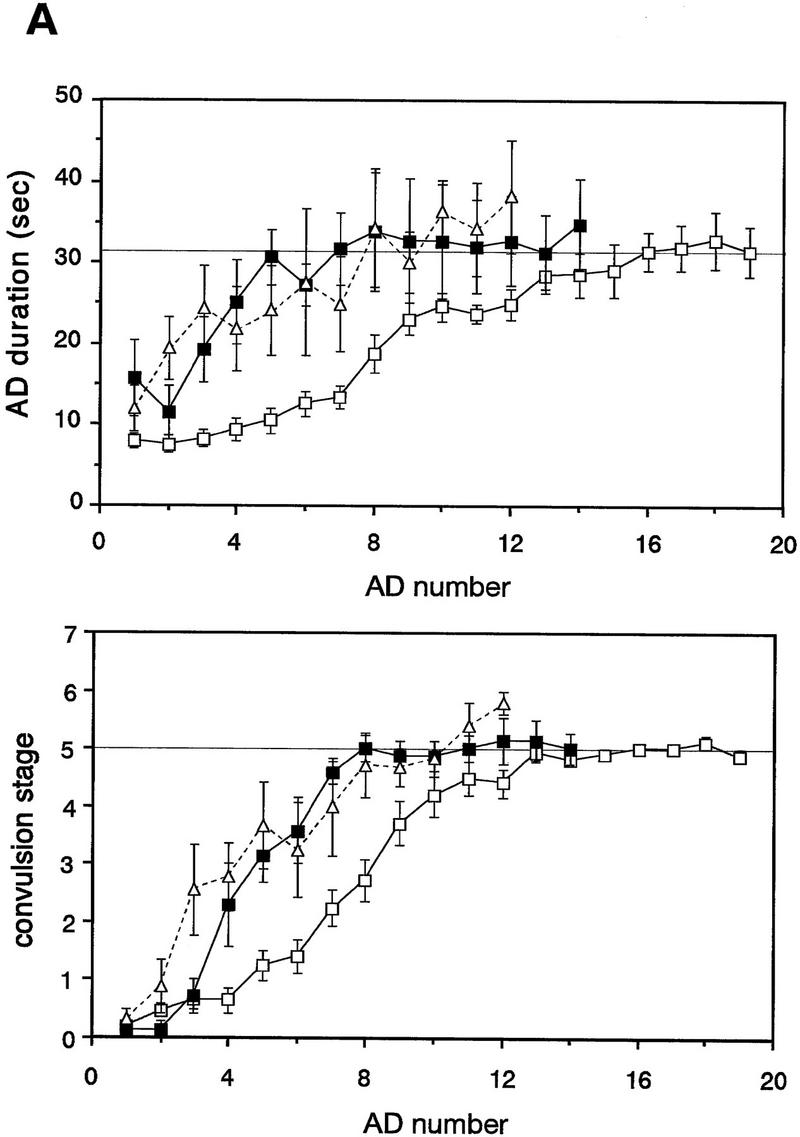


Amygdaloid kindling in fyn transgenic mice. (A) Once-daily stimulation of the amygdala was delivered to wild-type mice (wt, □, n = 9) and fyn transgenic mice which did not show convulsion greater than stage 1 after the initial stimulation (N8, █, n = 7; M27 and 78, ▵, n = 9). AD was recorded from the ipsilateral amygdala. Values represent the mean ± s.e.m. The thin line in the upper graph indicates the mean of AD duration at the nineteenth stimulation in wild-type mice (31.7 sec). The AD duration and convulsion stage progressed more rapidly in a line expressing native Fyn N8 and mice expressing mutant Fyn M27 and 78, as compared with wild-type mice. (B) Each bar represents the mean of the AD number required for full kindling ± s.e.m. A decrease in the AD number was significant in the lines N8 and M27 and 78. (*) P < 0.01 (Fisher’s PLSD test). (C) The severity of kindling was compared among the group of mice which were fully kindled. The white bar represents the mean of AD duration ± s.e.m.; the hatched bar represents the mean of convulsion stage ± s.e.m. Whereas the AD duration was not different significantly from that in wild-type mice, convulsion stage was significantly higher in kindled mice expressing mutant Fyn. (*) P < 0.01 (Fisher’s PLSD test).
In spite of the acceleration of kindling, the severity of fully generalized seizure in line N8 was similar to that in wild-type mice [stage 5.0 ± 0.1 in wild-type (n = 17), stage 5.0 ± 0.2 in N8 (n = 7), Fig. 4C]. The duration of AD was not significantly longer than that in wild-type mice, and convulsions more serious than stage 5 was rare in line N8. By contrast, convulsion stage was significantly higher in kindled mice expressing mutant Fyn [stage 6.0 ± 0.5 in M27 and 78 (n = 9), ANOVA F(3,33) = 3.34, P = 0.031; wild-type vs. M27 and 78, P = 0.0085, post hoc Fisher’s PLSD test]. Convulsions more severe than stage 5 were elicited in 4 out of 9 kindled mice expressing mutant Fyn. Two of these mice had stage 8 seizures, comprising tonic extension and death.
Once mice were kindled fully, the susceptibility to seizure persisted for at least 1 month. We found no significant reduction in the electrographic and behavioral responses to the stimulation after the resting period of 1–2 months (data not shown). During this period, a number of kindled mice expressing mutant Fyn died from spontaneous seizure.
TYROSINE PHOSPHORYLATION OF MULTIPLE PROTEINS IS ENHANCED IN THE FOREBRAINS OF FYN TRANSGENIC MICE
We demonstrated previously that multiple proteins were significantly less phosphorylated in the brains of fyn knockout mice (Kojima et al. 1997; see also Grant et al. 1995), suggesting that they are phosphorylated by Fyn preferentially. Using anti-PY antibody in RIPA extracts from several brain regions of line N8, we first examined changes in the level of PY-containing proteins by immunoblotting. As shown in Figure 5A, at least three protein bands (designated as PY180, PY120, and PY100 by their relative molecular sizes), as well as an ∼60-kD protein that was identified as phosphorylated Fyn, were hyperphosphorylated in the forebrain, including cerebral cortex, hippocampus, and amygdala/piriform cortex complex, the regions where the highest level of Fyn protein was observed. Weak but significant enhancement of tyrosine phosphorylation was observed in the olfactory bulb, diencephalon, and midbrain, in which the Fyn protein was expressed moderately. By contrast, the enhanced tyrosine phosphorylation was not observed obviously in the medulla oblongata or pons, or in the cerebellum, where the transgenic Fyn protein was expressed at a low level. Thus, the regional pattern of the enhancement of tyrosine phosphorylation was consistent with the regional distribution of transgenic Fyn protein.
We next examined changes in tyrosine phosphorylation of these hyperphosphorylated proteins in the forebrains of individual fyn transgenic lines (Fig. 5B). Significant enhancement of tyrosine phosphorylation was observed in the forebrains from all the transgenic lines except line M58, in which mutant fyn transgene was expressed at the lowest level. We quantified the intensity of each band and normalized it to that in wild-type mice. As shown in Figure 5C, the relative level of PY180 and 100 was the highest in line N8, which expresses transgenic Fyn at the highest level; it was also high in other lines except with line M58, and it was low in the knockout mice. The phosphorylation of PY120 showed the same change, but we could not quantify accurately the intensity of the band because the signal of PY120 was weak and it was difficult to detect in wild-type mice. Interestingly, the relative level of these proteins was remarkably high in mutant fyn transgenic mouse lines, M27 and M78, despite the fact that the level of transgenic mutant Fyn and its kinase activity measured by in vitro kinase assay using the synthetic substrate peptide were relatively low in these lines. We also analyzed the relationship between the levels of PY proteins and the Fyn protein. When the relative levels of PY180 and 100 were plotted against the relative level of Fyn protein, the enhancement of their phosphorylation was in parallel to the relative Fyn level; it was higher in mutant Fyn than that in native Fyn (Fig. 5D). This result indicates that the in vivo mutant Fyn is able to phosphorylate the endogenous substrates more effectively than the native Fyn.
TYROSINE PHOSPHORYLATION OF THE NR2 subunits is enhanced by overexpression of fyn
A major 180-kD PY-containing protein in the postsynaptic density fraction, Gp180, has been identified as NR2B (Moon et al. 1994). Furthermore, the NR2A (ε1) and 2B (ε2) proteins have shown to be phosphorylated by Fyn in vitro (Suzuki and Okumura-Noji 1995). We therefore investigated whether one of the hyperphosphorylated proteins in fyn transgenic mice, PY180, was identical to the NR2 subunit. We immunoprecipitated NR2 subunit from SDS-solubilized P2 membrane prepared from each group of animals (wild-type, fyn knockout, and several lines of fyn transgenic mice). As shown in Figure 6A, we found a significant change in tyrosine phosphorylation of the NR2B in fyn knockout mice and both native and mutant fyn transgenic lines except with line M58 [ANOVA F(7, 29) = 9.58266, P = 0.0001]. The relative ratio of phosphorylated NR2B to total NR2B, as compared to that of wild-type mice, was reduced significantly in fyn knockout mice [0.4 ± 0.1 (n = 5), P = 0.0034, two-tailed, one-group t-test] and it was increased significantly in mice overexpressing either native Fyn or mutant Fyn [2.4 ± 0.3 in N8 (n = 5), P = 0.001; 1.6 ± 0.3 in N39 (n = 5), P = 0.0178; 2.0 ± 0.2 in N92 (n = 4), P = 0.0143; 2.0 ± 0.3 in M27 (n = 5), P = 0.0097; 2.3 ± 0.2 in M78 (n = 4), P = 0.0066]. Similar to the change in tyrosine phosphorylation of PY180, the level of enhancement of tyrosine phosphorylated NR2B was parallel to the relative amount of Fyn protein. The bands of NR2B comigrated with that of PY180 on SDS-PAGE and PY180 was immunoprecipitated with anti-PY antibody and then reacted with anti-NR2B antibody (data not shown). Thus, PY180, which is one of the major PY-containing proteins and is hyperphosphorylated in both mice expressing native and mutant Fyn, and is hypophosphorylated in fyn knockout mice, seems to be identical to the NR2B.
Figure 6.

Change in tyrosine phosphorylation of NR2 subunits in fyn transgenic mice. (A) The NR2B was immunoprecipitated from SDS-solubilized P2 membrane (500 μg of protein) prepared from each group of mouse brain by anti-NR2B antibody, then subjected to SDS-PAGE/immunoblotting with ether anti-PY antibody (αPY, top) or anti-NR2B antibody (αNR2B, bottom). The intensity of the band was determined by densitometric analysis of the fluorographic image. Relative ratio of the level of phosphorylated NR2B to the total level of NR2B was increased significantly in all the fyn transgenic lines except with line M58 and decreased in fyn knockout mice (fyn−), as compared to that in wild-type mice (wt). (*) P < 0.05, (**) P < 0.01, (***) P < 0.005 (two-tailed, one-group t-test). (B) The NR2A was immunoprecipitated from SDS-solubilized P2 membrane (500 μg protein) prepared from each group of mouse brain by anti-NR2A antibody and subjected to SDS-PAGE/immunoblotting with ether anti-PY antibody (αPY, top) or anti-NR2A antibody (αNR2A, bottom). The intensity of the band was determined as described above. Relative ratio of the level of phosphorylated NR2A to the total level of NR2A was significantly increased in lines N8 and 92, as compared to that in wild-type mice (wt). *P < 0.05 (two-tailed, one-group t-test).
We also observed significant change in tyrosine phosphorylation of NR2A, another subunit of the NMDA receptor [ANOVA F(6,21) = 5.4804, P = 0.0015, Fig. 6B]. Significant enhancement was observed in lines N8 and N92 [2.0 ± 0.2 in N8 (n = 4), P = 0.0174; 1.5 ± 0.1 in N92 (n = 4), P = 0.0102, two-tailed, one group t-test]. Thus, this subunit also capable of being phosphorylated by Fyn in vivo, as reported in vitro (Suzuki and Okumura-Noji 1995). However, this enhancement was smaller than that observed in NR2B and we could not find significant level of enhancement in line N39 or in the mutant fyn transgenic lines. A striking difference in tyrosine phosphorylation of the NR2A from that of the NR2B was observed in fyn knockout mice: Tyrosine phosphorylation of the NR2A was not affected by the disruption of Fyn, whereas tyrosine phosphorylation of the NR2B was reduced by half in these mice.
ACCELERATION OF KINDLING IS SUPPRESSED BY MK-801
The NMDA receptor is known to be involved in development of kindling (Gilbert 1988; McNamara 1988; Sato et al. 1988; Holmes et al. 1990; Croucher et al. 1995). To evaluate whether kindling in line N8 is also mediated by the NMDA receptor activity, we administered MK-801 systemically, an activity-dependent noncompetitive NMDA receptor antagonist, 1–2 hr before electrical stimulation from days 2–11. MK-801 (1 mg/kg intraperitoneally) induced a stereotyped motor syndrome consisting of increased locomotor activity, lateral head turning, jumping, and ataxia (Tricklebank et al. 1989). This behavioral response was observed 15 min after injection and lasted up to 2–3 hr. Kindling was retarded by preadministration of MK-801 in wild-type mice, as reported previously in rat (Gilbert 1988; McNamara 1988; Sato et al. 1988). As shown in Figure 7, administration of MK-801 also suppressed kindling in line N8. The AD duration and convulsion stage after the 10th stimulation were not significantly different from that in wild-type mice [17.1 ± 5.4 sec and stage 2.0 ± 0.8 in N8 (n = 8), 18.9 ± 3.1 sec and stage 1.4 ± 0.4 in wild-type mice (n = 9), respectively]. This effect was reversible: Mice could be subsequently kindled by once-daily electrical stimulation without MK-801.
Figure 7.
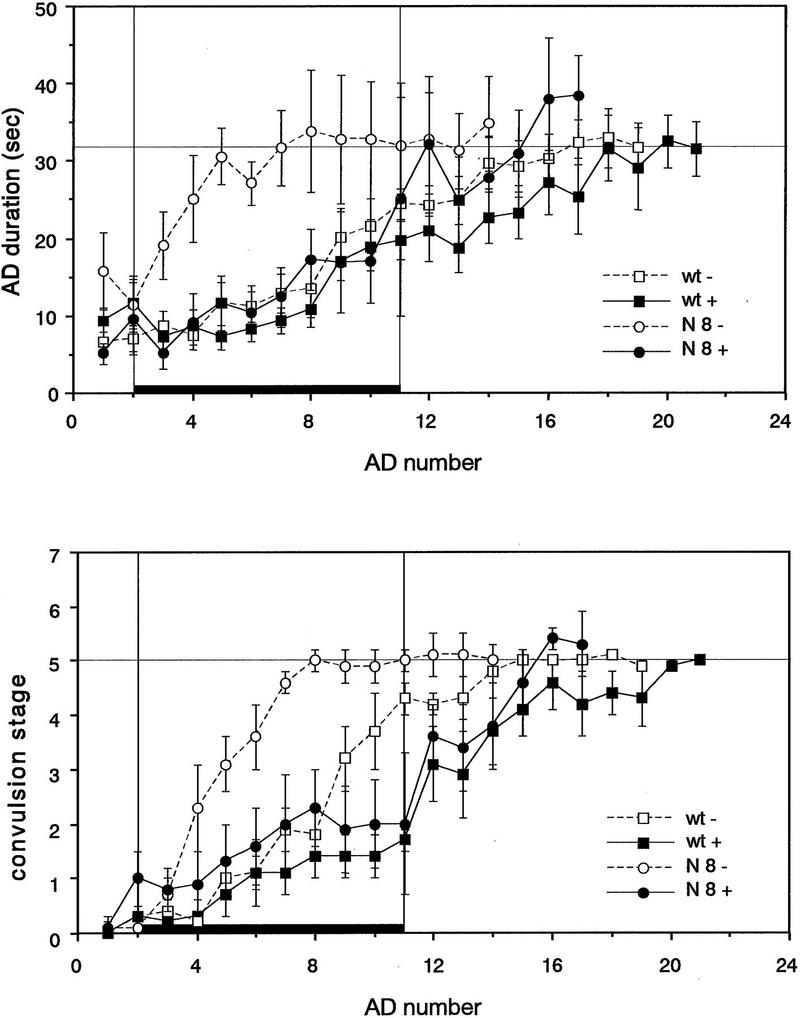
Effect of MK-801 on kindling development in fyn transgenic mice. MK-801 was systemically administered prior to once-daily stimulation of the amygdala in wild-type mice (wt +, █, n = 9) and fyn transgenic line N8 (N8 +, •, n = 8) from days 2–11. The period of the drug treatment is represented by thick bars. Values represent the means of the AD duration and convulsion stage ± s.e.m. Values in control animals which were not treated with the drug are also represented (wt −, □, n = 7; N8−, ○, n = 7). MK-801 reversibly suppressed the development of kindling in the line N8, as well as that in wild-type mice.
Discussion
To obtain a better understanding of the physiological role of the Fyn protein in LTP and in behavior, we have examined the physiological and biochemical effects in the adult brain of overexpressing either native Fyn or the constitutively active mutant. The spatial and temporal expression pattern of fyn transgenes driven by the CaMKIIα promoter is essentially similar to CaMKIIα rather than the endogenous Fyn. In the adult brain, the fyn transgenes were expressed at high levels in forebrain, presumably specific to neurons, whereas the endogenous Fyn is expressed more widely in both neurons and glial cells (Umemori et al. 1992; Yagi et al. 1993). Another important difference between endogenous and transgenic Fyn is in their time course of expression. Whereas the endogenous Fyn is expressed highly in the embryonic brain and localized in developing axons and growth cones (Bare et al. 1993; Bixby and Jhabvala 1993), the fyn transgene does not turn on until late in development. Transgenic fyn mRNA is expressed only at a very low level at postnatal day 1 and increases gradually during first 10–14 postnatal days (Kojima et al. 1997).
Consistent with the idea that Fyn has an important role on neural development, the disruption of Fyn leads to abnormalities in brain anatomy (Grant et al. 1992). Thus, it was difficult to determine in these knockout mice whether there was a direct connection between Fyn and adult brain functions such as hippocampal LTP and spatial learning. Previous studies showed that introduction of the native fyn transgene under the CaMKIIα promoter rescued the LTP phenotype but not the morphological abnormalities in the hippocampus (Kojima et al. 1997). Although we did not determine the precise time-point of transgene expression in each line, no lines of fyn transgenic mice show overt anatomical abnormalities, suggesting that the fyn transgene expression did not affect neural development. Thus, the fyn transgene expressed under the CaMKIIα promoter allows us to distinguish between the physiological and developmental roles of Fyn.
Another drawback of the conventional knockout approaches is that compensatory response of the related molecules may mask phenotypes of the knockout mice or cause secondary defects in fyn knockout mice. The elevation of Src kinase activity may compensate for the lack of Fyn. We pointed out previously the possibility that Src compensates the deficit in LTP in fyn knockout mice <15 weeks of age (Kojima et al. 1997). We did not find any changes in the level of Src kinase activity in fyn transgenic mice. Thus, we can exclude the possibility that secondary change in Src kinase activity affects phenotypes observed in fyn transgenic mice.
We found that mice expressing mutant Fyn were prone to sudden death and exhibited higher seizure activity. Disruption or overexpression of certain genes often leads to spontaneous seizure (for review, see Noebels 1996). Some genes are known to be related directly to neuronal excitability, such as potassium channels (Patil et al. 1995; Signorini et al. 1997) and a certain subunit of the GABAA receptor (Homanics et al. 1997). However, the molecular mechanism for epileptogenesis in most epileptic mutants including Synapsin I/II (Rosahl et al. 1995), CaMKIIα (Butler et al. 1995), and serotonin receptor 5-HT2C (Tecott et al. 1995), is still not clear.
How could Fyn regulate neuronal excitability? The findings that lines showing higher expression of mutant fyn transgene exhibited higher mortality and higher incidence of spontaneous seizure, and that spontaneous death was observed after postnatal 4 weeks, when the transgene expression reached its maximal level, suggest that severity of seizure activity is related to the level of expression of the activated Fyn. In contrast to mice expressing mutant Fyn, mice expressing native Fyn did not show such an obvious epileptic phenotype, even though the level of transgene expression in all the lines expressing native Fyn was much higher than in the remaining three lines expressing mutant Fyn. The differences in their phenotypes suggest that perhaps the CSK-type kinase exerts a regulatory mechanism that suppresses the effects of the overexpression of native Fyn. Although we do not know how the Fyn kinase activity is regulated in neurons, our data imply that constitutive dephosphorylation of the carboxy-terminal tyrosine residue in Fyn leads to enhancement of excitatory function or reduction of inhibitory function in the brain.
We found that at least three proteins were hyperphosphorylated in the brain of the transgenic mice overexpressing native and mutant Fyn: PY180, PY120, and PY100. These three proteins were conversely less phosphorylated in fyn knockout mice, suggesting that these proteins are substrates of Fyn in neurons. Despite clear differences in their phenotypes, we could not find qualitative differences in tyrosine phosphorylation between mice overexpressing native and mutant Fyn, with the exception of increased tyrosine-phosphorylated Fyn in mice overexpressing native Fyn. The finding that tyrosine phosphorylation of these three proteins was enhanced in both sets of transgenic mice suggests that epileptic phenotype observed in mice expressing mutant Fyn may not be explained simply by enhanced phosphorylation. Constitutive phosphorylation of substrates by transgenic mutant Fyn may secondarily cause other molecular defects related to neuronal excitability. In hippocampal slices, a decrease of GABAergic inhibition has been found in mice expressing mutant Fyn, but not in mice expressing native Fyn (Lu et al. 1998). This effect might result in higher seizure activity observed in mice expressing mutant Fyn.
Though without an obvious epileptic phenotype, one transgenic line overexpressing native Fyn, N8, also exhibited accelerated kindling as compared with wild-type mice. This effect seems to be distinct from higher seizure susceptibility in mice expressing mutant Fyn, because the majority of the line N8 did not show convulsive response at the initial stimulation. Excepting the rate of kindling, these mice showed the similar aspects of kindling phenomena to that in wild-type mice, in progression of the AD duration and convulsive response, in severity of kindled seizure, and in stability of seizure susceptibility. By contrast, line N39 expressing transgenic Fyn at relatively low level was normal and did not show an acceleration of kindling. Thus, the rate of kindling is likely to correlate with the native Fyn protein level and with the phosphorylation levels of the three PY proteins. Although we cannot rule out the inverse possibility that the physiological change underlying the acceleration of kindling in line N8 is sufficient to induce the enhancement of tyrosine phosphorylation of these PY proteins, tyrosine phosphorylation by Fyn may affect the seizure susceptibility in kindling. The finding by Cain et al. (1995) that kindling is retarded in fyn knockout mice, and the finding in the present study that the PY proteins are hypophosphorylated in the knockout mice, support this idea.
Pharmacological studies suggest that NMDA receptor activity is involved in molecular mechanism for kindling (Gilbert 1988; McNamara 1988; Sato et al. 1988; Holmes et al. 1990; Croucher et al. 1995). The finding that administration of MK-801 suppressed the development of kindling in line N8, as well as wild-type mice, suggests that the accelerated kindling observed in mice overexpressing Fyn is also mediated by the NMDA receptor activity.
The NMDA receptor is phosphorylated by several protein kinases including protein kinase C (Chen and Huang 1992; Tingley et al. 1993), CaMKII (Omkumar et al. 1996), and PTKs. The NR2A and NR2B subunits have been shown to be targets for tyrosine phosphorylation (Moon et al. 1994; Lau and Huganir 1995; Suzuki and Okumura-Noji 1995) and to be able to bind directly to SH2 domains (Gurd and Bissoon 1997). The channel activity of the NMDA receptor is regulated by tyrosine phosphorylation (Wang and Salter 1994). Yu et al. (1997) have shown that activation of endogenous Src increases the activity of NMDA receptor and that Src interacts directly with the NMDA receptor. Furthermore, Zheng et al. (1998) have reported recently that Src potentiates the NMDA receptor currents by the relief of zinc inhibition. We found that tyrosine phosphorylation of the NR2 subunits was enhanced by overexpression of Fyn. Thus, Fyn also phosphorylates these subunits in vivo. Our data also suggest that the NR2B, but not the NR2A, is phosphorylated preferentially by Fyn rather than Src, because upregulation of Src seems not to compensate for hypophosphorylation of the NR2B in fyn knockout mice. The amino acid sequence of the carboxy-terminal intracellular domain, which is supposed to be phosphorylated by Src and Fyn, exhibits relatively low homology between the NR2A and NR2B, suggesting that tyrosine phosphorylation of these two modulatory subunits of the NMDA receptor may be differentially regulated.
In a coexpression experiment using cultured HEK 293 embryonic kidney cells, Köhr and Seeburg (1996) suggest that an increase in the NMDA receptor current induced by Src and Fyn is mediated by tyrosine phosphorylation of NR2A, but not NR2B. The contribution of tyrosine phosphorylation of these two NR2 subunits on the NMDA receptor activity might be different, and tyrosine phosphorylation of NR2B may not alter channel properties directly. This idea is consistent with the finding that the NMDA current is normal in fyn knockout hippocampus (Grant et al. 1992), which exhibits hypophosphorylation of NR2B. Rather, tyrosine phosphorylation of NR2B may alter the intracellular signaling machinery in which the NMDA receptor is involved. The NMDA receptor exists at the postsynaptic membrane as a complex composed of numerous number of the associated proteins including the PDZ-containing proteins, such as PSD-95 (also referred as SAP90), chapsin-110, and SAP102, which are thought to regulate clustering of NMDA receptor (for review, see Kornau et al. 1997), as well as the actin-binding proteins, α-actinin-2 (Wyszynski et al. 1997) and spectrin (Wechsler and Teichberg 1998). Fyn might have an important role in transducing extracellular signals to the submembraneous cytoskeletal network via tyrosine phosphorylation of the NR2B and in clustering the NMDA receptor to the activated synapses.
In this paper, we provide evidence that tyrosine phosphorylation by Fyn might be important for the rate of kindling. We have observed recently a reduction of the threshold for LTP induction in hippocampus from mice overexpressing mutant Fyn (. Enhancement of tyrosine phosphorylation of the NR2B has been reported in the dentate gyrus after LTP induction (Lu et al. Rosenblum et al. 1996; Rostas et al. 1996) and in the insular cortex after exposure to a novel taste (Rosenblum et al. 1997). Thus, through tyrosine phosphorylation of NR2B and other substrates, Fyn tyrosine kinase might regulate the effectiveness of learning-related forms of neuronal plasticity.
Acknowledgments
We would like to thank R.M. Perlmutter for providing the fynB cDNA pmBF and N. Nakanishi for providing the plasmid vectors, pNN265 and pNN279, T. Ozaki for technical advises for recording of electroencephalogram, H. Kuzume for help in performing in situ hybridization and in photographic works, Harriet Ayers for typing the manuscript. We would also like to thank C. Pittenger, D. Bartsch, D. Winder, M. Mayford, and Y. Yamagata for their helpful comments on the manuscript. This work was supported by the Howard Hughes Medical Institute, by the National Institute for Aging and by Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
References
- Abe K, Saito H. Tyrosine kinase inhibitors, herbimycin A and lavendustin A, block formation of long-term potentiation in the dentate gyrus in vivo. Brain Res. 1993;621:167–170. doi: 10.1016/0006-8993(93)90315-e. [DOI] [PubMed] [Google Scholar]
- Bare DJ, Lauder JM, Wilkie MB, Maness PF. p59fyn in rat brain is localized in developing axonal tracts and subpopulations of adult neurons and glia. Oncogene. 1993;8:1429–1436. [PubMed] [Google Scholar]
- Bixby JL, Jhabvala P. Tyrosine phosphorylation in early embryonic growth cones. J Neurosci. 1993;13:3421–3432. doi: 10.1523/JNEUROSCI.13-08-03421.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxall AR, Lancaster B. Tyrosine kinases and synaptic transmission. Eur J Neurosci. 1998;10:2–7. doi: 10.1046/j.1460-9568.1998.00009.x. [DOI] [PubMed] [Google Scholar]
- Boxall AR, Lancaster B, Garthwaite J. Tyrosine kinase is required for long-term depression in the cerebellum. Neuron. 1996;16:805–813. doi: 10.1016/s0896-6273(00)80100-2. [DOI] [PubMed] [Google Scholar]
- Butler LS, Silva AJ, Abeliovich A, Watanabe Y, Tonegawa S, McNamara JO. Limbic epilepsy in transgenic mice carrying a Ca2+/calmodulin-dependent kinase II α-subunit mutation. Proc Natl Acad Sci. 1995;92:6852–6855. doi: 10.1073/pnas.92.15.6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain DP, Grant SGN, Saucier D, Hargreaves EL, Kandel ER. Fyn tyrosine kinase is required for normal amygdala kindling. Epilepsy Res. 1995;22:107–114. doi: 10.1016/0920-1211(95)00029-1. [DOI] [PubMed] [Google Scholar]
- Chen L, Huang L-YM. Protein kinase C reduces Mg2+ block of NMDA receptor channels as a mechanism of modulation. Nature. 1992;356:521–523. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- Cooke MP, Perlmutter RM. Expression of a novel form of the fyn proto-oncogene in hematopoietic cells. New Biol. 1989;1:66–74. [PubMed] [Google Scholar]
- Croucher MJ, Cotterell KL, Bradford HF. Amygdala kindling by repeated focal N-methyl-d-aspartate administration: Comparison with electrical kindling. Eur J Pharmacol. 1995;286:265–271. doi: 10.1016/0014-2999(95)00459-6. [DOI] [PubMed] [Google Scholar]
- Gilbert ME. The NMDA receptor antagonist, MK-801, suppresses limbic kindling and kindled seizures. Brain Res. 1988;463:90–99. doi: 10.1016/0006-8993(88)90530-6. [DOI] [PubMed] [Google Scholar]
- Grant SGN, O’Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science. 1992;258:1903–1910. doi: 10.1126/science.1361685. [DOI] [PubMed] [Google Scholar]
- Grant SGN, Karl KA, Kiebler MA, Kandel ER. Focal adhesion kinase in the brain: Novel subcellular localization and specific regulation by Fyn tyrosine kinase in mutant mice. Genes & Dev. 1995;9:1909–1921. doi: 10.1101/gad.9.15.1909. [DOI] [PubMed] [Google Scholar]
- Gurd JW, Bissoon N. The N-methyl-d-aspartate receptor subunits NR2A and NR2B bind to the SH2 domains of phospholipase C-γ. J Neurochem. 1997;69:623–630. doi: 10.1046/j.1471-4159.1997.69020623.x. [DOI] [PubMed] [Google Scholar]
- Hogan B, Costantini F, Lacy E. Manipulating the mouse embryo, a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1986. Introduction of new genetic information into the developing mouse embryo; pp. 151–182. [Google Scholar]
- Holmes KH, Bilkey DK, Laverty R, Goddard GV. The N-methyl-d-aspartate antagonists aminophosphonovalerate and carboxypiperazinephosphonate retard the development and expression of kindled seizures. Brain Res. 1990;506:227–235. doi: 10.1016/0006-8993(90)91255-f. [DOI] [PubMed] [Google Scholar]
- Homanics GE, DeLorey TM, Firestone LL, Quinlan JJ, Handforth A, Harrison NL, Krasowski GE, Rick CE, Korpi ER, Makela R, Brilliant MH, Hagiwara N, Ferguson C, Snyder K, Olsen RW. Mice devoid of gamma-aminobutyrate type A receptor beta3 subunit have epilepsy, cleft palate, and hypersensitive behavior. Proc Natl Acad Sci. 1997;94:4143–4148. doi: 10.1073/pnas.94.8.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas EA, Kaczmarek LK. Regulation of potassium channels by protein kinases. Curr Opin Neurobiol. 1996;6:318–323. doi: 10.1016/s0959-4388(96)80114-0. [DOI] [PubMed] [Google Scholar]
- Kojima N, Wang J, Mansuy IM, Grant SGN, Mayford M, Kandel ER. Rescuing impairment of long-term potentiation in fyn-deficient mice by introducing Fyn transgene. Proc Natl Acad Sci. 1997;94:4761–4765. doi: 10.1073/pnas.94.9.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornau H-C, Seeburg PH, Kennedy MB. Interaction of ion channels and receptors with PDZ domain proteins. Curr Opin Neurobiol. 1997;7:368–373. doi: 10.1016/s0959-4388(97)80064-5. [DOI] [PubMed] [Google Scholar]
- Köhr G, Seeburg PH. Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. J Physiol. 1996;492:445–452. doi: 10.1113/jphysiol.1996.sp021320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau L-F, Huganir RL. Differential tyrosine phosphorylation of N-methyl-d-aspartate receptor subunits. J Biol Chem. 1995;270:20036–20041. doi: 10.1074/jbc.270.34.20036. [DOI] [PubMed] [Google Scholar]
- Lu Y-F, Kojima N, Tomizawa K, Moriwaki A, Matsushita M, Obata K, Matsui H. Enhanced synaptic transmission and reduced threshold for LTP induction in fyn transgenic mice. Eur J Neurosci. 1998;11:66–73. doi: 10.1046/j.1460-9568.1999.00407.x. [DOI] [PubMed] [Google Scholar]
- McNamara JO, Russell RD, Rigsbee L, Bonhaus DW. Anticonvulsant and antiepileptogenic actions of MK-801 in the kindling and electroshock models. Neuropharmacology. 1988;27:563–568. doi: 10.1016/0028-3908(88)90176-1. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Yagi T, Tateishi K, Niki H. Susceptibility to drug-induced seizures of Fyn tyrosine kinase-deficient mice. NeuroReport. 1996;7:2723–2726. doi: 10.1097/00001756-199611040-00063. [DOI] [PubMed] [Google Scholar]
- Moon IS, Apperson ML, Kennedy MB. The major tyrosine-phosphorylated protein in the postsynaptic density fraction is N-methyl-d-aspartate receptor subunit 2B. Proc Natl Acad Sci. 1994;91:3954–3958. doi: 10.1073/pnas.91.9.3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss SJ, Gorrie GH, Amato A, Smart TG. Modulation of GABAA receptors by tyrosine phosphorylation. Nature. 1995;377:344–348. doi: 10.1038/377344a0. [DOI] [PubMed] [Google Scholar]
- Noebels JL. Targeting epilepsy genes. Neuron. 1996;16:241–244. doi: 10.1016/s0896-6273(00)80042-2. [DOI] [PubMed] [Google Scholar]
- O’Dell TJ, Kandel ER, Grant SGN. Long-term potentiation in the hippocampus is blocked by tyrosine kinase inhibitors. Nature. 1991;353:558–560. doi: 10.1038/353558a0. [DOI] [PubMed] [Google Scholar]
- Omkumar RV, Kiely MJ, Rosenstein AJ, Min K-T, Kennedy MB. Identification of a phosphorylation site for calcium/calmodulin-dependent protein kinase II in the NR2B subunit of the N-Methyl-d-aspartate receptor. J Biol Chem. 1996;271:31670–31678. doi: 10.1074/jbc.271.49.31670. [DOI] [PubMed] [Google Scholar]
- Patil N, Cox DR, Bhat D, Faham M, Myers RM, Peterson AS. A potassium channel mutation in weaver mice implicates membrane excitability in granule cell differentiation. Nat Genet. 1995;11:126–129. doi: 10.1038/ng1095-126. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroenceph Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC. Essential functions of synapsin I and II in synaptic vesicle regulation. Nature. 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- Rosenblum K, Schul R, Meiri N, Hadari YR, Zick Y, Dudai Y. Modulation of protein phosphorylation in rat insular cortex after conditioned taste aversion training. Proc Natl Acad Sci. 1995;92:1157–1161. doi: 10.1073/pnas.92.4.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum K, Dudai Y, Richter-Levin G. Long-term potentiation increases tyrosine phosphorylation of the N-methyl-d-aspartate receptor subunit 2B in rat dentate gyrus in vivo. Proc Natl Acad Sci. 1996;93:10457–10460. doi: 10.1073/pnas.93.19.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum K, Berman DE, Hazvi S, Lamprecht R, Dudai Y. NMDA receptor and the tyrosine phosphorylation of its 2B subunit in taste learning in the rat insular cortex. J Neurosci. 1997;17:5129–5135. doi: 10.1523/JNEUROSCI.17-13-05129.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostas JAP, Brent VA, Voss K, Errington ML, Bliss TVP, Gurd JW. Enhanced tyrosine phosphorylation of the 2B subunit of the N-methyl-d-aspartate receptor in long-term potentiation. Proc Natl Acad Sci. 1996;93:10452–10456. doi: 10.1073/pnas.93.19.10452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Morimoto K, Okamoto M. Anticonvulsant action of a non-competitive antagonist of NMDA receptors (MK-801) in the kindling model of epilepsy. Brain Res. 1988;463:12–20. doi: 10.1016/0006-8993(88)90521-5. [DOI] [PubMed] [Google Scholar]
- Signorini S, Liao YJ, Duncan SA, Jan LY, Stoffel M. Normal cerebellar development but susceptibility to seizures in mice lacking G protein-coupled, inwardly rectifying K+ channel GIRK2. Proc Natl Acad Sci. 1997;94:923–927. doi: 10.1073/pnas.94.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein PL, Lee H-M, Rich S, Soriano P. pp59fyn mutant mice display differential signaling in thymocytes and peripheral T cells. Cell. 1992;70:741–750. doi: 10.1016/0092-8674(92)90308-y. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Okumura-Noji K. NMDA receptor subunits ε1 (NR2A) and ε2 (NR2B) are substrates for Fyn in the postsynaptic density fraction isolated from the rat brain. Biochem Biophys Res Comm. 1995;216:582–588. doi: 10.1006/bbrc.1995.2662. [DOI] [PubMed] [Google Scholar]
- Swope SL, Qu Z, Huganir RL. Phosphorylation of the nicotinic acetylcholine receptor by protein tyrosine kinases. Ann NY Acad Sci. 1995;757:197–214. doi: 10.1111/j.1749-6632.1995.tb17476.x. [DOI] [PubMed] [Google Scholar]
- Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH, Dallman MF, Julius D. Eating disorder and epilepsy in mice lacking 5-HT2C serotonin receptors. Nature. 1995;374:542–546. doi: 10.1038/374542a0. [DOI] [PubMed] [Google Scholar]
- Tingley WG, Roche KW, Thompson AK, Huganir RL. Regulation of NMDA receptor phosphorylation by alternative splicing of the C-terminal domain. Nature. 1993;364:70–73. doi: 10.1038/364070a0. [DOI] [PubMed] [Google Scholar]
- Tricklebank MD, Singh L, Oles RJ, Preston C, Iversen SD. The behavioural effects of MK-801: A comparison with antagonists acting non-competitively and competitively at the NMDA receptor. Eur J Pharmacol. 1989;167:127–135. doi: 10.1016/0014-2999(89)90754-1. [DOI] [PubMed] [Google Scholar]
- Umemori H, Wanaka A, Kato H, Takeuchi M, Tohyama M, Yamamoto T. Specific expressions of Fyn and Lyn, lymphocyte antigen receptor-associated tyrosine kinases, in the central nervous system. Mol Brain Res. 1992;16:303–310. doi: 10.1016/0169-328x(92)90239-8. [DOI] [PubMed] [Google Scholar]
- Valenzuela CF, Machu TK, McKernan RM, Whiting P, VanRenterghem BB, McManaman JL, Brozowski SJ, Smith GB, Olsen RW, Harris RA. Tyrosine kinase phosphorylation of GABAA receptors. Mol Brain Res. 1995;31:165–172. doi: 10.1016/0169-328x(95)00048-w. [DOI] [PubMed] [Google Scholar]
- Wang YT, Salter MW. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature. 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- Wechsler A, Teichberg VI. Brain spectrin binding to the NMDA receptor is regulated by phosphorylation, calcium and calmodulin. EMBO J. 1998;17:3931–3939. doi: 10.1093/emboj/17.14.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M. Competitive binding of α-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- Yagi T, Shigetani N, Okado T, Tokunaga T, Ikawa Y, Aizawa S. Regional localization of Fyn in adult brain; studies with mice in which fyn gene was replaced by lacZ. Oncogene. 1993;8:3343–3351. [PubMed] [Google Scholar]
- Yu X-M, Askalan R, Keil GJ, II, Salter MW. NMDA channel regulation by channel-asociated protein tyrosine kinase Src. Science. 1997;275:674–678. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]
- Zheng F, Gingrich MB, Traynelis SF, Conn PJ. Tyrosine kinase potentiates NMDA receptor currents by reducing tonic zinc inhibition. Nature Neurosci. 1998;1:185–191. doi: 10.1038/634. [DOI] [PubMed] [Google Scholar]