

Non-technical summary
Cystic fibrosis is a genetic disease caused by the malfunction of a chloride channel called cystic fibrosis transmembrane conductance regulator (CFTR). The most common disease-associated mutation is the deletion of the phenylalanine residue at position 508 (ΔF508), which result in channels with poor membrane expression and defective function. Opening of CFTR channels is controlled by ATP binding at two intracellular domains, called nucleotide-binding domains (NBDs), and subsequent NBD dimerization. Our previous studies revealed that ΔF508-CFTR channels open very infrequently, raising the possibility that the mutation perturbs NBD dimerization although the mutation is not located near the dimer interface. In this paper, we employed a functional assay to assess the stability of the NBD dimer. Our data suggest that the ΔF508 mutation significantly destabilizes the NBD dimer, supporting the hypothesis that the mutation disrupts the dimer interface. Our results provide structural insights that are potentially useful for drug design.
Abstract
Abstract
The cystic fibrosis transmembrane conductance regulator (CFTR) is a chloride channel that belongs to the ATP binding cassette (ABC) superfamily. The deletion of the phenylalanine 508 (ΔF508-CFTR) is the most common mutation among cystic fibrosis (CF) patients. The mutant channels present a severe trafficking defect, and the few channels that reach the plasma membrane are functionally impaired. Interestingly, an ATP analogue, N6-(2-phenylethyl)-2′-deoxy-ATP (P-dATP), can increase the open probability (Po) to ∼0.7, implying that the gating defect of ΔF508 may involve the ligand binding domains, such as interfering with the formation or separation of the dimeric states of the nucleotide-binding domains (NBDs). To test this hypothesis, we employed two approaches developed for gauging the stability of the NBD dimeric states using the patch-clamp technique. We measured the locked-open time induced by pyrophosphate (PPi), which reflects the stability of the full NBD dimer state, and the ligand exchange time for ATP/N6-(2-phenylethyl)-ATP (P-ATP), which measures the stability of the partial NBD dimer state wherein the head of NBD1 and the tail of NBD2 remain associated. We found that both the PPi-induced locked-open time and the ATP/P-ATP ligand exchange time of ΔF508-CFTR channels are dramatically shortened, suggesting that the ΔF508 mutation destabilizes the full and partial NBD dimer states. We also tested if mutations that have been shown to improve trafficking of ΔF508-CFTR, namely the solubilizing mutation F494N/Q637R and ΔRI (deletion of the regulatory insertion), exert any effects on these newly identified functional defects associated with ΔF508-CFTR. Our results indicate that although these mutations increase the membrane expression and function of ΔF508-CFTR, they have limited impact on the stability of both full and partial NBD dimeric states for ΔF508 channels. The structure–function insights gained from this mechanism may provide clues for future drug design.
Introduction
Cystic fibrosis transmembrane conductance regulator (CFTR) is the only member in the ATP-binding cassette (ABC) transporter superfamily known to function as an ion channel (Riordan et al. 1989; Bear et al. 1992). CFTR is mostly expressed in the apical membrane of epithelial cells and helps maintain the fluid and electrolyte balance across the cell membrane. Impairment of CFTR function causes cystic fibrosis (CF), the most prevalent lethal genetic disease among Caucasians (Bobadilla et al. 2002; Cutting 2005). Although more than 1600 mutations have been found in patients with CF (http://www.genet.sickkids.on.ca/cftr/app), the deletion of phenylalanine at position 508 (ΔF508) is the most common one, associated with ∼70% of CF alleles (Zielenski & Tsui 1995). It is generally accepted that the Δ508 mutation causes a protein folding defect, resulting in most channels being degraded intracellularly and very few reaching the plasma membrane (Cheng et al. 1990; Denning et al. 1992; Lukacs et al. 1993; Sato et al. 1996). Moreover, those few channels that actually reach the plasma membrane are functionally impaired (Dalemans et al. 1991; Haws et al. 1996; Hwang et al. 1997; Ostedgaard et al. 2007), with an open probability (Po) ∼15 times lower than that of WT-CFTR (Miki et al. 2010).
The structure of CFTR is similar, but more complex, than that of other members in the ABC superfamily. In addition to the canonical domains, namely two transmembrane domains (TMDs) and two nucleotide-binding domains (NBDs), CFTR possesses a unique regulatory domain (R domain). Once phosphorylated by protein kinase A (PKA) in the R domain, CFTR functions as an ATP-gated chloride channel (reviewed by Hwang & Sheppard, 2009). Using mutant cycle analysis, Vergani et al.(2005) provided convincing evidence supporting the idea that ATP binding induces the formation of an NBD dimer, which precedes gate opening. Subsequent hydrolysis of the ATP molecule bound in NBD2 leads to channel closure (Vergani et al. 2005; Mense et al. 2006, Zhou et al. 2006). Surprisingly, channel closure does not require complete separation of the NBD dimer. It was shown recently that closed channels enter a stable partial NBD dimer state, with the head of NBD1 and the tail of NBD2 (i.e. ATP-binding site 1) still connected by the bound ATP, while the ATP-binding site 2 (the head of NBD2 and the tail of NBD1) is separated (Tsai et al. 2010b). This partial NBD dimer, which lasts for tens of seconds, allows another ATP molecule to bind to the ATP-binding site 2 and initiate a new gating cycle.
Although the ΔF508 mutation induces severe processing and functional defects, the crystal structures of NBD1 (Lewis et al. 2005, 2010) with the ΔF508 mutation show little difference in protein conformation except changes in the surface topography near the ΔF508 mutation site. It is therefore proposed that deletion of F508 causes only a local disturbance rather than affect the overall structure of NBD1. Because in the crystal structure of other ABC transporters, the equivalent residue of F508 resides at the interface between the NBDs and the TMDs, it was also suggested that ΔF508 may affect the interaction between these domains (Lewis et al. 2005; Thibodeau et al. 2005). Indeed, biochemical studies by Serohijos et al. (2008) suggest that F508 might face the coupling helix of the fourth intracellular loop (ICL4), since cysteines introduced at the F508 position and at several positions in the ICL4 could be cross-linked. Moreover, cross-linking C508 and C1068 residues inhibited channel gating (Serohijos et al. 2008).
In contrast, our previous studies have provided evidence that the ΔF508 mutation may affect the function of the NBDs. We found that the ATP analogue N6-(2-phenylethyl) 2′-deoxy-ATP (P-dATP) increases the Po of ΔF508 by ∼15-fold, to ∼0.7 (Miki et al. 2010), a value very similar to the Po of WT-CFTR gated by P-dATP. This remarkable increase in the Po of ΔF508-CFTR is achieved by a dramatic increase in the opening rate and also by a prolongation of the open time. As P-dATP acts on both ATP binding sites (i.e. NBD1 and NBD2) to rectify the gating defect associated with the ΔF508 mutation (Miki et al. 2010), these data support the notion that the gating machinery of CFTR might be defective in ΔF508 channels.
To further explore the possibility of a mutational defect on the function of the NBDs, we decided to use two different functional assays we have recently established to study the effects of the ΔF508 mutation on the stability of full and partial NBD dimer states. First, as the lifetime of the locked-open state induced by the phosphate analogue pyrophosphate (PPi) reflects the stability of the full NBD dimer state (Tsai et al. 2009), we can gauge the effect of the ΔF508 mutation on NBD dimers by comparing the locked-open time between WT and ΔF508 channels. Second, the ‘ligand-exchange protocol’ (Tsai et al. 2010b), wherein ATP and the high-affinity analogue N6-(2-phenylethyl)-ATP (P-ATP) are suddenly exchanged, allows us to monitor how long the nucleotides remain bound at each binding site. As dissociation of the more tightly bound ATP in ATP-binding site 1 reports the stability of the aforementioned partial NBD dimer state (Tsai et al. 2010b), we can use the ligand-exchange protocol to assess the effect of the ΔF508 mutation on the partial NBD dimer state. Since channel gating is coupled to NBD dimer formation/separation, it is interesting to see if this basic mechanism is perturbed in ΔF508-CFTR. In this study, results from these two assays point to a new functional defect associated with the ΔF508 mutation. We believe that unravelling the molecular mechanism for this functional defect could aid our fundamental understanding of the malfunction of the most common pathogenic mutation associated with CF.
Methods
Cell culture and transient expression system
Chinese hamster ovary (CHO) cells were grown at 37°C in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Cells were cultured in 35 mm tissue culture dishes for 1 day before transfection. CFTR cDNA was cotransfected with pEGFP-C3 (Clontech, Palo Alto, CA, USA), encoding green fluorescent protein, using PolyFect transfection reagent (Qiagen, Valencia, CA, USA). Cells expressing CFTR were placed at 27°C for at least 2 days before electrophysiology experiments were performed.
CFPAC-1 cells (purchased from ATCC) were grown at 37°C in Iscove's modified Dulbecco's medium supplemented with 10% fetal bovine serum. Cells were cultured in 35 mm tissue culture dishes for 1 day before virus infection.
Mutagenesis
All mutant constructs were generated using the QuickChange XL kit (Stratagene, La Jolla, CA, USA) according to manufacturer's protocols. All the CFTR cDNA was sequenced to confirm the mutation (DNA core, University of Missouri).
Adenovirus infection
Adenoviruses expressing ΔF508 CFTR (H5.040.CMV.deltaF508) and GFP (H5′.040CMVEGFP) were purchased from the Gene Therapy Program Vector Core in the Department of Medicine at the University of Pennsylvania. Approximately 106 cells were co-infected with H5′.040CMVEGFP and H5.040.CMV.deltaF508 (ratio1:5) at 1000 viral particles per cell in 2 ml medium at 37°C overnight. Cells were grown at 27°C for 2–3 days before harvested for electrophysiological experiments.
Electrophysiological recordings
CFTR channel currents were recorded using an EPC9 or EPC10 amplifier (HEKA, Lambrecht/Pfalz, Germany) at room temperature. The pipettes were prepared from borosilicate capillary glass using a Flaming/Brown-type micropipette puller (P97, Sutter Instrument Co., Novato, CA, USA) and then fire-polished with a homemade microforge. The resistance of pipettes in the bath solution was 2–4 MΩ. Glass chips carrying the transfected cells were transferred to a chamber located on the stage of an inverted microscope (IX51, Olympus). After the seal resistance was >40 GΩ, the membrane was excised. CFTR channels were first activated with 2.75 mm ATP and 25 U ml−1 PKA until the current reached the steady state. All test solutions contained 10 U ml−1 PKA to maintain the phosphorylation level. The data were filtered at 100 Hz with an eight-pole Bessel filter (LPF-8, Warner Instruments, LLC, Hamden, CT, USA), and digitized to a computer at a sampling rate of 500 Hz. The membrane potential was held at −60 mV and the inward current was inverted for clear data presentation.
All inside-out patch experiments were performed with a fast solution exchange perfusion system (SF-77B, Warner Instruments). The dead time of solution change is ∼30 ms (Tsai et al. 2009).
Chemicals
The pipette solution contained (in mm): 140 N-methyl-d-glucamine chloride (NMDG-Cl), 2 MgCl2, 5 CaCl2 and 10 Hepes (pH 7.4 with NMDG). Cells were perfused with a bath solution containing (in mm): 145 NaCl, 5 KCl, 2 MgCl2, 1 CaCl2, 5 glucose, 5 Hepes and 20 sucrose (pH 7.4 with NaOH). For inside-out configuration, the perfusion solution contained (in mm): 150 NMDG-Cl, 2 MgCl2, 10 EGTA and 8 Tris (pH 7.4 with NMDG).
MgATP, PPi and PKA were purchased from Sigma-Aldrich. N6-(2-Phenylethyl)-ATP (P-ATP) and N6-(2-phenylethyl)-2′-deoxy ATP (P-dATP) were purchased from Biolog Life Science Institute (Bremen, Germany). PPi and MgATP were stored in 200 mm and 250 mm stock solution, respectively, at −20°C. P-ATP and P-dATP were stored in 10 mm stock at −70°C. All chemicals were diluted to the concentration indicated in each figure using the inside-out perfusion solution and the pH was adjusted to 7.4 with NMDG. For solutions containing PPi, an equal concentration of MgCl2 was added to the solution.
Data analysis and statistics
Steady-state mean currents were calculated with Igor Pro program (Wavemetrics, Lake Oswego, OR, USA) after baseline subtraction. Current relaxations were fitted with single or double exponential functions using a Levenberg–Marquardt-based algorithm with the Igor Pro program. Ensemble currents were generated by adding 8–14 raw traces.
Results are shown as means ± SEM. Student's t test was performed for statistical analysis using Microsoft Excel. P < 0.05 was considered significant.
Results
Effect of ΔF508 mutation on the stability of the locked-open state
To examine the effect of ΔF508 mutation on the stability of the NBD dimer, we used the non-hydrolysable analogue PPi to lock ΔF508-CFTR channels into an open state. Our previous studies (Tsai et al. 2009) demonstrated that PPi, by binding to NBD2, emptied after ATP hydrolysis and dissociation of the hydrolytic products, locks open CFTR by forming a stable NBD dimer state. The stability of this state can be gauged by measuring the relaxation time constant of the current decay upon removal of ligands. Figure 1A and C shows that application of 1 mm ATP and 4 mm PPi to WT-CFTR increases the ATP-induced macroscopic current by 2.04 ± 0.09-fold (n = 8). Upon wash-out of the ligands, the current decays mono-exponentially with a time constant of 31.28 ± 3.2 s (n = 8) (Fig. 1D). In patches containing fewer channels, stepwise closings of the locked-open channels can be readily seen after the ligands were removed (Fig. 1A). Interestingly, for ΔF508-CFTR (Fig. 1B), the same treatment increases the macroscopic current by 2.88 ± 0.13-fold (n = 7) (Fig. 1C), but the locked-open time (i.e. the relaxation time constant) is ∼10-fold shorter than that of WT channels (3.29 ± 0.43 s, n = 5) (Fig. 1D). This drastically shortened locked-open time can be discerned more clearly in patches with fewer channels. Unlike WT-CFTR, where long-lasting openings were seen after ligand removal, ΔF508-CFTR channels closed rapidly once ATP and PPi were washed out (Fig. 1B). These results suggest that deletion of the phenylalanine at position 508 significantly decreases the stability of NBD dimers. As the locked-open time for ΔF508-CFTR is shorter than that of WT channels, it may seem odd that PPi increases ΔF508-CFTR macroscopic currents slightly more than it does WT-CFTR currents (Fig. 1C). This is likely to be due to the much lower Po of ΔF508-CFTR with a very long mean closed time (Dalemans et al. 1991; Haws et al. 1996; Hwang et al. 1997; Ostedgaard et al. 2007; Miki et al. 2010). Thus, a slight prolongation of the open time results in a significantly larger effect on Po for ΔF508-CFTR.
Figure 1. Difference in locked-open time induced by PPi in WT-CFTR and ΔF508-CFTR.
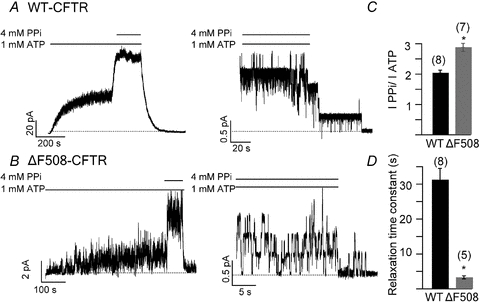
Representative traces of macroscopic (left) and microscopic (right) WT-CFTR (A) and ΔF508-CFTR (B) currents elicited by 1 mm ATP and 4 mm PPi. The dotted line represents zero current. The bars above the traces represent the presence of different ligands to the membrane patch (same in following figures). C, current increase induced by PPi for WT (n = 8) and ΔF508-CFTR (n = 7) (P < 0.01). D, summary of locked-open times induced by PPi for WT-CFTR (n = 7) and ΔF508-CFTR (n = 5). *P < 0.01 versus WT-CFTR. The number above each bar represents the n number for each experiment (same in following figures).
To verify these results, we employed an alternative approach to produce locked-open channels. We introduced the E1371S mutation, which is known to demolish ATPase activity in ABC proteins including CFTR (Aleksandrov et al. 2000; Vergani et al. 2003; Bompadre et al. 2005), into WT and ΔF508 backgrounds. For E1371S channels, the relaxation time constant of the current decay after ATP washout is ∼110 s (Fig. 2A and C, Zhou et al. 2006), whereas that of ΔF508/E1371S channels is only 32.45 ± 4.07 s (n = 4) (Fig. 2B and C). Although it is unclear why the ΔF508 mutation shows less effect on the stability of NBD dimer under the E1371S background, the shortening of the locked-open time seen in ΔF508/E1371S channels is consistent with the idea that the culprit is a destabilization of the NBD dimer rather than a lower affinity or efficacy of PPi.
Figure 2. Comparison of the locked-open time of E1371S- and ΔF508/E1371S-CFTR.
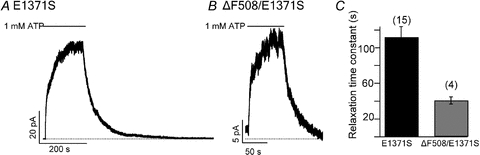
Representative traces of non-hydrolytic E1371S-CFTR (A) and ΔF508/E1371S-CFTR (B) in the presence of 1 mm ATP. C, summary of the locked-open times for E1371S-CFTR (n = 15) and ΔF508/E1371S-CFTR (n = 4) (P < 0.01).
Tight binding of nucleotides in NBD1 prolongs the channel locked-open time
In a previous report (Tsai et al. 2010a), we demonstrated that the locked-open time of WT-CFTR induced by PPi is prolonged by replacing ATP with the high affinity ATP analogue N6-phenylethyl-ATP (P-ATP), or by introducing ‘gain-of-function’ mutations to the ATP-binding site 1 (mutations which increase the Po of CFTR, such as W401F and H1348G) as the locked-open state reflects an NBD dimer with ATP-binding site 1 occupied by ATP and ATP-binding site 2 by PPi (Tsai et al. 2009). In Fig. 3, we show that the gain-of-function mutations W401F and H1348G (Fig. 3A) and P-ATP (Fig. 3B) also prolong the locked-open time of ΔF508-CFTR channels. Compared to ΔF508-CFTR, the double mutant W401F/ΔF508-CFTR (ΔF508/DM) prolonged the locked-open time by ∼2-fold, and the triple mutant W401F/H1348G/ΔF508-CFTR (ΔF508/TM) by ∼4-fold. Moreover, the locked-open time of each mutant was further prolonged when P-ATP, instead of ATP, was used as the ligand, suggesting that the effect of P-ATP and the gain-of-function mutations are additive. Despite these manoeuvres, the maximal locked-open time (P-ATP together with two gain-of-function mutations) remains ∼2/3 of that of WT-CFTR channels locked open by ATP and PPi (20 s versus 31 s in WT-CFTR). Nonetheless, the observation that manipulations of ligand interactions with ATP-binding site 1 result in similar effects on the locked-open time for both WT- and ΔF508-CFTR channels suggests a common structural basis for the locked-open state of these two channels, namely the conformation of the NBD dimer.
Figure 3. Increase of the locked-open time for ΔF508-CFTR channels by gain-of-function mutations or the high affinity ATP analogue P-ATP.
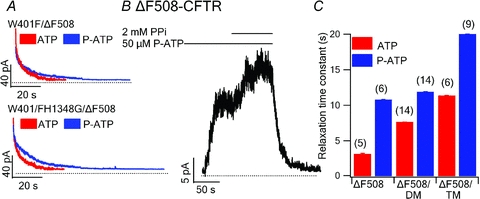
A, relaxations of ensemble current traces for various mutations. Upper, red, ΔF508/DM locked open with 1 mm ATP and 2 mm PPi (ensemble from 14 traces). Upper, blue, ΔF508/DM locked open with 50 μm P-ATP and 2 mm PPi (ensemble from 14 traces). Lower, red, ΔF508/TM locked open with 1 mm ATP and 2 mm PPi (ensemble from 8 traces). Lower, blue, ΔF508/TM locked open with 50 μm P-ATP and 2 mm PPi (ensemble from 9 traces). B, a representative trace of ΔF508-CFTR locked open with 50 μm P-ATP and 2 mm PPi (n = 6). C, summary of PPi locked-open times for each construct (ΔF508/DM: W401F/ΔF508-CFTR, ΔF508/TM: W401F/H1348G/ΔF508-CFTR).
ATP/P-ATP ligand exchange time in ATP-binding site 1 is accelerated in ΔF508-CFTR
The shortened locked-open time with ΔF508-CFTR channels suggests the intriguing possibility that the binding of the ATP molecule in NBD1 may not be as stable as in WT channels. Tsai et al. (2010b) demonstrated the existence of a partial NBD dimer state, wherein ATP in ATP-binding site 2 has been hydrolysed and the hydrolytic products have been released, but one ATP remains tightly bound in ATP-binding site 1 for tens of seconds. The stability of this partial NBD dimer can be examined by conducting ligand exchange experiments (Tsai et al. 2010b). When the ligand is switched from ATP to P-ATP, there is a fast current rising phase, which represents the ATP/P-ATP exchange in the ATP-binding site 2, followed by a slow current rising phase representing the ATP/P-ATP exchange in the ATP-binding site 1. The tightness of ligand binding in ATP-binding site 1 can be examined quantitatively by measuring the time constant of the slow phase upon ligand exchange in the ATP-binding site 1.
In WT-CFTR, the ligand exchange time at ATP-binding site 1 is ∼40 s (42.18 ± 9.63 s, n = 6, Fig. 4A and C). However, in ΔF508-CFTR, the current reached the steady state soon after the perfusion solution was switched from ATP to P-ATP (Fig. 4B). As a result, the ligand exchange time for ΔF508-CFTR is dramatically shortened (3.27 ± 0.53 s, n = 9, Fig. 4C). Thus, the ΔF508 mutation not only destabilizes the full NBD dimer (Figs 1 and 2), it also dramatically shortens the ligand dwell time in ATP-binding site 1, presumably by destabilizing the partial NBD dimer state (Fig. 4C). As expected, the time constant of the fast rising phase is similar between WT-CFTR and ΔF508-CFTR, suggesting that the rate of hydrolysis and sequential separation at ATP-binding site 2 is nearly identical in both constructs.
Figure 4. ATP/P-ATP ligand exchange for WT-CFTR and ΔF508-CFTR.
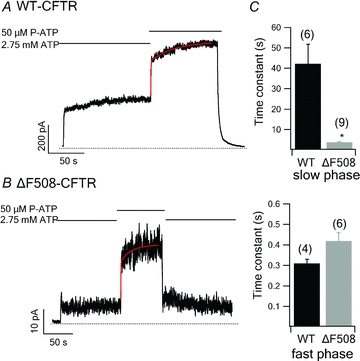
Representative current traces of WT-CFTR (A) and ΔF508-CFTR (B) upon ATP/P-ATP exchange. The current rising phase was fitted with a double exponential function. C, time constants (τ) of the slow (upper panel) and fast (lower panel) current raising phases after the ligand is switched from ATP to P-ATP. *P < 0.01 versus WT- CFTR.
The results described above were obtained from CHO cells transiently expressing CFTR. However, in physiological conditions, CFTR channels are predominantly expressed in epithelial cells. Therefore, we also carried out similar experiments with ΔF508-CFTR channels expressed in human CFPAC-1 epithelial cells. The results are consistent with those observed in CHO cells (online supplemental material, Fig. S1).
Expression improving mutations improve dimer stability
ΔF508-CFTR exhibits defective processing and trafficking; thus only a small proportion of the channels mature and traffic from the endoplasmic reticulum to the cell surface (Cheng et al. 1990; Denning et al. 1992; Lukacs et al. 1993; Sato et al. 1996). Several recent reports have identified mutations and deletions that improve the trafficking, maturation and function of ΔF508 channels (DeCarvalho et al. 2002; Pissarra et al. 2008; Aleksandrov et al. 2010). We introduced into ΔF508-CFTR the ‘solubilizing mutations’ F494N/Q637R (Pissarra et al. 2008) and the regulatory insertion deletion (ΔRI, deletion of residues 404–435) (Aleksandrov et al. 2010) to test whether they have any effects on the ΔF508-CFTR gating defects described above.
We first tested the F494N/Q637R and ΔRI mutations on the WT background and found that both the solubilizing mutations and ΔRI significantly prolong the locked-open time and ligand exchange time. As seen in Figs 5A and 6A, in either case, the current relaxation upon removal of ATP and PPi was significantly slower compared with that for WT-CFTR (F494N/Q637R-CFTR: τ = 86.14 ± 12.61s, n = 6; ΔRI-CFTR: τ = 75.33 ± 14.36 s, n = 7). Figure 7 summarizes the results. For ligand exchange experiments, these mutations also significantly prolong the second phase of current changes upon switching the ligand from ATP to P-ATP (F494N/Q637R-CFTR: τ = 76.41 ± 12.31 s, n = 6; ΔRI-CFTR: τ = 81.78 ± 6.66 s, n = 7) (Figs 5A, 6A and 7). Thus, for unknown reason, these mutations stabilize the NBD dimer as well as the partial NBD dimer states. We next engineered these mutations into the ΔF508 background. We found that although the locked-open time (F494N/Q637R/ΔF508-CFTR: τ = 5.95 ± 0.36 s, n = 8; ΔRI/ΔF508-CFTR: τ = 5.52 ± 0.45 s, n = 11) and ligand exchange time (F494N/Q637R/ΔF508-CFTR: τ = 8.44 ± 1.3s, n = 6; ΔRI/ΔF508-CFTR: τ = 8.95 ± 1.75 s, n = 4) of F494N/Q637R/ΔF508 and ΔF508/ΔRI channels are prolonged (Figs 5B, 6B and 7), they are still much shorter than those of WT channels. As these mutations improve these gating parameters to a similar degree for WT- and ΔF508-CFTR, their effects may not be specific for channels carrying the ΔF508 mutation.
Figure 5. Effects of ‘solubilizing mutations’, F494N/Q637R, on WT- and ΔF508-CFTR channels.
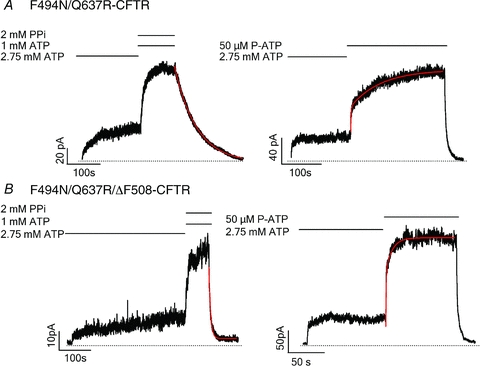
A, representative current traces of F494N/Q637R-CFTR channels locked opened by 1 mm ATP and 2 mm PPi (left) and ATP/P-ATP ligand exchange (right). B, representative current traces of F494N/Q637R/ΔF508-CFTR channels locked opened by 1 mm ATP and 2 mm PPi (left) and ATP/P-ATP ligand exchange (right). The current decay upon ligand wash-out was fitted with a single exponential function, while the current rising phase was fitted with a double exponential function.
Figure 6. Effects of deletion of the regulatory insertion (ΔRI) on WT- and ΔF508-CFTR channels.
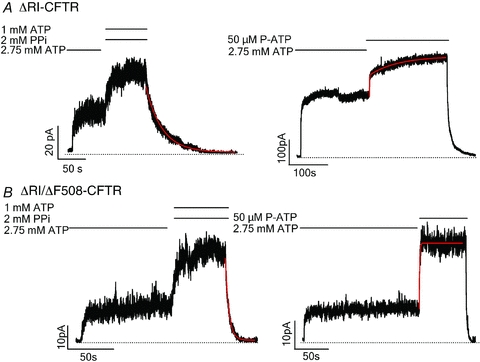
A, representative current traces of ΔRI-CFTR locked open by 1 mm ATP and 2 mm PPi (left) and ATP/P-ATP ligand exchange (right). B, representative current traces of ΔRI/ΔF508-CFTR locked open by 1 mm ATP and 2 mm PPi (left) and ATP/P-ATP ligand exchange (right). The current decay upon ligand wash-out was fitted with a single exponential function, while the current rising phase was fitted with a double exponential function.
Figure 7. Summary of effects of solubilizing mutations or ΔRI on locked-open time and the time constant of the slow phase for current rise upon ATP/P-ATP ligand exchange for different WT-CFTR constructs (A) and ΔF508-CFTR constructs (B).
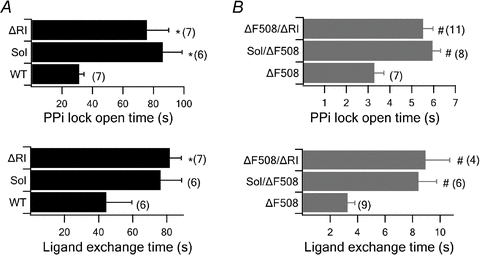
*P < 0.01 versus WT- CFTR. #P < 0.01 versusΔF508-CFTR. sol: solubilizing mutation, F494N/Q637R. The number next to each bar represents the n number for each experiment.
Besides prolonging the PPi locked-open time and the ligand exchange time, F494N/Q637R/ΔF508 and ΔF508/ΔRI have been previously shown to improve the function of ΔF508-CFTR (Pissarra et al. 2008; Aleksandrov et al. 2010). Here we used the high affinity ATP analogue P-dATP to boost the Po of these mutant channels. To our surprise, we found that P-dATP still increases the current dramatically for both compound mutants: 6.96 ± 0.17-fold increase for ΔRI/ΔF508-CFTR (n = 5), and 12.36 ± 1.21-fold increase for F494N/Q637R-CFTR (n = 8) (Fig. 8). Previously we found that ΔF508-CFTR channels assume a Po of ∼0.7 in the presence of P-dATP, similar to those of WT channels under the same conditions (Miki et al. 2010). While P-dATP increased the mean macroscopic current of ΔF508-CFTR channels by 19-fold, WT currents barely doubled in the presence of P-dATP (Miki et al. 2010). Thus, these microscopic and macroscopic results allow us to accurately calculate the Po of ΔF508 to be ∼15 times smaller than the Po of WT channels at saturating [ATP] (0.03 vs 0.45; Miki et al. 2010). Since P-dATP does not alter single channel conductance (Miki et al. 2010), the increase in the macroscopic current induced by P-dATP in F494N/Q637R/ΔF508 and ΔF508/ΔRI (12- and 7-fold, respectively) suggests that the Po of these mutant channels is still lower than that of WT channels when ATP is used as the ligand. Thus, although the exact Po for these compound mutants is not determined, we can safely conclude that these expression-improving mutations do not completely restore the function of the ΔF508 channel in our expression system.
Figure 8. Effect of P-dATP on F494N/Q637R/ΔF508-CFTR and ΔRI/ΔF508-CFTR.
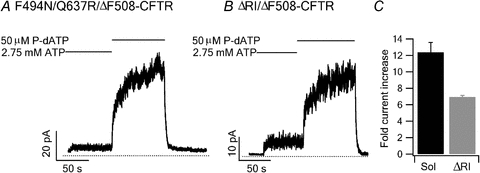
Representative current traces of F494N/Q637R/ΔF508-CFTR (A) and ΔRI/ΔF508-CFTR (B) in the presence of 50 μm P-dATP. C, current increase induced by P-dATP for F494N/Q637R/ΔF508-CFTR (n = 8) and ΔRI/ΔF508-CFTR (n = 5).
Discussion
In the current study, we used the locked-open time induced by PPi and the ligand exchange protocol to gauge the effects of the ΔF508 mutation on the stability of both the full and partial NBD dimer states of CFTR channels. Our results strongly suggest that the ΔF508 mutation dramatically destabilizes both. We also tested mutations that improve the stability of both NBD dimers states as well as mutations that had been shown to increase the surface expression of ΔF508-CFTR on these functional parameters. All the mutants that we tested improved the function of ΔF508-CFTR although their effects on WT channels are similar.
ΔF508 is the most common mutation found in patients with CF. Two decades of studies have revealed several defects associated with ΔF508-CFTR: poor membrane expression and impaired channel function. Early studies reported that the deletion of F508 severely impeded channel maturation (Cheng et al. 1990). The majority of ΔF508 channels are retained in the ER rather than traffic to the cell membrane (Ward et al. 1995). This effect can be observed in Western blot, where the amount of mature form of ΔF508-CFTR (band C) is dramatically reduced compared to WT-CFTR (see Fig. S2; Pissarra et al. 2008; Aleksandrov et al. 2010). The mutation also affects the stability of the protein in the plasma membrane since ΔF508 channels are removed from the membrane at a faster rate than WT channels (Lukacs et al. 1993). The mechanism responsible for this reduction of the surface pool is not well understood and it has been attributed to different processes such as recycling deficiencies due to misfolding (Sharma et al. 2004) and accelerated endocytosis (Swiatecka-Urban et al. 2005), which might be related to peripheral protein quality control (Okiyoneda et al. 2010). Thus, with less membrane insertion and more retrieval, the channel density in the cell membrane is much less for ΔF508 than WT-CFTR. Despite these biochemical anomalies, the crystal structure of isolated NBD1/ΔF508 showed surprisingly little difference when compared to that of WT NBD1. Lewis et al. (2010) reported that the structural difference between WT and ΔF508 is minor and localized to areas adjacent to ΔF508. It is therefore proposed that enhanced dynamics of nearby residues could affect chaperone interactions with mutant proteins (Lewis et al. 2010).
Even when those few ΔF508 channels actually traffic to the plasma membrane, their function is gravely impaired. In several studies in native ΔF508-CFTR-expressing tissues, including nasal epithelium, airway epithelium, pancreatic ducts and intestine, no or very little cAMP induced Cl− current was found (Knowles et al. 1983; Welsh & Lietdke 1986; Gray et al. 1988; Riordan et al. 1989). Our recent studies in excised patches also revealed that ΔF508-CFTR channels exhibit a very low Po in response to ATP (Miki et al. 2010; cf. Dalemans et al. 1991; Haws et al. 1996; Hwang et al. 1997; Ostedgaard et al. 2007). A previous study by Schultz et al. (1999) reported a rightward shift of the ATP dose response for ΔF508-CFTR, but we do not observe that effect (Wang et al. 2000). We took advantage of the high affinity ATP analogue P-dATP to accurately measure the Po of ΔF508 channel expressed in CHO cells. Our results suggest that the Po for ΔF508-CFTR is ∼0.03, about 15-fold less than that of WT channels. Interestingly, even though the chemical difference between ATP and P-dATP is fairly minor and both nucleotides act on the nucleotide-binding sites, P-dATP brings the gating activity of ΔF508-CFTR to levels comparable to those of WT channels (Miki et al. 2010). That such a minor modification of the ligand significantly restores the function of ΔF508-CFTR suggests an underlying molecular defect within the NBD that results in a poor response to ATP. As the mutation does not affect the apparent affinity of the channel to ATP (Wang et al. 2000), the simplest possibility is a slowed NBD dimer formation following ATP binding (see below for detail). Alternatively, the ΔF508 mutation may perturb coupling between NBD and TMD (Serohijos et al. 2008), and/or interfere with the function of the RI domain (Aleksandrov et al. 2010).
The hypothesis of defective coupling was proposed in Serohijos et al. (2008) by constructing a homology model of CFTR based on the crystal structure of SAV1866, a bacterial ABC transporter. Based on this homology model, F508 is close to the NBD-TMD interface. Cross-linking experiments indeed showed proximity between the F508 region of NBD1 and ICL4 as well as between NBD2 and ICL2. Electrophysiological recordings further demonstrated that cross-linking the F508 region with residues in ICL4 inhibits channel activity. Therefore, the authors suggested that the deletion of F508 could interfere with the coupling between the NBDs and TMDs, resulting in channels with poor function.
A more recent report (Aleksandrov et al. 2010) proposed that the regulatory insertion (RI, amino acids 404–435) may play a role in the pathogenesis of the ΔF508 mutation as removal of the RI improves both surface expression and gating of ΔF508-CFTR. In our study, although we confirmed that deletion of RI indeed somewhat improves the expression of the ΔF508 channels (Fig. S2), both the PPi-induced locked-open time and the ligand exchange time of ΔRI/ΔF508-CFTR channels are still much shorter than that of WT-CFTR, suggesting that the removal of RI does not completely restore dimer stability. Moreover, we found that application of P-dATP to ΔRI/ΔF508-CFTR channels increases the current by ∼7-fold, suggesting that the Po of this compound mutant is still much lower than WT channels (Fig. 8B).Similarly, the solubilizing mutations F494N and Q637R also partially restore the membrane expression and function of ΔF508 channels (Figs 5B, 7B, 8 and S2; also, see Pissarra et al. 2008).
By employing two assays that more directly gauge the states of the NBDs, the current study provides more insights about the basis for the functional perturbations associated with the ΔF508 mutation. Interestingly, however, although P-dATP brings the Po of ΔF508-CFTR back to normal levels, it failed to prolong the PPi locked-open time and the ligand exchange time of ΔF508-CFTR to WT-CFTR values (data not shown). If we accept the premise that these two functional assays accurately estimate the stability of the NBD dimer states, these results directly point to a defect in the NBDs of ΔF508-CFTR, even though little structural perturbation is seen in the crystal structure of NBD1 carrying the ΔF508 mutation. However, it should be noted that our interpretation is based on a crucial assumption, that is, the gating of ΔF508-CFTR follows similar mechanisms that have been proposed for WT-CFTR. In other words, the opening/closing of the gate for ΔF508-CFTR, like that of WT-CFTR, is coupled to the formation and partial separation of the NBD dimer (Tsai et al. 2009, 2010a,b). Then, our interpretation that the ΔF508 mutation destabilizes both full and partial NBD dimers should be valid. As in an NBD dimer, two NBDs are connected mostly by the ligand–NBD interactions and to a lesser degree by the NBD–NBD interactions, we speculate that a simple explanation for the destabilization effect observed in ΔF508 channels is that the mutation creates a distortion which is transmitted to the dimer interface and thus weakens the dimer stability.
Can this hypothesis also account for the decreased opening rate manifested in ΔF508-CFTR? Results from thermodynamic studies of CFTR gating may provide a tentative answer. Csanady et al. (2006), by carefully measuring gating parameters at different temperatures, concluded that opening of the channel by NBD dimerization is associated with a large increase of entropy presumably due to dehydration at the dimer interface and dispersion of relatively ordered water molecules to the disordered bulk. Using this energetic argument, we hypothesize that the structural changes induced by the ΔF508 mutation at the NBD dimer interface may hinder NBD dimerization (thus a slower opening rate), whereas P-dATP, with a hydrophobic benzene ring added to the adenine moiety and the removal of 2′-hydroxyl group at the ribose ring, may facilitate NBD dimerization by lowering the energetic barrier between open and closed states. More studies are needed to verify or disprove this hypothesis.
Even if the aforementioned hypothesis is correct, the fact that F508 is located far from the NBD dimer interface indicates that any changes in the NBD dimer interface has to be allosteric in nature. Since the NBDs and other parts of the CFTR protein must move in a concerted way during the gating motion, it is entirely possible that the instability of the NBD dimer states demonstrated in the current study is secondary to alterations in those regions involved in the coupled movements. Indeed the crystal structure of ΔF508/NBD1 (Lewis et al. 2010) does not reveal major structural perturbation in regions that presumably form contacts with bound ATP and NBD2. It is, however, important to point out that the solved crystal structure is a monomeric form. Moreover as the crystal structure provides only a snapshot of a protein, it is not possible to predict what conformational changes the NBDs may undergo during a gating cycle. It is equally important to note that due to the exponential relationship between the distribution of different kinetic states and the changes in free energy between these states, a slight alteration in free energy due to minor structural changes, which may not be readily seen in the crystal structure, could be enough to lower the Po from 0.4 to 0.03 or shorten the locked open time from 30 s to 3 s. For example, a 10-fold difference in the closing rate only reflects a ΔΔG of ∼5 kJ mol−1, a value smaller than the average strength of a hydrogen bond. Therefore, a dramatic gating defect can occur without severe disturbance in structure. Of note, a previous study by Roxo-Rosa et al. (2006) suggested the possibility that the ΔF508 mutation may cause a structural instability in NBD1 which might weaken the binding energy for stable NBD dimer formation. Interestingly, recent studies on the defective folding mechanism and thermal instability of isolated NBD1 constructs suggest that the ΔF508 mutation destabilizes the native state and accelerates the formation of a partially-folded intermediate state (Protasevich et al. 2010; Wang et al. 2010). How this structural instability of an isolated NBD1 relates to the instabilities revealed by our functional studies using the whole protein remains unclear.
Irrespective of which structural mechanism may account for the gating defects associated with ΔF508-CFTR, a better understanding of the cause underlying the gating defect of the mutant channel may prove helpful in the development of new CFTR potentiators (i.e. compounds that increase the activity of channels). Our latest reports demonstrated that strengthening the ligand binding in ATP-binding site 1 either through mutations (Tsai et al. 2010a) or using ATP analogues (Miki et al. 2010) significantly improves the activity of ΔF508-CFTR, underscoring the possibility that targeting the NBD dimer interface could be a promising approach for drug design.
In conclusion, our study demonstrates a new functional defect associated with ΔF508-CFTR channels. The full and partial NBD dimers of ΔF508-CFTR are less stable than those of WT-CFTR. Elucidating the structural basis for these functional defects of ΔF508-CFTR may open a new track for future drug design.
Acknowledgments
We thank Cindy Chu and Haruna Miki for their technical assistance. This work was supported by the National Institutes of Health (R01-DK55835 (T-C.H.), R01-HL53455 (T-C.H.), K01-DK075408 (S.G.B.) and P30-GM092456, and the Cystic Fibrosis Foundation (BOMPAD06G0, S.G.B.). This investigation was conducted in a facility constructed with support from the Research Facilities Improvement Program (C06 RR-016489–01).
Glossary
Abbreviations
- ABC
ATP binding cassette
- CFTR
cystic fibrosis transmembrane conductance regulator
- ER
endoplasmic reticulum
- ICL
intracellular loop
- NBD
nucleotide-binding domain
- PKA
protein kinase A
- P-ATP
N6-(2-phenylethyl)-ATP
- P-dATP
N6-(2-phenylethyl)-2′-deoxy-ATP
- PPi
pyrophosphate
- RI
regulatory insertion
Author contributions
K.J.: conception and design of the experiments; collection, analysis and interpretation of data; drafting and revising the article. M.L.: collection, analysis and interpretation of data. T-C.H.: conception and design of the experiments; drafting and revising the article. S.G.B.: conception and design of the experiments; collection, analysis and interpretation of data; drafting and revising the article. All authors approved the final version of this manuscript. All experiments were performed in Dalton Cardiovascular Research Center, University of Missouri.
Supplementary material
Figure S1
Figure S2
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Aleksandrov AA, Chang X, Aleksandrov L, Riordan JR. The non-hydrolytic pathway of cystic fibrosis transmembrane conductance regulator ion channel gating. J Physiol. 2000;528:259–265. doi: 10.1111/j.1469-7793.2000.00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksandrov AA, Kota P, Aleksandrov LA, He L, Jensen T, Cui L, Gentzsch M, Dokholyan NV, Riordan JR. Regulatory insertion removal restores maturation, stability and function of ΔF508 CFTR. J Mol Biol. 2010;401:194–210. doi: 10.1016/j.jmb.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear CE, Li CH, Kartner N, Bridges RJ, Jensen TJ, Ramjeesingh M, Riordan JR. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR) Cell. 1992;68:809–818. doi: 10.1016/0092-8674(92)90155-6. [DOI] [PubMed] [Google Scholar]
- Bobadilla JL, Macek M, Jr, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations – correlation with incidence data and application to screening. Hum Mutat. 2002;19:575–606. doi: 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- Bompadre SG, Cho JH, Wang X, Zou X, Sohma Y, Li M, Hwang TC. CFTR gating II: Effects of nucleotide binding on the stability of the open states. J Gen Physiol. 2005;125:377–394. doi: 10.1085/jgp.200409228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Csanady L, Nairn AC, Gadsby DC. Thermodynamics of CFTR channel gating: a spreading conformational change initiates an irreversible gating cycle. J Gen Physiol. 2006;128:523–533. doi: 10.1085/jgp.200609558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutting GR. Modifier genetics: cystic fibrosis. Annu Rev Genomics Hum Genet. 2005;6:237–260. doi: 10.1146/annurev.genom.6.080604.162254. [DOI] [PubMed] [Google Scholar]
- Dalemans W, Barbry P, Champigny G, Jallat S, Dott K, Dreyer D, Crystal RG, Pavirani A, Lecocq JP, Lazdunski M. Altered chloride ion channel kinetics associated with the ΔF508 cystic fibrosis mutation. Nature. 1991;354:526–528. doi: 10.1038/354526a0. [DOI] [PubMed] [Google Scholar]
- DeCarvalho AC, Gansheroff LJ, Teem JL. Mutations in the nucleotide-binding domain 1 signature motif region rescue processing and functional defects of cystic fibrosis transmembrane conductance regulator ΔF508. J Biol Chem. 2002;277:35896–35905. doi: 10.1074/jbc.M205644200. [DOI] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Gray MA, Greenwell JR, Argent BE. Secretin-regulated chloride channel on the apical plasma membrane of pancreatic duct cells. J Membr Biol. 1988;105:131–142. doi: 10.1007/BF02009166. [DOI] [PubMed] [Google Scholar]
- Haws CM, Neponucemo IB, Krouse ME, Wakelee H, Law T, Xia Y, Nguyen H, Wine JJ. ΔF508-CFTR channels: kinetics, activation by forskolin, and potentiation by xanthines. Am J Physiol Cell Physiol. 1996;270:C1544–C1555. doi: 10.1152/ajpcell.1996.270.5.C1544. [DOI] [PubMed] [Google Scholar]
- Hwang TC, Sheppard DN. Gating of the CFTR Cl− channel by ATP-driven nucleotide-binding domain dimerisation. J Physiol. 2009;587:2151–2161. doi: 10.1113/jphysiol.2009.171595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang TC, Wang F, Yang IC, Reenstra WW. Genistein potentiates wild-type and ΔF508-CFTR channel activity. Am J Physiol Cell Physiol. 1997;273:C988–C998. doi: 10.1152/ajpcell.1997.273.3.C988. [DOI] [PubMed] [Google Scholar]
- Knowles MR, Stutts MJ, Spock A, Fischer N, Gatzy JT, Boucher RC. Abnormal ion permeation through cystic fibrosis respiratory epithelium. Science. 1983;221:1067–1070. doi: 10.1126/science.6308769. [DOI] [PubMed] [Google Scholar]
- Lewis HA, Wang C, Zhao X, Hamuro Y, Conners K, Kearins MC, Lu F, Sauder JM, Molnar KS, Coales SJ, Maloney PC, Guggino WB, Wetmore DR, Weber PC, Hunt JF. Structure and dynamics of NBD1 from CFTR characterized using crystallography and hydrogen/deuterium exchange mass spectrometry. J Mol Biol. 2010;396:406–430. doi: 10.1016/j.jmb.2009.11.051. [DOI] [PubMed] [Google Scholar]
- Lewis HA, Zhao X, Wang C, Sauder JM, Rooney I, Noland BW, Lorimer D, Kearins MC, Conners K, Condon B, Maloney PC, Guggino WB, Hunt JF, Emtage S. Impact of the ΔF508 mutation in first nucleotide-binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. J Biol Chem. 2005;280:1346–1353. doi: 10.1074/jbc.M410968200. [DOI] [PubMed] [Google Scholar]
- Lukacs GL, Chang XB, Bear C, Kartner N, Mohamed A, Riordan JR, Grinstein S. The ΔF508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem. 1993;268:21592–21598. [PubMed] [Google Scholar]
- Mense M, Vergani P, White DM, Altberg G, Nairn AC, Gadsby DC. In vivo phosphorylation of CFTR promotes formation of a nucleotide-binding domain heterodimer. EMBO J. 2006;25:4728–4739. doi: 10.1038/sj.emboj.7601373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki H, Zhou Z, Li M, Hwang TC, Bompadre SG. Potentiation of disease-associated cystic fibrosis transmembrane conductance regulator mutants by hydrolyzable ATP analogs. J Biol Chem. 2010;285:19967–19975. doi: 10.1074/jbc.M109.092684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T, Barriere H, Bagdany M, Rabeh WM, Du K, Hohfeld J, Young JC, Lukacs GL. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science. 2010;329:805–810. doi: 10.1126/science.1191542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostedgaard LS, Rogers CS, Dong Q, Randak CO, Vermeer DW, Rokhlina T, Karp PH, Welsh MJ. Processing and function of CFTR-ΔF508 are species-dependent. Proc Natl Acad Sci U S A. 2007;104:15370–15375. doi: 10.1073/pnas.0706974104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pissarra LS, Farinha CM, Xu Z, Schmidt A, Thibodeau PH, Cai Z, Thomas PJ, Sheppard DN, Amaral MD. Solubilizing mutations used to crystallize one CFTR domain attenuate the trafficking and channel defects caused by the major cystic fibrosis mutation. Chem Biol. 2008;15:62–69. doi: 10.1016/j.chembiol.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Protasevich I, Yang Z, Wang C, Atwell S, Zhao X, Emtage S, Wetmore D, Hunt JF, Brouillette CG. Thermal unfolding studies show the disease causing F508del mutation in CFTR thermodynamically destabilizes nucleotide-binding domain 1. Protein Sci. 2010;19:1917–1931. doi: 10.1002/pro.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Roxo-Rosa M, Xu Z, Schmidt A, Neto M, Cai Z, Soares CM, Sheppard DN, Amaral MD. Revertant mutants G550E and 4RK rescue cystic fibrosis mutants in the first nucleotide-binding domain of CFTR by different mechanisms. Proc Natl Acad Sci U S A. 2006;103:17891–17896. doi: 10.1073/pnas.0608312103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Ward CL, Krouse ME, Wine JJ, Kopito RR. Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J Biol Chem. 1996;271:635–638. doi: 10.1074/jbc.271.2.635. [DOI] [PubMed] [Google Scholar]
- Schultz BD, Frizzell RA, Bridges RJ. Rescue of dysfunctional ΔF508-CFTR chloride channel activity by IBMX. J Membr Biol. 1999;170:51–66. doi: 10.1007/s002329900537. [DOI] [PubMed] [Google Scholar]
- Serohijos AW, Hegedus T, Aleksandrov AA, He L, Cui L, Dokholyan NV, Riordan JR. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci U S A. 2008;105:3256–3261. doi: 10.1073/pnas.0800254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Pampinella F, Nemes C, Benharouga M, So J, Du K, Bache KG, Papsin B, Zerangue N, Stenmark H, Lukacs GL. Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J Cell Biol. 2004;164:923–933. doi: 10.1083/jcb.200312018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiatecka-Urban A, Brown A, Moreau-Marquis S, Renuka J, Coutermarsh B, Barnaby R, Karlson KH, Flotte TR, Fukuda M, Langford GM, Stanton BA. The short apical membrane half-life of rescued ΔF508-cystic fibrosis transmembrane conductance regulator (CFTR) results from accelerated endocytosis of ΔF508-CFTR in polarized human airway epithelial cells. J Biol Chem. 2005;280:36762–36772. doi: 10.1074/jbc.M508944200. [DOI] [PubMed] [Google Scholar]
- Thibodeau PH, Brautigam CA, Machius M, Thomas PJ. Side chain and backbone contributions of Phe508 to CFTR folding. Nat Struct Mol Biol. 2005;12:10–16. doi: 10.1038/nsmb881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MF, Jih KY, Shimizu H, Li M, Hwang TC. Optimization of the degenerated interfacial ATP binding site improves the function of diseases related mutant cystic fibrosis transmembrane conductance regulator (CFTR) channels. J Biol Chem. 2010a;285:37663–37671. doi: 10.1074/jbc.M110.172817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MF, Li M, Hwang TC. Stable ATP binding mediated by a partial NBD dimer of the CFTR chloride channel. J Gen Physiol. 2010b;135:399–414. doi: 10.1085/jgp.201010399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MF, Shimizu H, Sohma Y, Li M, Hwang TC. State-dependent modulation of CFTR gating by pyrophosphate. J Gen Physiol. 2009;133:405–419. doi: 10.1085/jgp.200810186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P, Nairn AC, Gadsby DC. On the mechanism of MgATP-dependent gating of CFTR Cl− channels. J Gen Physiol. 2003;121:17–36. doi: 10.1085/jgp.20028673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P, Lockless SW, Nairn AC, Gadsby DC. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature. 2005;433:876–880. doi: 10.1038/nature03313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Protasevich I, Yang Z, Seehausen D, Skalak T, Zhao X, Atwell S, Spencer Emtage J, Wetmore DR, Brouillette CG, Hunt JF. Integrated biophysical studies implicate partial unfolding of NBD1 of CFTR in the molecular pathogenesis of F508del cystic fibrosis. Protein Sci. 2010;19:1932–1947. doi: 10.1002/pro.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Zeltwanger S, Hu S, Hwang TC. Deletion of phenylalanine 508 causes attenuated phosphorylation-dependent activation of CFTR chloride channels. J Physiol. 2000;524:637–648. doi: 10.1111/j.1469-7793.2000.00637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- Welsh MJ, Liedtke CM. Chloride and potassium channels in cystic fibrosis airway epithelia. Nature. 1986;322:467–470. doi: 10.1038/322467a0. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Wang X, Liu HY, Zou X, Li M, Hwang TC. The two ATP binding sites of cystic fibrosis transmembrane conductance regulator (CFTR) play distinct roles in gating kinetics and energetics. J Gen Physiol. 2006;128:413–422. doi: 10.1085/jgp.200609622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielenski J, Tsui LC. Cystic fibrosis: genotypic and phenotypic variations. Annu Rev Genet. 1995;29:777–807. doi: 10.1146/annurev.ge.29.120195.004021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.