

Non-technical summary
Deep brain stimulation (DBS) refers to a neurosurgical technique where chronically implanted electrodes serve to deliver electrical impulses to highly defined brain regions in neuropsychiatric disorders including Parkinson's disease (PD), depression and obsessive–compulsive disorders. Despite its broad acceptance as a safe and effective treatment for advanced PD, DBS has remained enigmatic with respect to its underlying mechanism(s). In general terms, DBS is capable of reinstating regular electrical activity in the complex neuronal networks that exhibit aberrant firing properties in PD, but how this effect is achieved at the network and cellular level is still highly debated. Our study focuses on a crucial, but hitherto neglected, issue in this controversy, namely the propagation of impulses within and away from the site of electrical stimulation. We propose that DBS overburdens the capacity of axons to transmit signals, thereby filtering and abating the pathological activity in the brain motor loops of PD patients.
Abstract
Abstract
Deep brain stimulation (DBS) has been established as an effective surgical therapy for advanced Parkinson's disease (PD) and gains increasing acceptance for otherwise intractable neuropsychiatric diseases such as major depression or obsessive–compulsive disorders. In PD, DBS targets predominantly the subthalamic nucleus (STN) and relieves motor deficits only at high frequency (>100 Hz). In contrast to the well-documented clinical efficacy of DBS, its underlying principle remains enigmatic spawning a broad and, in part, contradictory spectrum of suggested synaptic and non-synaptic mechanisms within and outside STN. Here we focused on a crucial, but largely neglected issue in this controversy, namely the axonal propagation of DBS within and away from STN. In rat brain slices preserving STN projections to substantia nigra (SN) and entopeduncular nucleus (EP, the rodent equivalent of internal globus pallidus), STN-DBS disrupted synaptic excitation onto target neurons through an unexpected failure of axonal signalling. The rapid onset and, upon termination of DBS, recovery of this effect was highly reminiscent of the time course of DBS in the clinical setting. We propose that DBS-induced suppression of axonal projections from and to STN serves to shield basal ganglia circuitry from pathological activity arising in or amplified by this nucleus.
Introduction
Progressive neurodegeneration of dopaminergic neurons in substantia nigra compacta (SNc) leads to perturbed subthalamic nucleus (STN) neuronal firing associated with abnormally synchronized bursts and oscillations in basal ganglia loops (Rivlin-Etzion et al. 2006; Hammond et al. 2007). The aberrant activity occurs mainly in the β-frequency range (15–30 Hz) and responds to dopamine replacement therapy or deep brain stimulation (DBS) of STN (Kuhn et al. 2006, 2008; Wingeier et al. 2006; Steigerwald et al. 2008). Because DBS mimics the therapeutic benefits of lesioning in the STN and all other brain areas targeted so far, it has been suggested that the high frequency required for the beneficial effects of DBS acts universally and causes a reversible suppression of neuronal activity. The proposed functional silencing of pathological activity in the STN and associated aberrant oscillations might be achieved through non-synaptic mechanisms including depolarization block of Na+ channels (Beurrier et al. 2001), axonal conduction block (Iremonger et al. 2006; Jensen & Durand, 2009), antidromic effects (Li et al. 2007), mixed effects on cell somata (inhibition of intrinsic firing) and axons (excitation of target regions) (McIntyre et al. 2004), and adenosinergic inhibition (Bekar et al. 2008). Furthermore, synaptic mechanisms might play a role such as neurotransmitter depletion (Iremonger et al. 2006) or stimulation of STN afferents (Gradinaru et al. 2009). Detailed biophysical examination showed that STN neurons are particularly well tailored to follow high frequency stimulation (HFS) (Do & Bean, 2003; Garcia et al. 2003), and electrophysiological and neurochemical studies revealed increased activation of STN output regions and enhanced transmitter release therein during HFS (Windels et al. 2000; Hashimoto et al. 2003; Maurice et al. 2003; Galati et al. 2006). Most interestingly, STN-HFS has been reported to increase striatal dopamine efflux (Bruet et al. 2001; Meissner et al. 2003; Lee et al. 2006; Lacombe et al. 2007; Walker et al. 2009), although the clinical relevance of this observation has been challenged (Hilker et al. 2003). To explore the relationship between DBS and dopamine efflux in terms of synaptic connectivity, we examined whether STN-HFS is capable of driving SNc dopaminergic neurons using whole-cell and field potential recordings in rat parasagittal brain slices. We demonstrate that HFS disrupts the synaptic excitation of various STN target neurons and ascribe this predominantly to a failure of axonal signalling.
Methods
Slice preparation and solutions
Procedures for slice preparation were carried out according to the guidelines and with the approval of the local governments of Schleswig-Holstein and Mittelfranken. The experiments comply with the policies and regulations of The Journal of Physiology given by Drummond(2009).
To preserve the main synaptic connectivity in the basal ganglia circuits, parasagittal brain slices (350 μm thick) were cut at an angle of 10–15 deg from juvenile Wistar rats (15–20 days old) (Beurrier et al. 2006; Ammari et al. 2009), which were deeply anaesthetized with ketamine or halothane prior to decapitation. The slices were initially maintained in artificial cerebrospinal fluid (aCSF) containing (in mm): 125 NaCl, 3 KCl, 0.2 CaCl2, 3.8 MgCl2, 1.25 NaH2PO4, 25 NaHCO3 and 10 d-glucose, bubbled with 95% O2–5% CO2 (pH 7.4). The solution was ice-cold for cutting and warmed to 35°C for 20 min immediately thereafter. Slices were then incubated in aCSF containing (in mm) 125 NaCl, 3 KCl, 1 CaCl2, 3 MgCl2, 1.25 NaH2PO4, 25 NaHCO3 and 10 d-glucose (pH 7.4) at room temperature (23 ± 1oC) for at least 2 h before individual slices were transferred to the recording chamber that was mounted on the stage of an upright microscope (Zeiss Axioskop). Brain regions of interest and individual neurons therein were visualized by means of Dodt infrared gradient contrast in conjunction with a contrast-enhanced CCD camera (Hamamatsu Photonics, Herrsching, Germany). Unless otherwise stated, recordings were performed in aCSF at 33 ± 1oC using a submerged chamber equipped with a gravity-driven perfusion system (flow rate 2–3 ml min−1). All solutions were constantly gassed with 95% O2–5% CO2. Drugs and chemicals were obtained from Tocris (Cologne, Germany) and Sigma (Deisenhofen, Germany).
Whole-cell recording
Whole-cell recordings of visually identified neurons in substantia nigra compacta (SNc) were performed in normal aCSF containing (in mm): 125 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 25 NaHCO3 and 10 d-glucose (pH 7.4). Patch pipettes were filled with (in mm) 135 potassium gluconate, 5 Hepes, 3 MgCl2, 5 EGTA, 2 Na2ATP, 0.3 NaGTP, 4 NaCl (pH 7.3). In some experiments, pipette solution also contained biocytin (1%) for later reconstruction of neuronal morphology. Electrode resistance ranged from 3 to 5 MΩ when filled with internal solution. Series resistance in whole-cell configuration was about 10–25 MΩ and was compensated by 60–80%. Constant current pulses (pulse width 60 μs, 200–1000 μA) were delivered every 10 s to a bipolar tungsten stimulating electrode positioned in STN to evoke postsynaptic currents (PSCs) in SNc neurons. Dopaminergic neurons were identified by their typical electrophysiological profile (Lacey et al. 1989) and were voltage-clamped at a holding potential of –70 mV after liquid junction potential was corrected. Monosynaptic glutamatergic excitatory postsynaptic currents (EPSCs) were pharmacologically isolated by perfusing slices with the GABAA receptor antagonists bicuculline (30 μm) or picrotoxin (100 μm). Synaptic currents were filtered at 1–2 kHz and sampled at 10 kHz using an Axopatch 200 amplifier in conjunction with Digidata 1200 interface and pCLAMP 9.2 software (all from Molecular Devices, Sunnyvale, CA, USA). After establishing baseline recording for 5–10 min at 0.1 Hz, PSCs/EPSCs were subjected to train stimuli at different frequencies (10, 50 and 130 Hz) for 10 s. Each data point before and after stimulus train represents the average of six consecutive recordings evoked at 0.1 Hz.
Field potential recording
Extracellular recording pipettes were filled with modified aCSF, in which bicarbonate was replaced with Hepes to avoid pH change. Slices were perfused with normal aCSF that also contained picrotoxin (100 μm) to obtain pure field excitatory postsynaptic potentials (fEPSPs). Field potentials (fibre volley, FV, and fEPSP) in SNc were elicited by constant current pulses (pulse width 90 μs, stimulus strength at 100–300 μA, every 10 s) delivered to a bipolar tungsten electrode located in STN. Single-unit activity was recorded from SNc neurons which typically exhibit spontaneous rhythmic firing with constant inter-spike intervals in the slice preparation (Kang & Futami, 1999). SNc neurons were considered to be orthodromically connected to STN when STN stimulation was able to reliably trigger a spike outside their rhythmic firing pattern (Kang & Futami, 1999). To isolate fibre volleys in SNc and other STN downstream structures, synaptic responses were abrogated by a combination of low calcium aCSF (0.2 mm CaCl2/ 3.8 mm MgCl2) and the ionotropic glutamate receptor antagonist kynurenic acid (KA, 2 mm). The same extracellular solution was also used for single unit recordings from STN neurons antidromically activated through electrical stimulation in STN. Extracellular spikes were regarded as antidromic if they could follow each stimulus with constant latency. Extracellular signals were filtered at 1 kHz (for field potentials) or 5 Hz (for single unit discharge) and sampled at 10–20 kHz using a Multiclamp 700B amplifier in conjunction with Digidata 1440A interface and pCLAMP 10 software (all from Molecular Devices).
Histology
After the experiments with biocytin (1%) in the pipette solution, slices were fixed in 4% paraformaldehyde in 0.1 m phosphate buffer overnight, cryoprotected and sectioned at 60 μm intervals. The cryostat sections were directly mounted onto glass slides and incubated in avidin–biotin conjugated horseradish peroxidase (ABC standard Elite Kit, PK-4000, Vector Laboratories, Inc., Burlingame, CA, USA). For visualization of the tracer, sections were reacted with diaminobenzidine and hydrogene peroxidase (DAB Kit SK-4100, Vector), followed by counterstaining with haematoxylin QS (H-3404, Vector). Finally the neurons were embedded in Fluoromount and pictures were taken with a digital camera (ColourView II) on a Zeiss Axioscope. Camera lucida drawings of the cell body and processes were done with the aid of a drawing tube (Zeiss) attached to the microscope by using a 40× objective magnification lens.
Data analysis
Data were analysed off-line with Clampfit (Molecular Devices). The negative peaks were used to determine amplitudes and latencies of PSCs/EPSCs, FVs and fEPSPs. Single unit discharges were analysed using threshold spike detection and template matching under visual inspection. Discharge rate was calculated from the detected event channel of unit activity. The decline of the amplitudes of EPSCs and FVs during HFS were fitted to obtain a monoexponential decay time constant using Origin 8 software (OriginLab Corp., Northampton, MA, USA).
Statistical analysis
Data are expressed as means ± SEM. Statistical comparisons of data were performed using Student's t test or analysis of variance (ANOVA), as appropriate. Significance was assumed for P < 0.05.
Results
STN-HFS silences synaptic transmission onto SNc dopaminergic neurons recorded in whole-cell voltage-clamp mode
In whole-cell recordings from SNc dopaminergic neurons, single STN stimuli reliably evoked a mainly AMPA receptor-mediated postsynaptic current (PSC) that was enhanced upon blockade of GABAA receptors (Fig. 1A). We then tested PSC responses during 10 s long trains of STN stimulation at 130 Hz (HFS, therapeutic frequency of DBS), at 50 Hz (ineffective in PD patients) and at 10 Hz (worsening of motor symptoms). During HFS, PSCs were only able to follow the first few stimuli before their amplitude rapidly declined (Fig. 1B and C). Upon cessation of HFS, PSC responses quickly recovered to baseline (Fig. 1D). When we delivered HFS in the presence of the GABAA receptor antagonist, picrotoxin (100 μm), we obtained a virtually identical time course and extent of synaptic suppression (Fig. 2A), thus excluding an appreciable contribution of fast GABAergic inhibition. The decay time constant of the EPSCs summarized in Fig. 2A was 0.1 ± 0.02 s (n = 5).
Figure 1. STN-HFS silences synaptic transmission onto SNc dopaminergic neurons recorded in whole-cell voltage-clamp mode.
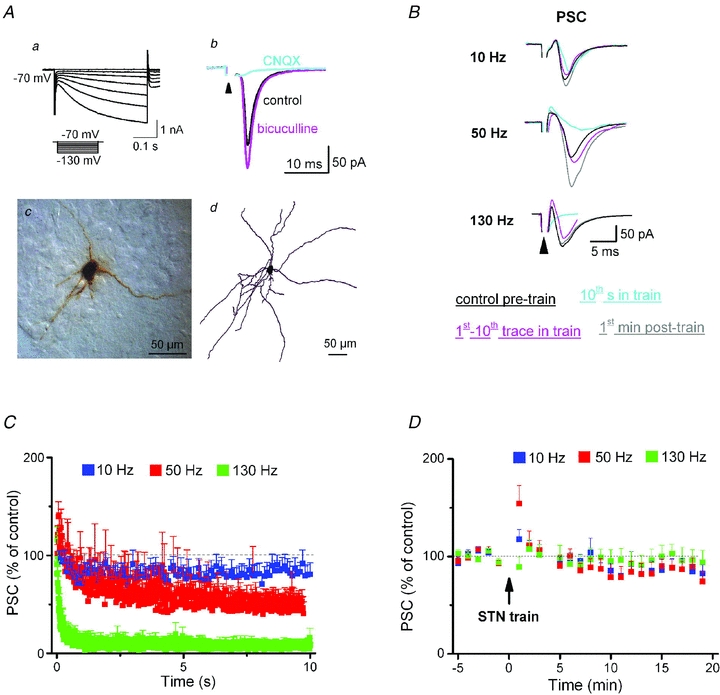
Aa and b, dopaminergic neurons in SNc were identified by the characteristic activation of pronounced hyperpolarization-activated current (Ih) in response to hyperpolarizing voltage commands (a); they responded to single STN stimuli (indicated by arrowhead; 60 μs, 200 μA, stimulus artifact truncated) with brief postsynaptic current (PSC, b). PSC amplitude increased during superfusion with the GABAA receptor antagonist bicuculline (30 μm) and disappeared in the presence of the AMPA receptor antagonist CNQX (20 μm) (b). Ac and d, example of a biocytin-labelled SNc neuron in cryostat section (c) and after reconstruction using Neurolucida (d). B, superimposed traces from different neurons illustrate the frequency-dependent effect of STN trains (10 s long) on PSC amplitudes. C, plot of changes in normalized PSC amplitudes during stimulus trains demonstrates time- and frequency-dependent effects of STN stimulation (10 Hz, n = 10; 50 Hz, n = 10; 130 Hz, n = 9). Averaged PSC amplitudes at the end of train were 82 ± 11%, 48 ± 11% and 12 ± 8% of control for 10 Hz, 50 Hz and 130 Hz, respectively. Note that 50 Hz and 130 Hz trains caused an initial transient increase in PSC amplitude. D, plot of grouped data before and after STN trains shows rapid recovery of PSC amplitude to pre-train levels after cessation of trains. STN was stimulated at 0.1 Hz before and after train, with pulse width of 60 μs and intensity of 200–1000 μA. Each data point in D represents the average of 6 consecutive recordings.
Figure 2. Rapid rundown of excitatory postsynaptic currents (EPSCs) in voltage-clamped SNc dopaminergic neurons during STN-HFS.
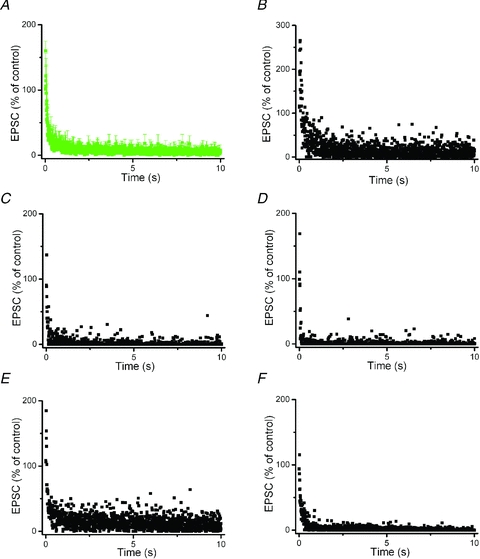
EPSCs were recorded in the presence of GABAA receptor antagonist picrotoxin (100 μm). A, averaged normalized EPSC responses from the five neurons shown in B–F. B–F, changes of EPSC responses during STN-HFS train (130 Hz, 10 s) in five individual neurons. EPSC responses were normalized to their amplitude before STN-HFS. Note incomplete suppression of EPSCs during HFS, with sporadic large-amplitude responses. Mean EPSC amplitude at the end of HFS train was 5 ± 2% of control (n = 5).
Despite the pronounced synaptic decline during HFS that is evident from the plots of Fig. 1C and Fig. 2A, we wondered if, once in a while, single EPSCs might have escaped from the overall depression. We therefore replotted in Fig. 2B–F the five recordings that were averaged to give Fig. 2A. From an inspection of the individual records it becomes evident that the synaptic currents exhibited a remarkable degree of variability during HFS, with intermittent responses of considerable amplitude, as if signal transfer had briefly, though incompletely, recovered.
As in SN reticulata (SNr) (Shen & Johnson, 2008), additional experiments ruled out that presynaptic suppression of glutamate release by GABAB heteroceptors or metabotropic glutamate receptors was responsible for the rapid suppression of EPSCs during HFS trains. As shown in Fig. 3A, both the GABAB receptor antagonist CGP 55845 (2 μm, n = 9) and the mGluRII antagonist LY 341495 (100 nm, n = 7) did not alleviate the synaptic depression during HFS. Unlike the GABAB receptor antagonist, the mGluRII antagonist transiently enhanced EPSCs immediately after termination of HFS (Fig. 3B). To determine whether this effect was mediated presynaptically or postsynaptically, we employed a paired-pulse protocol, in which two identical stimuli (inter-pulse interval of 25 ms) were delivered to STN and resulted in a facilitated second response (inset). The transient increase in the 1st EPSC in the presence of the mGluRII antagonist was accompanied by a reduction in paired-pulse ratio (the factor obtained by dividing the amplitude of the 2nd EPSC by that of the 1st EPSC) from 1.50 ± 0.14 in control to 1.08 ± 0.06 at the 1st minute after HFS cessation (n = 7; P < 0.05, paired t test). This is indicative of a presynaptic site of action and suggests that mGluRII on glutamatergic terminals serve to suppress the sudden, but transient, overshoot in EPSC amplitude when HFS is terminated. Similar effects were also obtained with antagonists for mGluRIa (CPCCOEt, 100 μm, n = 8) and mGluRIII (CPPG, 100 μm, n = 10) (data not shown).
Figure 3. Effects of GABAB receptor and metabotropic glutamate receptor antagonists on EPSCs of SNc dopaminergic neurons during and after STN-HFS.
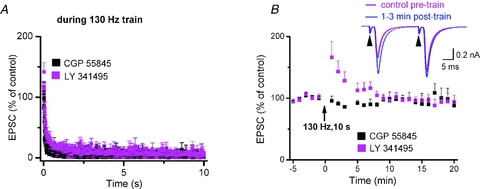
A, the GABAB receptor antagonist CGP 55845 (2 μm, n = 9) and the mGluRII antagonist LY 341495 (100 nm, n = 7) failed to relieve the synaptic depression during HFS train. B, grouped data before and after HFS-trains indicate rapid recovery of EPSC after termination of HFS. Note that mGluRII antagonist transiently enhanced EPSCs immediately after termination of HFS. In some experiments, we employed a paired-pulse protocol, in which two identical stimuli (inter-pulse interval of 25 ms) were delivered to STN and resulted in a facilitated second response (inset). The transient increase in the 1st EPSC in the presence of mGluRII antagonist was accompanied by reduction in paired-pulse ratio.
Effects of STN-HFS on single unit activity and field potentials in SNc
The rapid HFS-induced disconnection of SNc neurons from STN predicts that their intrinsic rhythmicity should become equally fast independent of STN input. In fact, in single-unit recordings from rhythmically firing SNc neurons (1.8 ± 0.1 Hz, n = 13) orthodromically connected to STN, HFS produced only a brief and transient increase in discharge rate before baseline firing resumed (Fig. 4A and B). This is highly reminiscent of the failure of HFS in the thalamocortical preparation to produce more than an initial brief irritation of spontaneous firing in pyramidal cells (Meeks et al. 2005). We therefore propose that HFS functionally uncouples STN neurons from their target neurons, and argue therefore against the notion that HFS would exert a continuous synaptic drive onto SNc dopaminergic neurons by enhancing STN axonal output.
Figure 4. Effects of STN-HFS on single unit activity and field potentials in SNc.
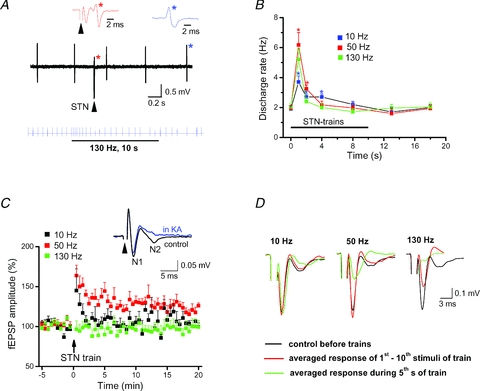
All recordings were made in the presence of picrotoxin (100 μm). A, upper black trace of original recording illustrates single unit activity of a rhythmically discharging SNc dopaminergic neuron that was orthodromically activated by single STN stimulation. Note the out-of-rhythm discharge (red star) after STN stimulation (arrowhead; 90 μs, 200 μA). Coloured traces above depict the waveforms of unit discharge (like-coloured stars) at expanded time scale. Lower blue trace of spike events demonstrates transient increase of single-unit activity when STN-HFS begins. Note that the neuron quickly resumed baseline firing pattern despite sustained delivery of HFS train. B, plot quantifies brief transient increases in firing rate after start of stimulus trains at different frequencies (10 Hz, n = 6; 50 Hz, n = 6; 130 Hz, n = 5; *P < 0.05). C, electrical stimulation in STN evoked biphasic field potentials in SNc, consisting of an early axonal component (N1) termed fibre volley (FV) and a late synaptic component (N2). Latencies to peak were 3.0 ± 0.1 ms for N1 and 7.5 ± 0.5 ms for N2 (n = 11). The latter N2 reflects field EPSP as indicated by its sensitivity to the unselective ionotropic glutamate receptor antagonist kynurenic acid (KA, 2 mm; inset). Plot depicts normalized N2 component before and after STN trains of different frequencies (10 Hz, n = 7; 50 Hz, n = 9; 130 Hz, n = 11). Both 10 Hz and 50 Hz trains induced short-term potentiation, but only stimulation at 50 Hz was able to induce long-term potentiation of field EPSP (122 ± 4% of control, averaged over 16th–20th min post train, n = 9). Note that HFS (130 Hz) failed to induce either form of plasticity. STN was stimulated at 0.1 Hz before and after train, with pulse width of 90 μs and intensity of 100–300 μA. D, averaged field potentials recorded in SNc before and at two time points during 10 s stimulus trains of different frequencies delivered to STN (stimulus intensity 200 μA). Note strong and virtually complete suppression of fibre volley (N1) in the course of 130 Hz stimulation.
Tetanic stimulation of the glutamatergic projection can induce synaptic plasticity in dopaminergic neurons (Overton et al. 1999; Nugent et al. 2008), but it is questionable whether STN-HFS would produce similar effects given the rapid decline of postsynaptic responses. Since whole-cell recording might perturb the molecular machinery required for long-term synaptic changes, we recorded SNc field potentials elicited by electrical stimulation in STN. SNc field potentials exhibited an early component (N1) that reflects axonal action potentials and is termed fibre volley (FV), and a subsequent component (N2) that arises from field EPSPs (fEPSPs) and is sensitive to the glutamate receptor antagonist, kynurenic acid (KA, Fig. 4C, inset). In contrast to 10 Hz stimulation, which produced short-term potentiation, and to 50 Hz stimulation, which produced both short-term and long-term potentiation of fEPSPs, HFS failed to induce either form of plasticity (Fig. 4C), suggesting that therapeutic effects of DBS are not mediated by long-term changes in synaptic plasticity.
Reversible axonal failure during STN-HFS
Close examination of individual field potentials during HFS revealed an unexpected decline of axonal excitability (N1, Fig. 4D). This observation offers the intriguing possibility that the loss of synaptic transmission during HFS is secondary to the failure of axonal impulses to reach presynaptic terminals. To elaborate on the behaviour of axonal action potentials during HFS, we isolated N1 by suppressing synaptic potentials with KA, picrotoxin and reduced extracellular calcium (0.2 mm). Application of a 10 s stimulus train at 10 Hz produced a slight increase of FV latency without reducing FV amplitude, whereas stimulation at 50 Hz and 130 Hz strongly diminished and virtually abrogated, respectively, FV responses (Fig. 5A–C). Magnitude and recovery of FV responses during and after HFS trains (Fig. 5A–D) closely resembled those of PSC responses in single SNc neurons (see above). The decay time constant of FVs during HFS was 1.57 ± 0.11 s, (n = 7) which is substantially slower than that of EPSCs (see above). Although we do not have an unambiguous measure of how to translate a change in FV into the corresponding change in EPSC, the much more rapid decline of EPSCs that clearly preceded the decay of FVs in the brief time window after the onset of HFS points to a mechanism other than axonal failure, most probably involving presynaptic neurotransmitter depletion. However, from a comparison of the time courses it is safe to conclude that within about 2 s of HFS, axonal failure emerged as the primary mechanism to halt signal transmission.
Figure 5. Reversible axonal failure during STN-HFS.
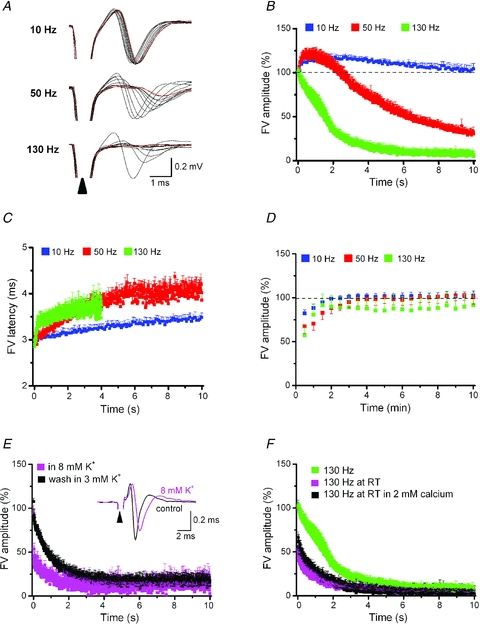
FVs were recorded in bath solution containing reduced calcium (0.2 mm), picrotoxin (100 μm) and kynurenic acid (2 mm) to abrogate synaptic potentials. A, selected FV responses (every 1 s) elicited by STN trains of 10, 50 or 130 Hz were superimposed to show gradual shift in latency and peak amplitude. Most leftward black trace and red trace indicate first and last FV response, respectively, in each sequence. Sample traces were collected in a single recording session with inter-train intervals of 30 min each. Plots of normalized FV amplitudes (B) and averaged FV latencies (C) recorded in SNc during 10 s stimulus trains delivered to STN at different frequencies (10 Hz, n = 5; 50 Hz, n = 5; 130 Hz, n = 8). Averaged FV amplitudes at the end of train were 103 ± 6%, 26 ± 5% and 8 ± 4% of control for 10 Hz, 50 Hz and 130 Hz, respectively. STN trains for 10 s at 10 Hz and 50 Hz increased FV latency from 3.0 ± 0.1 ms to 3.5 ± 0.1 ms (P = 0.01) and to 4.0 ± 0.2 ms (P = 0.02), respectively. In view of the progressive decline of FVs during HFS (130 Hz), latencies were only determined over 4 s (from 3.0 ± 0.1 ms to 3.5 ± 0.1 ms at 4th s, P = 0.002). D, amplitude of FVs recovered quickly to pre-train level after cessation of trains. E, raising [K+]o from 3 mm to 8 mm expedited FV rundown during STN-HFS in a reversible fashion (n = 5). Inset depicts superimposed FV responses to single STN stimulus in control and high [K+]o solution. Elevation of [K+]o increased latency to negative peak from 2.9 ± 0.2 ms to 3.4 ± 0.3 ms (P = 0.03) and reduced peak amplitude from 0.34 ± 0.03 mV to 0.23 ± 0.03 mV (P = 0.01; n = 5). F, lowering bath temperature to room temperature (RT) accelerated rundown of FV amplitude during HFS independent of extracellular calcium concentration (0.2 mm, n = 4; 2 mm, n = 6). For comparison, we replotted here the decay of FV amplitude at control (high) temperature already shown in B (green symbols each).
Why does HFS suppress axonal conduction of action potentials? HFS and high-frequent neuronal activity in the brain are known to engender substantial rises in extracellular potassium ([K+]o) (Heinemann & Gutnick, 1979; Shin et al. 2007). Elevated [K+]o might exert particular dramatic effects on action potential generation in myelinated axons, because diffusion is impaired and reuptake by Na+/K+ pumps will be more readily saturated. Indeed, computer simulations predict conduction block through firing-dependent [K+]o accumulation within the periaxonal space of myelinated axons (Bellinger et al. 2008). Since one cannot directly determine [K+]o levels of the myelinated axons of STN neurons, we explored how experimental elevation of [K+]o would affect axonal excitability. Increasing [K+]o to 8 mm enhanced latency and reduced amplitude of FVs under control conditions and significantly accelerated FV rundown during HFS (Fig. 5E). In 8 mm K+, the decay time constant of FVs during HFS was 0.79 ± 0.05 s (n = 5), whereas subsequent wash in 3 mm slowed the time constant to 1.09 ± 0.04 s (n = 5, P = 0.03). We next performed field potential recordings at room temperature which should compromise [K+]o clearance by slowing Na+/K+ pumps. Just as high [K+]o solution, low temperature produced a significantly faster decline of FVs during HFS compared to high temperature, the decay time constants being 1.08 ± 0.12 s (n = 5) and 1.57 ± 0.11 (n = 7), respectively (P = 0.03, Fig. 5F). To exclude that the reduction of extracellular Ca2+ to 0.2 mm (with Mg2+ elevated to 3.8 mm, see Methods) might have influenced axonal excitability during HFS, we repeated the experiment in 2 mm Ca2+ and obtained a virtually identical decline of FVs (Fig. 5F). Although temperature-dependent effects on channel kinetics might also play a role, the remarkable and highly similar facilitation of FV rundown during HFS by both high [K+]o and low temperature suggest that the excitability of STN axons is exquisitely sensitive to [K+]o changes. We therefore advance depolarization block by elevated [K+]o as a likely mechanism of axonal failure during DBS.
STN-HFS disrupts axonal conduction within STN and towards basal ganglia output nuclei
From a clinical point of view, it is important to demonstrate that STN-HFS also silences the projections to SNr and entopeduncular nucleus (EP). Neurons in these output nuclei of the basal ganglia respond to single STN stimulation with monosynaptic and/or complex EPSCs (Shen & Johnson, 2006; Ammari et al. 2010). Again, we pharmacologically isolated FVs to monitor solely axonal excitability. Strongly resembling our findings at the STN→SNc projection, STN-HFS produced an equally rapid and effective suppression of FVs in SNr and EP (Fig. 6A and B), indicating that these STN projections are also subjected to axonal block.
Figure 6. STN-HFS disrupts axonal conduction within STN and towards basal ganglia output nuclei.
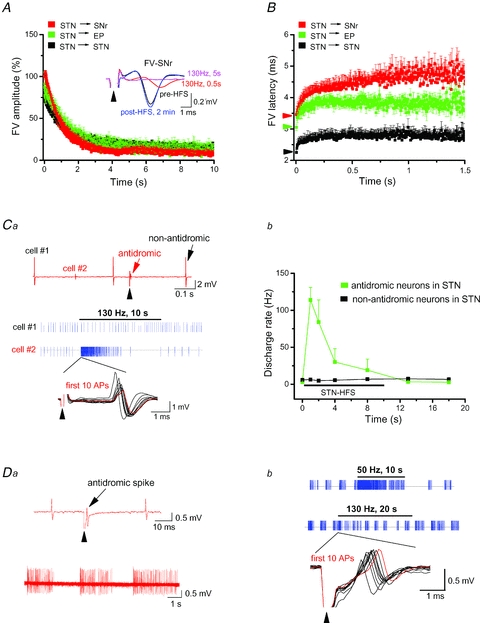
Plots of normalized FV amplitudes (A) and averaged FV latencies (B) during STN-HFS recorded within STN (n = 9), in SN reticulata (SNr, n = 7) and in entopeduncular nucleus (EP, n = 6). Averaged FV amplitudes at the end of HFS were 15 ± 2%, 8 ± 2% and 12 ± 2% of control for STN, SNr and EP, respectively. Inset in A depicts superimposed FVs recorded in SNr in response to STN stimulation (arrowhead, 90 μs, 200 μA) at different time points before, during and after HFS as indicated. Coloured arrowheads along the y-axis of B indicate control values for FV latency in like-coloured target regions. STN-HFS increased FV latency from 2.3 ± 0.1 ms to 2.8 ± 0.1 ms (P = 0.001) for STN, from 3.2 ± 0.1 ms to 3.8 ± 0.1 ms (P = 0.001) for EP and from 3.5 ± 0.2 ms to 4.5 ± 0.2 ms (P = 0.001) for SNr, respectively, measured at 1–1.5 s after switching on HFS trains. Note the different time scale along the x-axis in B. C and D single unit recordings of rhythmically firing STN neurons. Ca, impact of HFS on two simultaneously recorded, tonically firing neurons in STN. As indicated in the upper original trace, single STN stimulation (arrowhead, 90 μs, 200 μA) did not affect the spontaneous discharge of cell no. 1, but produced antidromic activation of cell no. 2. The spike event traces (blue, middle part) illustrate differential effect of STN-HFS on the firing pattern of the two cells. The first 10 action potentials of cell no. 2 at the beginning of HFS are superimposed and shown at expanded time scale below corresponding blue trace to demonstrate gradual shift in amplitude and latency of antidromic spike. Red trace represents last (10th) spike in this series. Cb, plot quantifying the transient effect of HFS on firing rate in antidromically activated STN neurons (n = 4, green). Non-antidromically activated STN neurons (n = 12, black) displayed unchanged activity during HFS. The antidromic spikes faithfully followed 10 Hz and 50 Hz during 10 s trains (data not shown). Da, STN neuron with burst firing pattern (lower trace) was antidromically activated by STN stimulation (arrowhead, 90 μs, 250 μA; upper trace). Db, STN-HFS transiently enhanced firing frequency during first few seconds (lower blue trace), in contrast to the massive increase with 50 Hz stimulation (upper blue trace). Recordings are from the burst-firing neuron of Da (similar data from two other burst-firing neurons are not illustrated). The first 10 action potentials at the beginning of HFS are superimposed and shown at expanded time scale below blue trace to demonstrate gradual shift in latency of antidromic spike. Trace in red represents last (10th) spike in this series.
Finally, we explored how HFS impacts the excitability of STN itself. FVs in STN arise from both incoming and outgoing fibres and were rapidly and effectively shut down by HFS (Fig. 6A and B) suggesting that DBS disconnects STN also from incoming signals. Indeed, PSCs in STN neurons declined quickly after switching on HFS (n = 7; data not shown). Does HFS also disrupt the intrinsic activity of STN neurons? To address this issue, we recorded spontaneous single-unit activity in tonic and burst firing STN neurons that were antidromically activated by STN stimulation. Tonically active neurons followed antidromic HFS only for a few seconds before resuming baseline firing or exhibiting decreased activity (Fig. 6C). Nearby STN neurons not antidromically activated remained largely unperturbed by STN-HFS (Fig. 6C). In bursting STN neurons displaying antidromic activation (Fig. 6D1), HFS for 20 s did not entirely abrogate burst firing, but produced a modified pattern with reduced burst duration and longer inter-burst intervals (Fig. 6D2).
Discussion
To the best of our knowledge, our study is the first to directly examine the effect of STN-HFS on axonal excitability by recording isolated fibre volleys. Our data strongly suggest that DBS overwhelms the axons’ capability to follow the high frequency (130 Hz) required for the therapeutic effects to occur. Loss of axonal action potential fidelity and conduction block during HFS was also reported from hippocampal and thalamocortical preparations (Meeks et al. 2005; Iremonger et al. 2006; Jensen & Durand, 2009) and might thus represent a common feature independent of the specific localization of the stimulation electrode. The concept of HFS-induced axonal failure might help to reconcile apparently conflicting views on how DBS abates pathological electrical activity in basal ganglia loops. The classical view that DBS exerts a local functional inhibition of STN neurons akin to surgical lesioning has been challenged more recently (reviewed in Deniau et al. 2010). For example, in a recent study in PD patients, Carlson et al. (2010) reported that DBS failed to silence surrounding STN neurons. This finding was also observed in our preparation and under our experimental conditions, where the delivery of HFS within the STN did not lead to a cessation of intrinsic firing of STN neurons (Fig. 6). It is worth noting though, that HFS was capable of modifying the pattern of intrinsic burst firing activity, an effect that might prove particularly beneficial in PD, in which abnormal burst firing seems to prevail and has been mechanistically linked to the characteristic motor symptoms (see Introduction).
Both clinical data and our own recordings in brain slices are therefore not compatible with the hypothesis that STN-HFS simply abrogates neuronal activity within the STN. At the same time, however, our data do not support the alternative view which posits that DBS drives axons both orthodromically and antidromically with high fidelity, thereby producing a broad spectrum of local and systems level effects that act in concert to re-establish more physiological patterns of activity in PD. In a sense, our findings refresh the classical hypothesis that likened DBS to tissue lesion, but we emphasize now depression of incoming and outgoing axons rather than a global silencing of local STN activity per se. We thus propose that DBS, while not necessarily overriding intrinsic activity at the site of stimulation, largely uncouples this site from upstream and downstream regions by inhibiting the pathways of neuronal communication.
Regarding the mechanisms of conduction block during HFS, a previous modelling study (Bellinger et al. 2008) and our experimental data suggest that accumulation of K+ in the submyelin space and concomitant axonal depolarization move Na+ channels towards inactivated states. Assuming Na+ inactivation as the leading cause of axonal failure, the extent to which HFS affects signal conduction will undoubtedly depend on a number of anatomical and physiological factors, including the geometrical and biophysical properties of the axonal fibres, the constraints of the submyelin space and the extent of K+ dispersion it allows, as well as the spatial and temporal efficacy of uptake systems which remove K+ from that space. It is worth noting that the development of axonal depression is not a self-perpetuating process, since, with Na+ channel progressively accumulating in inactivated states, axonal activity is progressively shut down and further K+ efflux ceases. As a consequence, neuronal excitability might at least partially recover, so that some stimuli will be propagated again and give rise to synaptic responses in the target regions.
Because intermittent synaptic responses remain indeed detectable during HFS (Fig. 2), STN target regions are not entirely disconnected during DBS. Although we advance axonal failure as an important mechanism in DBS, we are well aware that the ratio between failing and transmitted stimuli during DBS might display appreciable variations between tissue of different brain regions, between tissue of different species, and, importantly, between normal and diseased tissue. Our data are therefore not contradictory to electrophysiological and neurochemical studies in the intact brain of animals or patients, in which DBS was found to exert excitatory or inhibitory effects in target regions (for a recent review, see Deniau et al. 2010 and the references therein). Rather than simply producing a complete and lasting conduction block, we see axonal failure as a significant mechanism of DBS to filter pathological activity to and from STN, thereby allowing basal ganglia networks to resume regular firing patterns. Axonal failure offers a surprisingly simple and broadly applicable mechanism of DBS that readily explains major clinical features such as the requirement of high stimulus frequencies, the rapid onset and loss of therapeutic benefits with the make and break of stimulation, and the apparent lack of long-lasting effects.
As DBS is now being increasingly extended to treat drug-refractory neuropsychiatric disorders such as major depression, obsessive–compulsive disorder (OCD), Gilles-de-la-Tourette syndrome and addiction, it remains to be determined whether axonal failure emerges as a generally applicable principle to account for the effects of HFS in other brain regions. Whereas DBS relieves motor symptoms of PD almost instantaneously, the improvement in OCD or depression develops over several weeks, suggesting that the latter cases require plastic changes (Krack et al. 2010). This observation, however, does not necessarily argue against an involvement of axonal failure, since, in terms of clinical outcome, neuropsychiatric DBS, like STN-DBS, mimics the effect of neurosurgical lesion in different target regions (Krack et al. 2010). It is therefore well conceivable that axonal failure also participates in the beneficial effects of DBS in neuropsychiatric diseases, the major difference being that, in PD, an immediate effect on the perturbed basal ganglia connectivity predominates, whereas in depression and OCD, compensatory adaptations in target and projection regions have to occur to gradually readjust the dysfunctional circuits.
Acknowledgments
We thank Didier Gremelle for technical assistance. Funding was from the intramural research funds of the Universities of Kiel and Erlangen-Nürnberg.
Glossary
Abbreviations
- DBS
deep brain stimulation
- EP
entopeduncular nucleus
- EPSC
excitatory postsynaptic current
- EPSP
excitatory postsynaptic potential
- FV
fibre volley
- HFS
high frequency stimulation
- mGluR
metabotropic glutamate receptor
- PD
Parkinson's disease
- PSC
postsynaptic current
- SNc
substantia nigra compacta
- SNr
substantia nigra reticulata
- STN
subthalamic nucleus
Author contributions
F.Z. and C.A. designed the experiments and interpreted the data together with F.S. and J.V.; F.Z., K.L. and B.N.-B. performed experiments and carried out data analysis, with the help of F.S.; C.A. wrote the manuscript, with contributions from F.Z. and J.V. All authors approved the final version of the manuscript. All experiments were performed in the Physiological Institutes of Kiel and Erlangen.
References
- Ammari R, Lopez C, Bioulac B, Garcia L, Hammond C. Subthalamic nucleus evokes similar long lasting glutamatergic excitations in pallidal, entopeduncular and nigral neurons in the basal ganglia slice. Neuroscience. 2010;166:808–818. doi: 10.1016/j.neuroscience.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Ammari R, Lopez C, Fiorentino H, Gonon F, Hammond C. A mouse juvenile or adult slice with preserved functional nigro-striatal dopaminergic neurons. Neuroscience. 2009;159:3–6. doi: 10.1016/j.neuroscience.2008.10.051. [DOI] [PubMed] [Google Scholar]
- Bekar L, Libionka W, Tian GF, Xu Q, Torres A, Wang X, Lovatt D, Williams E, Takano T, Schnermann J, Bakos R, Nedergaard M. Adenosine is crucial for deep brain stimulation-mediated attenuation of tremor. Nat Med. 2008;14:75–80. doi: 10.1038/nm1693. [DOI] [PubMed] [Google Scholar]
- Bellinger SC, Miyazawa G, Steinmetz PN. Submyelin potassium accumulation may functionally block subsets of local axons during deep brain stimulation: a modeling study. J Neural Eng. 2008;5:263–274. doi: 10.1088/1741-2560/5/3/001. [DOI] [PubMed] [Google Scholar]
- Beurrier C, Ben-Ari Y, Hammond C. Preservation of the direct and indirect pathways in an in vitro preparation of the mouse basal ganglia. Neuroscience. 2006;140:77–86. doi: 10.1016/j.neuroscience.2006.02.029. [DOI] [PubMed] [Google Scholar]
- Beurrier C, Bioulac B, Audin J, Hammond C. High-frequency stimulation produces a transient blockade of voltage-gated currents in subthalamic neurons. J Neurophysiol. 2001;85:1351–1356. doi: 10.1152/jn.2001.85.4.1351. [DOI] [PubMed] [Google Scholar]
- Bruet N, Windels F, Bertrand A, Feuerstein C, Poupard A, Savasta M. High frequency stimulation of the subthalamic nucleus increases the extracellular contents of striatal dopamine in normal and partially dopaminergic denervated rats. J Neuropathol Exp Neurol. 2001;60:15–24. doi: 10.1093/jnen/60.1.15. [DOI] [PubMed] [Google Scholar]
- Carlson JD, Cleary DR, Cetas JS, Heinricher MM, Burchiel KJ. Deep brain stimulation does not silence neurons in subthalamic nucleus in Parkinson's patients. J Neurophysiol. 2010;103:962–967. doi: 10.1152/jn.00363.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniau JM, Degos B, Bosch C, Maurice N. Deep brain stimulation mechanisms: beyond the concept of local functional inhibition. Eur J Neurosci. 2010;32:1080–1091. doi: 10.1111/j.1460-9568.2010.07413.x. [DOI] [PubMed] [Google Scholar]
- Do MT, Bean BP. Subthreshold sodium currents and pacemaking of subthalamic neurons: modulation by slow inactivation. Neuron. 2003;39:109–120. doi: 10.1016/s0896-6273(03)00360-x. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galati S, Mazzone P, Fedele E, Pisani A, Peppe A, Pierantozzi M, Brusa L, Tropepi D, Moschella V, Raiteri M, Stanzione P, Bernardi G, Stefani A. Biochemical and electrophysiological changes of substantia nigra pars reticulata driven by subthalamic stimulation in patients with Parkinson's disease. Eur J Neurosci. 2006;23:2923–2928. doi: 10.1111/j.1460-9568.2006.04816.x. [DOI] [PubMed] [Google Scholar]
- Garcia L, Audin J, D'Alessandro G, Bioulac B, Hammond C. Dual effect of high-frequency stimulation on subthalamic neuron activity. J Neurosci. 2003;23:8743–8751. doi: 10.1523/JNEUROSCI.23-25-08743.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science. 2009;324:354–359. doi: 10.1126/science.1167093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C, Bergman H, Brown P. Pathological synchronization in Parkinson's disease: networks, models and treatments. Trends Neurosci. 2007;30:357–364. doi: 10.1016/j.tins.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Elder CM, Okun MS, Patrick SK, Vitek JL. Stimulation of the subthalamic nucleus changes the firing pattern of pallidal neurons. J Neurosci. 2003;23:1916–1923. doi: 10.1523/JNEUROSCI.23-05-01916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann U, Gutnick MJ. Relation between extracellular potassium concentration and neuronal activities in cat thalamus (VPL) during projection of cortical epileptiform discharge. Electroencephalogr Clin Neurophysiol. 1979;47:345–347. doi: 10.1016/0013-4694(79)90285-2. [DOI] [PubMed] [Google Scholar]
- Hilker R, Voges J, Ghaemi M, Lehrke R, Rudolf J, Koulousakis A, Herholz K, Wienhard K, Sturm V, Heiss WD. Deep brain stimulation of the subthalamic nucleus does not increase the striatal dopamine concentration in parkinsonian humans. Mov Disord. 2003;18:41–48. doi: 10.1002/mds.10297. [DOI] [PubMed] [Google Scholar]
- Iremonger KJ, Anderson TR, Hu B, Kiss ZH. Cellular mechanisms preventing sustained activation of cortex during subcortical high-frequency stimulation. J Neurophysiol. 2006;96:613–621. doi: 10.1152/jn.00105.2006. [DOI] [PubMed] [Google Scholar]
- Jensen AL, Durand DM. High frequency stimulation can block axonal conduction. Exp Neurol. 2009;220:57–70. doi: 10.1016/j.expneurol.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Futami T. Arrhythmic firing in dopamine neurons of rat substantia nigra evoked by activation of subthalamic neurons. J Neurophysiol. 1999;82:1632–1637. doi: 10.1152/jn.1999.82.3.1632. [DOI] [PubMed] [Google Scholar]
- Krack P, Hariz MI, Baunez C, Guridi J, Obeso JA. Deep brain stimulation: from neurology to psychiatry? Trends Neurosci. 2010;33:474–484. doi: 10.1016/j.tins.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Kuhn AA, Kempf F, Brucke C, Gaynor DL, Martinez-Torres I, Pogosyan A, Trottenberg T, Kupsch A, Schneider GH, Hariz MI, Vandenberghe W, Nuttin B, Brown P. High-frequency stimulation of the subthalamic nucleus suppresses oscillatory beta activity in patients with Parkinson's disease in parallel with improvement in motor performance. J Neurosci. 2008;28:6165–6173. doi: 10.1523/JNEUROSCI.0282-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn AA, Kupsch A, Schneider GH, Brown P. Reduction in subthalamic 8–35 Hz oscillatory activity correlates with clinical improvement in Parkinson's disease. Eur J Neurosci. 2006;23:1956–1960. doi: 10.1111/j.1460-9568.2006.04717.x. [DOI] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. Two cell types in rat substantia nigra zona compacta distinguished by membrane properties and the actions of dopamine and opioids. J Neurosci. 1989;9:1233–1241. doi: 10.1523/JNEUROSCI.09-04-01233.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacombe E, Carcenac C, Boulet S, Feuerstein C, Bertrand A, Poupard A, Savasta M. High-frequency stimulation of the subthalamic nucleus prolongs the increase in striatal dopamine induced by acute l-3,4-dihydroxyphenylalanine in dopaminergic denervated rats. Eur J Neurosci. 2007;26:1670–1680. doi: 10.1111/j.1460-9568.2007.05747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KH, Blaha CD, Harris BT, Cooper S, Hitti FL, Leiter JC, Roberts DW, Kim U. Dopamine efflux in the rat striatum evoked by electrical stimulation of the subthalamic nucleus: potential mechanism of action in Parkinson's disease. Eur J Neurosci. 2006;23:1005–1014. doi: 10.1111/j.1460-9568.2006.04638.x. [DOI] [PubMed] [Google Scholar]
- Li S, Arbuthnott GW, Jutras MJ, Goldberg JA, Jaeger D. Resonant antidromic cortical circuit activation as a consequence of high-frequency subthalamic deep-brain stimulation. J Neurophysiol. 2007;98:3525–3537. doi: 10.1152/jn.00808.2007. [DOI] [PubMed] [Google Scholar]
- Maurice N, Thierry AM, Glowinski J, Deniau JM. Spontaneous and evoked activity of substantia nigra pars reticulata neurons during high-frequency stimulation of the subthalamic nucleus. J Neurosci. 2003;23:9929–9936. doi: 10.1523/JNEUROSCI.23-30-09929.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre CC, Grill WM, Sherman DL, Thakor NV. Cellular effects of deep brain stimulation: model-based analysis of activation and inhibition. J Neurophysiol. 2004;91:1457–1469. doi: 10.1152/jn.00989.2003. [DOI] [PubMed] [Google Scholar]
- Meeks JP, Jiang X, Mennerick S. Action potential fidelity during normal and epileptiform activity in paired soma-axon recordings from rat hippocampus. J Physiol. 2005;566:425–441. doi: 10.1113/jphysiol.2005.089086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner W, Harnack D, Reese R, Paul G, Reum T, Ansorge M, Kusserow H, Winter C, Morgenstern R, Kupsch A. High-frequency stimulation of the subthalamic nucleus enhances striatal dopamine release and metabolism in rats. J Neurochem. 2003;85:601–609. doi: 10.1046/j.1471-4159.2003.01665.x. [DOI] [PubMed] [Google Scholar]
- Nugent FS, Hwong AR, Udaka Y, Kauer JA. High-frequency afferent stimulation induces long-term potentiation of field potentials in the ventral tegmental area. Neuropsychopharmacology. 2008;33:1704–1712. doi: 10.1038/sj.npp.1301561. [DOI] [PubMed] [Google Scholar]
- Overton PG, Richards CD, Berry MS, Clark D. Long-term potentiation at excitatory amino acid synapses on midbrain dopamine neurons. NeuroReport. 1999;10:221–226. doi: 10.1097/00001756-199902050-00004. [DOI] [PubMed] [Google Scholar]
- Rivlin-Etzion M, Marmor O, Heimer G, Raz A, Nini A, Bergman H. Basal ganglia oscillations and pathophysiology of movement disorders. Curr Opin Neurobiol. 2006;16:629–637. doi: 10.1016/j.conb.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Shen KZ, Johnson SW. Subthalamic stimulation evokes complex EPSCs in the rat substantia nigra pars reticulata in vitro. J Physiol. 2006;573:697–709. doi: 10.1113/jphysiol.2006.110031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen KZ, Johnson SW. Complex EPSCs evoked in substantia nigra reticulata neurons are disrupted by repetitive stimulation of the subthalamic nucleus. Synapse. 2008;62:237–242. doi: 10.1002/syn.20488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DS, Samoilova M, Cotic M, Zhang L, Brotchie JM, Carlen PL. High frequency stimulation or elevated K+ depresses neuronal activity in the rat entopeduncular nucleus. Neuroscience. 2007;149:68–86. doi: 10.1016/j.neuroscience.2007.06.055. [DOI] [PubMed] [Google Scholar]
- Steigerwald F, Potter M, Herzog J, Pinsker M, Kopper F, Mehdorn H, Deuschl G, Volkmann J. Neuronal activity of the human subthalamic nucleus in the parkinsonian and nonparkinsonian state. J Neurophysiol. 2008;100:2515–2524. doi: 10.1152/jn.90574.2008. [DOI] [PubMed] [Google Scholar]
- Walker RH, Koch RJ, Moore C, Meshul CK. Subthalamic nucleus stimulation and lesioning have distinct state-dependent effects upon striatal dopamine metabolism. Synapse. 2009;63:136–146. doi: 10.1002/syn.20592. [DOI] [PubMed] [Google Scholar]
- Windels F, Bruet N, Poupard A, Urbain N, Chouvet G, Feuerstein C, Savasta M. Effects of high frequency stimulation of subthalamic nucleus on extracellular glutamate and GABA in substantia nigra and globus pallidus in the normal rat. Eur J Neurosci. 2000;12:4141–4146. doi: 10.1046/j.1460-9568.2000.00296.x. [DOI] [PubMed] [Google Scholar]
- Wingeier B, Tcheng T, Koop MM, Hill BC, Heit G, Bronte-Stewart HM. Intra-operative STN DBS attenuates the prominent beta rhythm in the STN in Parkinson's disease. Exp Neurol. 2006;197:244–251. doi: 10.1016/j.expneurol.2005.09.016. [DOI] [PubMed] [Google Scholar]