

Abstract
Perfusion of hippocampal slices with an inhibitor of nitric oxide (NO) synthase-blocked induction of long-term potentiation (LTP) produced by a one-train tetanus and significantly reduced LTP by a two-train tetanus, but only slightly reduced LTP by a four-train tetanus. Inhibitors of heme oxygenase, the synthetic enzyme for carbon monoxide (CO), significantly reduced LTP by either a two-train or four-train tetanus. These results suggest that NO and CO are both involved in LTP but may play somewhat different roles. One possibility is that NO serves a phasic, signaling role, whereas CO provides tonic, background stimulation. Another possibility is that NO and CO are phasically activated under somewhat different circumstances, perhaps involving different receptors and second messengers. Because NO is known to be activated by stimulation of NMDA receptors during tetanus, we investigated the possibility that CO might be activated by stimulation of metabotropic glutamate receptors (mGluRs). Consistent with this idea, long-lasting potentiation by the mGluR agonist tACPD was blocked by inhibitors of heme oxygenase but not NO synthase. Potentiation by tACPD was also blocked by inhibitors of soluble guanylyl cyclase (a target of both NO and CO) or cGMP-dependent protein kinase, and guanylyl cyclase was activated by tACPD in hippocampal slices. However, biochemical assays indicate that whereas heme oxygenase is constitutively active in hippocampus, it does not appear to be stimulated by either tetanus or tACPD. These results are most consistent with the possibility that constitutive (tonic) rather than stimulated (phasic) heme oxygenase activity is necessary for potentiation by tetanus or tACPD, and suggest that mGluR activation stimulates guanylyl cyclase phasically through some other pathway.
Long-term potentiation (LTP) is a sustained increase in synaptic efficacy that is thought to be one of the candidate mechanisms for memory storage in the hippocampus (for reviews, see Bliss and Collingridge 1993; Hawkins et al. 1993). In the CA1 region of hippocampus, the induction of LTP generally requires Ca2+ influx through postsynaptic N-methyl-d-aspartate (NMDA) glutamate receptor channels, but the maintenance of LTP is thought to involve, in part, a presynaptic enhancement of transmitter release. These results suggest that the postsynaptic cells must send one or more retrograde messengers to the presynaptic terminals. There is now evidence that several molecules may act as such retrograde messengers during LTP in hippocampus, including the soluble gases nitric oxide (NO) (Böhme et al. 1991; O’Dell et al. 1991; Schuman and Madison 1991; Haley et al. 1992; Zhou et al. 1993; Arancio et al. 1996) and carbon monoxide (CO) (Stevens and Wang 1993; Zhuo et al. 1993), as well as arachidonic acid (Williams et al. 1989), platelet-activating factor (Del Cero et al. 1990; Clark et al. 1992; Wieraszko et al. 1993), and several neurotrophins (Kang and Schuman 1995; Thoenen 1995; Korte et al. 1996). However, a number of questions remain concerning the possible roles of NO and CO. First, although several studies have found that inhibitors, targeted mutation, or adenovirus-mediated inhibition of NO synthase block the induction of LTP in CA1 and dentate gyrus (Böhme et al. 1991; O’Dell et al. 1991; Schuman and Madison 1991; Haley et al. 1992; Mizutani et al. 1993; Boulton et al. 1995; Doyle et al. 1996; Son et al. 1996; Kantor et al. 1996; Wu et al. 1997), other studies have found that inhibitors of NO synthase either do not block LTP (Kato and Zorumski 1993; Bannerman et al. 1994; Cummings et al. 1994) or block LTP only under some experimental circumstances and not others (Gribkoff and Lum-Ragan 1992; Chetkovich et al. 1993; Haley et al. 1993, 1996; Williams et al. 1993; O’Dell et al. 1994; Malen and Chapman 1997). Second, inhibitors of heme oxygenase (the synthetic enzyme for CO) can also block the induction of LTP in the CA1 region of hippocampus and dentate gyrus (Stevens and Wang 1993; Zhuo et al. 1993; Ikegaya et al. 1994), but there are concerns about the specificity of those inhibitors (Ignarro et al. 1984; Linden et al. 1993; Luo and Vincent 1994; Meffert et al. 1994; Okada 1996). Third, if, as the inhibitor studies suggest, NO and CO are both involved in LTP, it is not clear what their respective roles might be. One possibility is that NO and CO are activated under somewhat different circumstances. This possibility would be consistent with the finding that inhibitors of NO synthase do not block LTP under all circumstances, suggesting that the residual potentiation might be mediated by another messenger. Alternatively, CO may provide a tonic level of stimulation, whereas NO provides phasic stimulation during the induction of LTP. This possibility would be consistent with the fact that NO has a very short half-life, whereas CO is more stable.
In the present study, we have addressed these questions to try to clarify the possible roles of NO and CO in LTP.
Materials and Methods
ELECTROPHYSIOLOGY
Male 4- to 6-week-old Sprague-Dawley rats were housed and sacrificed in accordance with the guidelines of the Health Sciences Division of Columbia University. Transverse slices of hippocampus (400 μm) were rapidly prepared and maintained between 28 and 30°C in an interface chamber (Fine Science Tools, Foster City, CA), in which they were subfused with artificial cerebrospinal fluid (ACSF) consisting of: 124 mm NaCl, 4.0 mm KCl, 2.0 mm CaCl2, 2.0 mm MgSO4, 1.0 mm Na2HPO4, 24.1 mm NaHCO3, 10 mm glucose, bubbled with 95% O2 and 5% CO2. Slices were allowed to recover for at least 1.5 hr before experiments were performed. A bipolar tungsten stimulating electrode was placed in the stratum radiatum of the CA1 region or in the stratum pyramidale of the CA3 region. In some experiments, a second independent stimulating electrode was placed in the stratum radiatum of the CA1 region on the other side of the recording microelectrode. Before the start of the experiments, paired-pulse facilitation (50 ms interval) was tested to verify the independence of the two pathways. Extracellular field potentials were recorded with a glass microelectrode (3–12 mΩ filled with ACSF) placed in the stratum radiatum. The stimulation intensity was adjusted to give field EPSP amplitudes of 1.0–1.5 mV (which is ∼25% of maximum) so that the weak tetanus by itself would not produce LTP. The two pathways were stimulated alternately at 0.02 Hz and the initial slope of the EPSP was measured. If the recording was stable for at least 30 min, potentiation was produced in one pathway by use of one of four tetanic stimulation protocols; (1) one train of 100 Hz, 1 sec stimulation; (2) two trains of 100 Hz, 1 sec stimulation separated by 20 sec; (3) four trains of 100 Hz, 1 sec stimulation separated by 5 min; or (4) one train of 50 Hz, 0.5 sec stimulation (weak tetanus) during brief (2 min) perfusion with CO, tACPD, or 8-Br-cGMP. Current intensity during the tetanic stimulation was always the same as test intensity. Potentiation was measured in each experiment as the average EPSP slope 50–60 min post-tetanus as a percentage of the average baseline for 30 min pre-tetanus.
DRUG PREPARATION
Nω-nitro-arginine, AP3, and 8-Br-cGMP (Sigma, St. Louis, MO), tACPD (RBI, Natick, MA), and Rp-8-Br-cGMPS (Biolog, La Jolla, CA) were dissolved in ACSF immediately before each experiment. Zinc protoporphyrin IX, tin protoporphyrin IX, copper protoporphyrin, and zinc deuteroporphyrin IX-3,4-bis-glycol (Porphyrin Products, Logan, UT) and LY83583 (Biomol, Plymouth Meeting, PA) were dissolved in dimethyl sulfoxide (DMSO) immediately before each experiment and diluted to the desired concentration in ACSF by sonication (final concentration of DMSO was ≤0.05%). (+)-MCPG (Tocris Neuramin, Bristol, UK) was dissolved with the 1.1 equivalent of sodium hydroxide solution to give 100 mm stock solution and then diluted to the desired concentration in ACSF. Hemoglobin was prepared from methemoglobin as described by Martin et al. (1985). CO solution was prepared by bubbling the gas in distilled water until saturation and immediately diluted to the desired concentration in ACSF. Slices were perfused with the various inhibitors throughout the experiment, starting at least 30 min before the beginning of the baseline period. CO, tACPD, or 8-Br-cGMP were injected directly into the recording chamber for 2 min before the weak tetanic stimulation and then washed out over a period of 5–10 min.
cGMP ASSAY
Hippocampal slices were prepared as in the LTP experiments and rested at 30°C for 2 hr with continuous ACSF perfusion. The slices were pretreated with 200 μm IBMX for 20 min followed by 100 μm tACPD, 10 μm NMDA, and 300 μm SNP or ACSF for ∼2 min. The slices were then removed from the perfusion chamber and frozen within 10 sec. After the CA3 region was removed, the slices were homogenized in 6% trichloroacetic acid. Protein was precipitated by spinning at 2000g for 20 min, and then the supernatant was extracted four times with water-saturated ether and dried under vacuum. The amount of cGMP in each sample was measured by radioimmunoassay (NEN) following the manufacturer’s instructions. The precipitated protein was dissolved in 100 mm NaOH and 0.3% SDS and quantified with the BCA protein assay kit (Pierce). The cGMP level in each slice was normalized to protein. There were three slices per condition in each experiment, and the average cGMP level for the experimental slices was expressed as a percentage of the average level for the control slices in that experiment. All of the slices in one experiment came from the same animal.
HEME OXYGENASE ACTIVITY ASSAY
Following in vitro treatment, hippocampal slices were frozen rapidly in dry ice. Tissue samples from the CA1 region of the hippocampus were collected after removing the CA3 region and dentate gyrus. To obtain enough material to assay, three slices were pooled together. Tissue samples were shipped to Finland on dry ice for heme oxygenase activity measurements, which were performed blind to the experimental treatment. Enzyme activity was determined by use of a novel sensitive microassay that relies on the conversion of [14C]heme to [14C]bilirubin by the concerted activity of heme oxygenase, NADPH-cytochrome P-450 reductase and biliverdin reductase, as described previously (Laitinen and Juvonen 1995). Briefly, slices (3–4 per assay) were sonicated at 0°C in 50 μl of 0.1 m K-phosphate buffer (pH 7.5) containing 50 μm phenylmethyl sulfonyl fluoride. The homogenate was centrifugated at 14,000g for 1 min in an Eppendorf minifuge. Duplicate aliquots of the supernatant (5 μl/7–26 μg protein) were incubated in 0.1 m K-phosphate buffer at pH 7.5 (total volume 10 μl) containing [14C]heme (sp. act. 52.5 Ci/mole) and NADPH (2 mm). The final substrate concentration was 21.4 μm in all but one experiment in which 4.4 μm substrate concentration was used to test for possible liberation of endogenous competing substrates during strong tetanic stimulation. Reagent blanks contained buffer instead of NADPH. Following 15 min incubation at 37°C, the tubes were cooled to 0°C and 190 μl of ice-cold K-phosphate buffer was added. [14C]bilirubin was extracted into toluene and counted in a Wallac LKB 1214 Rackbeta with 95.5% counting efficiency. Heme oxygenase activity (reagent blanks subtracted) is expressed as picomoles of [14C]bilirubin formed/mg protein per hour and was corrected for the extraction efficiency (15.4 ± 0.4%, n = 14). The following criteria have been applied to validate this method for the measurement of heme oxygenase activity in the rat brain (Laitinen and Juvonen 1995). First, incubation of rat brain homogenate with [14C]heme yielded a single reaction product, indistinguishable from bilirubin by thin layer chromatography. Second, the reaction was totally dependent on the presence of NADPH, was not catalyzed by boiled tissue, and was inhibited by ZnPP at doses known to inhibit purified rat heme oxygenase (IC50 = 0.3 μm). Third, the reaction required the presence of NADPH-cytochrome P-450, as evidenced by the ability of an antibody against the reductase to inhibit the reaction in a dose-dependent fashion.
DATA ANALYSIS AND STATISTICS
All data are presented as the mean ± s.e.m. The field EPSPs are presented as percentage of baseline. Statistical comparisons were made by by use of Student’s t-tests for comparison of groups or paired data. In all cases, P < 0.05 was considered significant.
Results
EFFECTS OF AN INHIBITOR OF NO SYNTHASE ON LRP INDUCED BY DIFFERENT NUMBERS OF TETANI
NO synthase inhibitors have blocked the induction of LTP in some studies, but have failed to block LTP in other studies. These discrepancies might be partially explained by different experimental conditions, including the strength of the tetanic stimulation (Gribkoff and Lum-Ragan, 1992; Chetkovich et al. 1993; Haley et al. 1993; O’Dell et al. 1994; Malen and Chapman 1997). To investigate this possibility further, we systematically examined the effects of inhibiting NO synthase on LTP induced by different numbers of tetanic stimuli. We found that pretreatment with the NO synthase inhibitor Nω-nitro-arginine (100 μm) for at least 30 min completely abolished LTP produced by a one-train tetanus (Fig. 1A). Nω-nitro-arginine also significantly reduced LTP produced by a two-train tetanus (130.8 ± 23.9%, n = 10 compared with 199.4 ± 18.6%, n = 10 in normal saline, t = 2.27, P < 0.05) and completely blocked it in 6 of 10 experiments. But this inhibitor produced only a slight reduction of LTP that was not significant when a four-train tetanus was used (Fig. 1B). These results suggest that NO makes a substantial contribution to the induction of LTP produced with low or moderate tetanic stimulation, but that other messengers may contribute more to LTP induced by stronger tetanic stimulation.
Figure 1.
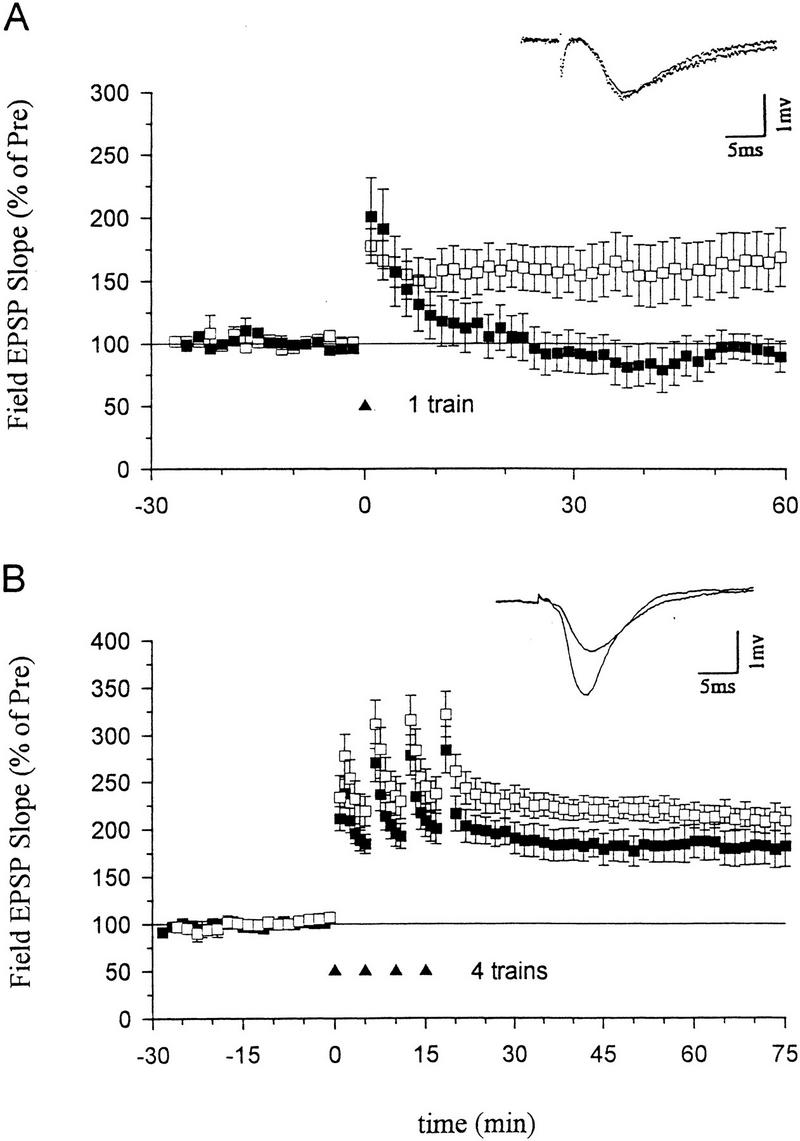
Effects of an inhibitor of nitric oxide synthase on LTP induced by a one- or four-train tetanus. (A) Nω-nitro-arginine (100 μm), a nitric oxide synthase inhibitor, blocked LTP induced by a one-train tetanus (100 Hz, 1 sec) (normal ACSF, average EPSP slope 50–60 min post-tetanus=162.7 ± 22.7% of the average baseline for 30 min pre-tetanus, n = 8; Nω-nitro-arginine, 94.1 ± 11.4%, n = 6, t = 2.42, P < 0.05 compared with ACSF-treated slices). (Inset) Representative recordings of the field EPSP before and 60 min after a one-train tetanus in a slice pretreated with Nω-nitro-arginine. (B) Nωq-nitro-arginine (100 μm) only slightly reduced ltp induced by a four-train tetanus (four 1-sec 100-hz trains delivered at an interval of 5 min; 181.0 ± 20.3%, n = 7) as compared with acsf-treated slices (210.5 ± 12.9%, n = 9, not significantly different). (inset) representative recordings of the field epsp before and 60 min after a four-train tetanus in a slice pretreated with nω-nitro-arginine. the average prevalues were 0.45 mv/msec (acsf), 0.40 (nω-nitro-arginine) (a) and 0.41 (acsf) 0.36 (nω-nitro-arginine) (b). (□acsf; (█) nitroarg.
TESTS OF INHIBITORS OF HEME OXYGENASE
Another candidate retrograde messenger is CO. Inhibitors of heme oxygenase (the synthetic enzyme for CO) block the induction of LTP, and CO paired with weak tetanic or low-frequency stimulation produces long-lasting potentiation (Stevens and Wang 1993; Zhuo et al. 1993). However, heme oxygenase inhibitors may also act by inhibiting other enzymes, including NO synthase (Meffert et al. 1994; Okada 1996). To examine whether inhibition of NO synthase, rather than heme oxygenase, might account for the block of LTP, we compared the effectiveness of inhibitors of heme oxygenase and NO synthase on LTP induced by different numbers of tetanic stimuli.
LTP induced by two trains of tetanus was significantly attenuated by the heme oxygenase inhibitors Tin protoporphyrin IX (SnPP, 10 μm) or zinc-deuteroporphyrin IX-2,4-bis-glycol (ZnBG, 10 μm) (Fig. 2A). In contrast, an inactive analog, copper protoporphyrin (CuPP, 10 μm), did not significantly affect LTP. LTP produced by a four-train tetanus was also significantly reduced by ZnBG (10 μm, Fig. 2B) or zinc protoporphyrin (ZnPP, 10 μm, 152.1 ± 17.5%, n = 6; t = 2.74, P < 0.05 compared with normal LTP induced by a four-train tetanus). Heme oxygenase inhibitors did not completely block the potentiation induced by a four-train tetanus, however. To test whether the remaining potentiation is mediated by NO, we pretreated slices with both Nω-nitro-arginine (100 μm) and ZnBG (10 μm). The added NO synthase inhibitor did not produce further reduction as compared with ZnBG alone (Fig. 2C). Thus, LTP induced by four trains of stimulation was only slightly reduced by an inhibitor of NO synthase, but was significantly reduced by inhibitors of heme oxygenase at a 10-fold lower dosage. Moreover, the combination of an NO synthase inhibitor and a heme oxygenase inhibitor had effects similar to the heme oxygenase inhibitor alone but not the NO synthase inhibitor alone. These results suggest that heme oxygenase inhibitors did not act by inhibiting NO synthase in these studies.
Figure 2.

Effects of inhibitors of heme oxygenase on ltp induced by a two- or four-train tetanus. (a) average potentiation of the field epsp by a two-train tetanus (100 hz for 1 sec each, separated by 20 sec) in normal acsf and in acsf containing 10 μm Tin protoporphyrin IX (SnPP), Zinc-deuteroporphyrin IX-2,4-bis-glycol (ZnBG), or Copper protoporphyrin (CuPP). Perfusion with SnPP, ZnBG, or CuPP started at least 30 min before the tetanus (normal LTP, 199.4 ± 18.6%, n = 10; 10 μm SnPP, 129.1 ± 3.8%, n = 7, t = 2.79, P < 0.05 compared with normal LTP; 10 μm ZnBG, 126.8 ± 8.3%, n = 6, t= 2.88, P < 0.05 compared with normal LTP; 10 μm CuPP, 188.1 ± 14.3%, n = 5). Results with SnPP and ZnBG were similar and have been pooled. (□) ACSF; (▴) CuPP; (█) 5nPP/ZmBG. (B) Average potentiation by a four-train tetanus in ACSF (210.5 ± 12.9%, n = 9) and in ACSF containing ZnBG (10 μm, 145.5 ± 14.2%, n = 6, t = 3.18, P < 0.01 compared with saline-treated slices). (□) ACSF; (█) ZmBG. (C) Average potentiation by a four-train tetanus in ACSF containing both ZnBG (10 μm) and Nω-nitro-arginine (100 μm) (143.1 ± 12.6%, n = 5). (▴) ZmBG + NitroARG. (D) ZnPP-IX (10 μm) or ZnBG (10 μm) did not block long-term enhancement produced by CO (100 nm) paired with weak tetanic stimulation (ZnPP-IX, 160.8 ± 19.9%, n = 5; ZnBG, 236.0 ± 22.7%, n = 5). (□) ZmBG/XmPP. Results with the two inhibitors were similar and have been pooled. The average prevalues were 0.37 mV/msec (normal LTP) (A), 0.38 (SnPP), 0.40 (ZnBG) and 0.39 (CuPP) (B); 0.45 (ACSF), 0.46 (ZnBG); 0.34 (C); and 0.29 (D).
Another potential concern is that inhibitors of heme oxygenase might also act by inhibiting soluble guanylyl cyclase (Ignarro et al. 1984; Luo and Vincent 1994), which could block the induction of LTP (Zhuo et al. 1994; Boulton et al. 1995; Son et al. 1998). To test this possibility, we examined enhancement by CO, which stimulates soluble guanylyl cyclase (J.T. Laitinen, K.S.M. Laitinen, L. Tuomiste, and M.M. Airaksinenm, unpubl; Maines 1993; Verma et al. 1993). If the heme oxygenase inhibitors act by directly inhibiting guanylyl cyclase, they should block the potentiation produced by CO. In the presence of 10 μm ZnPP or ZnBG, CO (100 nm) paired with weak stimulation still produced long-lasting potentiation (Fig. 2D). These observations suggest that ZnPP or ZnBG at a dose of 10 μm did not inhibit soluble guanylyl cyclase in these experiments, but rather acted by inhibiting heme oxygenase. These findings are consistent with recent studies showing that inhibitors of heme oxygenase at this dose have little effect on NO synthase or soluble guanylyl cyclase in endothelial or intestinal tissue (Zakhary et al. 1996, 1997).
EFFECTS OF HEME OXYGENASE INHIBITORS ON tACPD-INDUCED ENHANCEMENT
The results of the inhibitor experiments suggest that NO and CO may both be involved in LTP, which raises the question of what their respective roles might be. One possibility suggested by these results is that NO synthase and heme oxygenase are activated by different stimulation patterns that might engage different receptors and second messengers. In the hippocampus, NO synthase is activated by stimulation of NMDA glutamate receptors during tetanic stimulation (East and Garthwaite 1991; Chetkovich et al. 1993). Heme oxygenase is present in hippocampal pyramidal cells (Maines 1993; Verma et al. 1993), but it is not known whether heme oxygenase can be acutely activated during tetanic stimulation. Heme oxygenase appears to be activated by stimulation of metabotropic glutamate receptors (mGluRs) in other brain regions (Glaum and Miller 1993; Nathanson et al. 1995), and the mGluR agonist tACPD has been reported to produce long-lasting potentiation in the hippocampus (Otani and Ben-Ari 1991; Radpour and Thomson 1992; Bortolotto et al. 1994), suggesting the possibility that NO and CO could be activated in parallel by stimulation of NMDA and mGluRs, respectively. We tested that possibility by examining the effect of inhibitors of heme oxygenase on potentiation by tACPD.
Consistent with previous reports, we found that, when paired with weak tetanic stimulation of the presynaptic fibers (50 Hz, 0.5 sec), a short bath application of tACPD (20 μm, 2 min) produced a rapid enhancement of the EPSP that lasted more than 1 hr (Fig. 3A). Neither weak stimulation nor tACPD alone produced enhancement (Fig. 3A,B). Pretreatment with ZnPP (10 μm) or ZnBG (10 μm) completely abolished the long-lasting enhancement produced by tACPD paired with weak tetanic stimulation (Fig. 3C). As controls for specificity, ZnPP or ZnBG did not affect the baseline EPSP in a second, untetanized pathway in the same slice or post-tetanic potentiation (PTP) in the tetanized pathway. ZnPP or ZnBG also did not affect the decrementing potentiation 5 min after the weak tetanus (short-term potentiation or STP), suggesting that they did not affect NMDA receptors (Stevens and Wang 1993; Zhuo et al. 1993). Furthermore, the inactive analog CuPP did not affect the long-lasting enhancement produced by tACPD paired with weak stimulation (data not shown). Unlike ZnPP and ZnBG, the NO synthase inhibitor N℅q nitro-arginine (100 μm) also did not affect the enhancement by tACPD paired with weak tetanic stimulation (Fig. 4A). These results further support the idea that the heme oxygenase inhibitors did not act by inhibiting NO synthase in these studies, and suggest that CO is involved in potentiation by tACPD.
Figure 3.
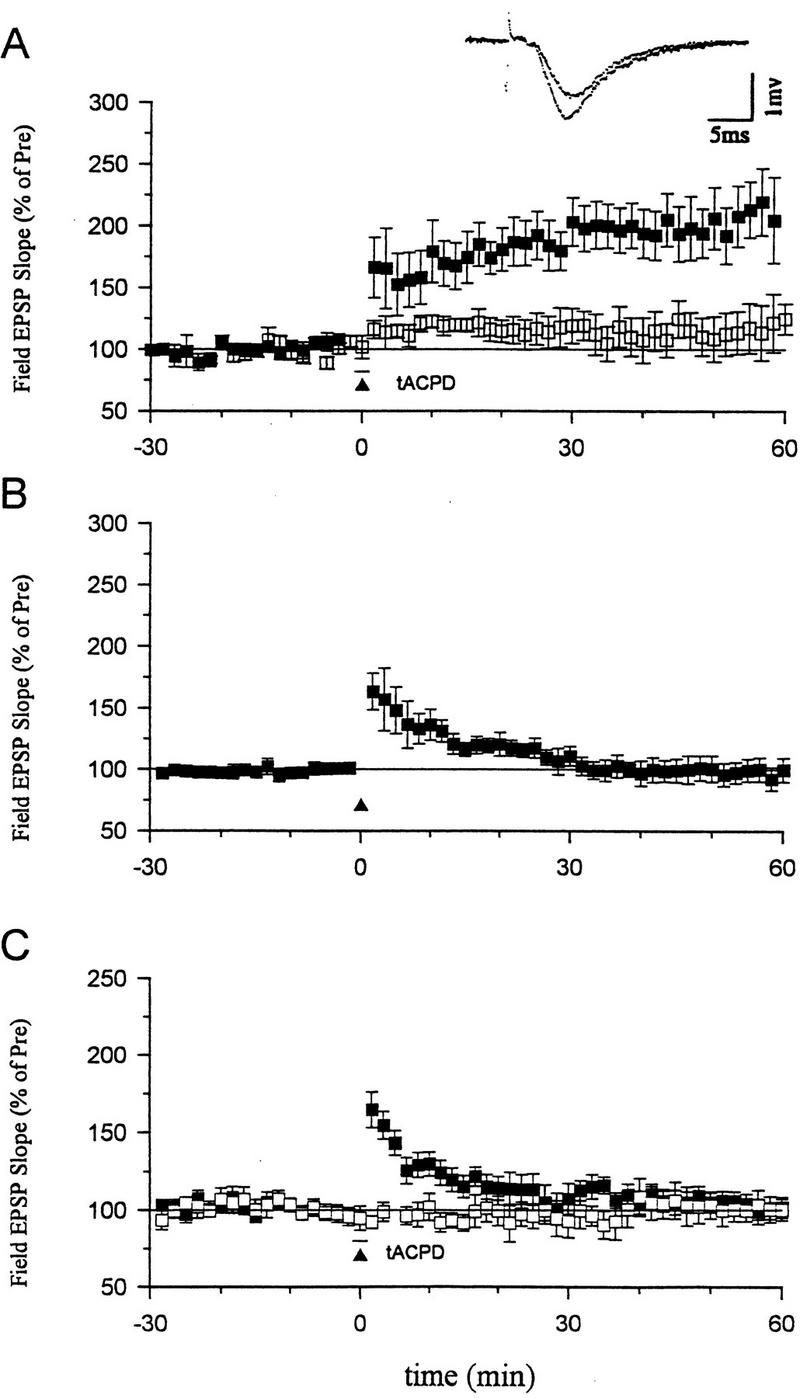
Effects of heme oxygenase inhibitors on long-term enhancement produced by tACPD. (A) tACPD (20 μm, horizontal bar) produced a rapid onset, long-lasting enhancement of the field EPSP (201.5 ± 22.4%, n = 8, t = 4.53, P < 0.01, comparing average EPSPs 50–60 min post-training and 30 min pre-training) when applied at the same time as weak tetanic stimulation of the presynaptic fibers (50 Hz, 0.5 sec; █; paired training). The EPSP at a control pathway in the same slice, which received tACPD alone, was not significantly potentiated (111.7 ± 14.8%). (Inset) Representative recordings of the field EPSP before and 60 min after tACPD paired training. (B) Weak stimulation (50 Hz, 0.5 sec) alone did not produce long-term enhancement (96.5 ± 6.7%, n = 7). (C) Heme oxygenase inhibitors (10 μm ZnPP-IX or ZnBG) completely blocked long-term enhancement produced by tACPD paired training (█) (ZnPP-IX, 113.9 ± 8.2%, n = 6; ZnBG, 89.8 ± 5.9%, n = 5). The EPSP at a second control pathway in the same slice was stable (□, tACPD alone) (100.7 ± 7.6%). Results with the two inhibitors were similar and have been pooled. The average prevalues were 0.26 mV/msec (tACPD paired), 0.27 (tACPD alone) (A); 0.26 (B); and 0.25 (paired), 0.30 (control) (C).
Figure 4.
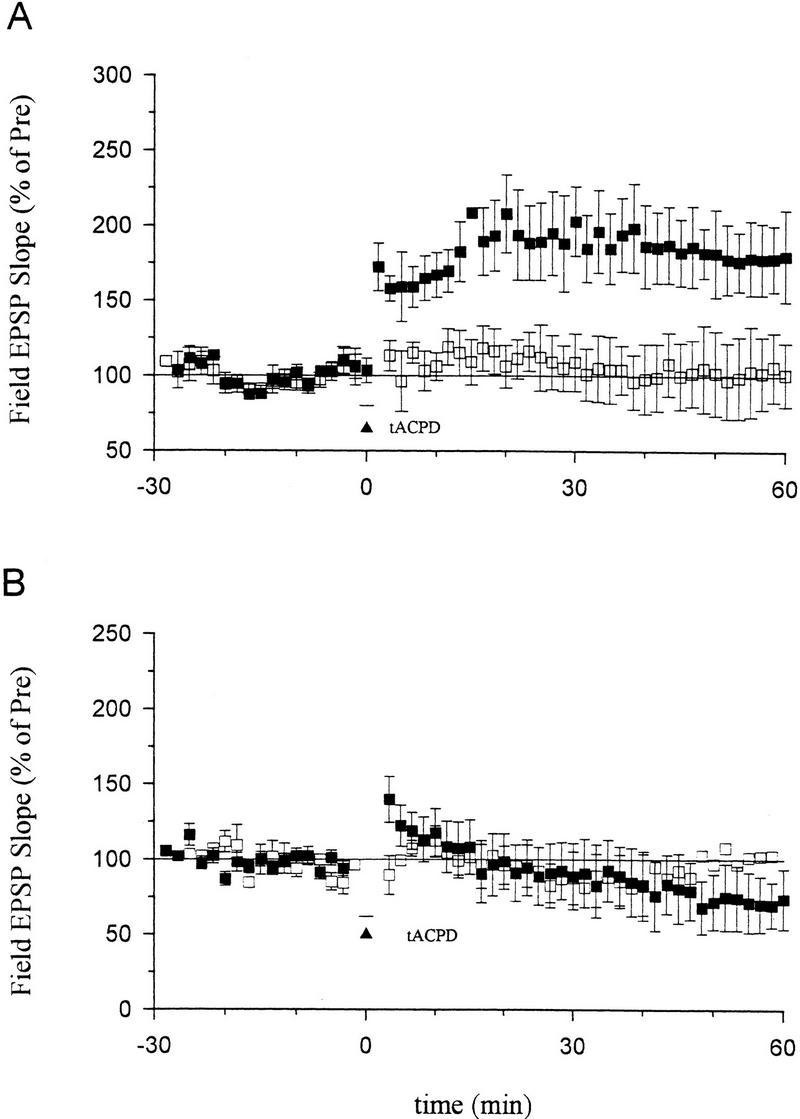
Effects of an NO synthase inhibitor or hemoglobin on long-term enhancement produced by tACPD paired training. (A) Nω-nitro-arginine (100 μm) did not block long-term enhancement by tACPD paired with weak stimulation (top; █, 172.4 ± 19.3%, n = 8, t = 3.75, P < 0.01). The EPSP at a second control pathway in the same slice (bottom), which received tACPD alone, was stable (□, 101.1 ± 24.5%). (B) Hemoglobin (20 μm) completely prevented long-term enhancement produced by tACPD paired training (█, 72.0 ± 19.5%, n = 7). The EPSP at a second control pathway in the same slice was stable (□, 104.3 ± 36%). The average prevalues were 0.30 mV/msec (paired), 0.26 (control) (A) and 0.28 (paired), 0.30 (control) (B).
If CO serves as one of the retrograde messengers during potentiation, it must diffuse from the postsynaptic cell to the presynaptic terminals through the extracellular space. Following bath application of the CO and NO binding protein hemoglobin (20 μm) for at least 0.5 hr, tACPD paired with weak tetanic stimulation failed to produce long-lasting potentiation, but rather produced slight depression (Fig. 4B). This depression seems unlikely to result from the nonspecific effects of hemoglobin, because the EPSP in a second (control) pathway in the same slice was stable. One possible explanation is that tACPD might also trigger other processes related to depression that do not require diffusion of CO or NO through the extracellular space (Baskys and Malenka 1991; Bolshakov and Siegelbaum 1994).
INVOLVEMENT OF SOLUBLE GUANYLYL CYCLASE IN tACPD-INDUCED ENHANCEMENT
CO activates soluble guanylyl cyclase and increases the production of cGMP in various regions of the central nervous system, including the hippocampus (J.T. Laitinen, K.S.M. Laitinen, L. Tuomisto, and M.M. Airaksinenn, unpubl.; Maine 1993; Verma et al. 1993). LY83583 and ODQ, two inhibitors of guanylyl cyclase, can block the induction of LTP (Zhuo et al. 1994; Boulton et al. 1995; Son et al. 1998). Conversely, membrane-permeable cGMP analogs paired with weak tetanic stimulation can produce long-lasting potentiation both in hippocampal slices (Haley et al. 1992; Zhuo et al. 1994; Son et al. 1998) and in dissociated cultures of hippocampal neurons (Arancio et al. 1995). Therefore, we investigated the relationship between mGluRs and cGMP during potentiation.
8-Br-cGMP paired with weak tetanic stimulation still produced significant long-term enhancement in the presence of the mGluR antagonists 2-amino-3-phosphonopropinate (AP3, 100 μm) or (+)-MCPG (500 μm) (Fig. 5A), suggesting that cGMP does not act by enhancing activation of some types of mGluRs. Conversely, pretreatment with the guanylyl cyclase inhibitor LY83583 (5 μm) abolished the enhancement produced by tACPD paired with weak tetanic stimulation (Fig. 5B), suggesting that cGMP acts downstream of the mGluRs. LY83583 did not affect the enhancement produced by 8-Br-cGMP paired with weak tetanic stimulation (Fig. 5C), indicating that LY83583 is relatively specific and did not inhibit processes downstream from soluble guanylyl cyclase. Furthermore, pretreatment with an inhibitor of cGMP-dependent protein kinase, Rp-8-Br-cGMPS (10 μm) also significantly reduced the enhancement produced by tACPD paired with weak tetanic stimulation (Fig. 5D). These results suggest that mGluRs produce potentiation by stimulating guanylyl cyclase and cGMP-dependent protein kinase.
Figure 5.

cGMP and cGMP-dependent protein kinase may act downstream of the mGluRs. (A) The enhancement by 8-Br-cGMP (100 μm, horizontal bar) paired with weak tetanic stimulation (▴) was not blocked by the mGluR antagonists AP3 (181.5 ± 14.9%, n = 5, t = 5.49, P < 0.01) or (+)-MCPG (159.7 ± 11.9%, n = 5, t = 5.02, P < 0.01). Results with the two antagonists were similar and have been pooled. (B) The enhancement produced by tACPD (20 μm) paired with weak tetanus was abolished by the guanylyl cyclase inhibitor LY83583 (100.1 ± 14.2%, n = 6). (C) The enhancement produced by 8-Br-cGMP (100 μm) paired with weak tetanus was not blocked by LY83583 (160.0 ± 10.0%, n = 6). (D) Rp-8-Br-cGMPS, an inhibitor of cGMP-dependent protein kinase, reduced the enhancement produced by tACPD (20 μm) paired with weak tetanus (120.6 ± 16.7%, n = 6, t = 2.50, P < 0.05 compared with tACPD paired training in ACSF; Fig. 3A). The average prevalues were 0.30 mV/msec (A), 0.32 (B), 0.42 (C), and 0.32 (D).
As another test for activation of guanylyl cyclase by mGluRs, we carried out an in vitro biochemical assay of cGMP in hippocampal slices. Application of tACPD (100 μm) produced an increase in cGMP (Fig. 6A). In agreement with previous reports (East and Garthwaite 1991; Chetkovich et al. 1993), NMDA or the NO donor sodium nitroprusside (SNP) also increased the level of cGMP in hippocampal slices.
Figure 6.
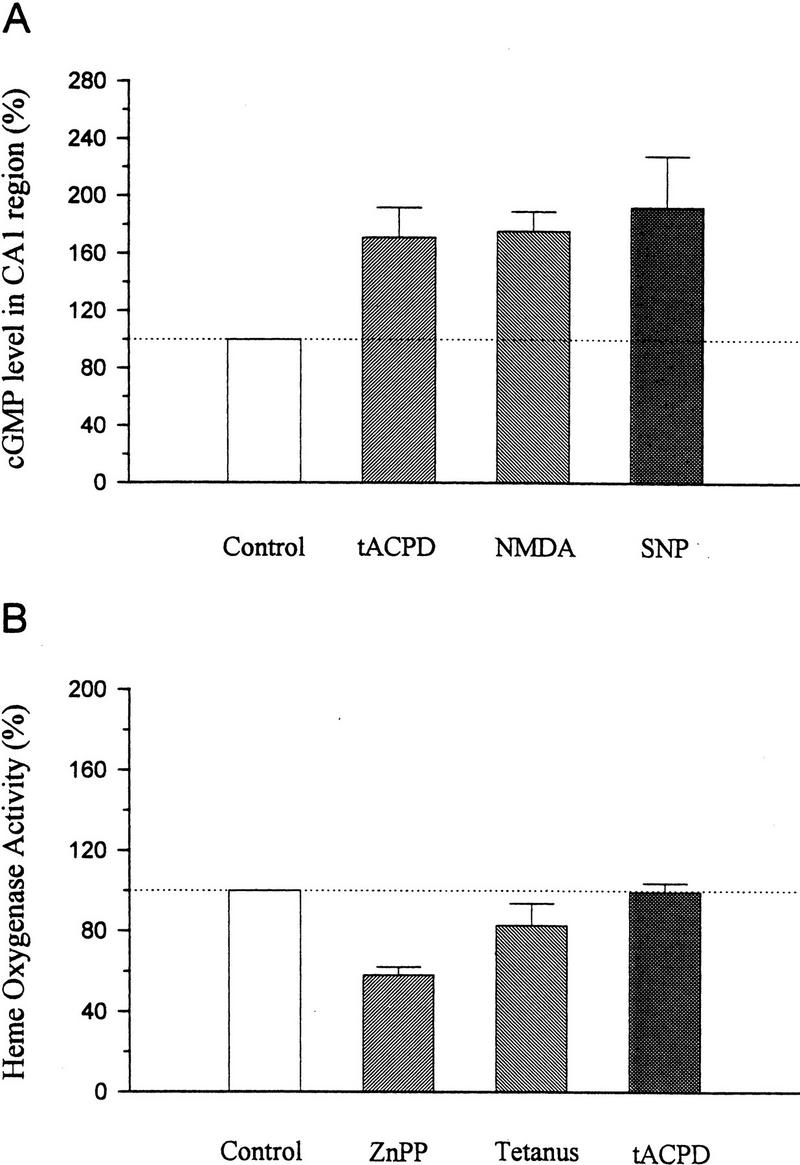
Biochemical assays. (A) Radioimmunoassay of cGMP in the CA1 region of hippocampus treated with vehicle (control), tACPD, NMDA, or SNP.The cGMP level was increased by 100 μm tACPD (170.8 ± 20.9%, n = 6, t = 3.39, P < 0.05), 10 μm NMDA (175.0 ± 13.9%, n = 3), or 300 μm SNP (192.2 ± 35.7%, n = 3). Data are presented as the percentage of the cGMP level in vehicle-treated slices from the same experiment. (B) Heme oxygenase activity in slices treated with vehicle (control), ZnPP, strong tetanus, or tACPD. Pretreatment with ZnPP (10 μm) for at least 30 min significantly decreased heme oxygenase activity to 58.3 ± 3.8% of control values (n = 4, t = 10.97, P < 0.01). Strong tetanus did not affect heme oxygenase activity, measured 1 min after tetanic stimulation, with 21.4 μm substrate concentration (82.7 ± 10.9%, n = 4). Application of tACPD (100 μm) for either 2 or 5 min did not significantly affect heme oxygenase activity (tACPD 2 min, 93.7 ± 5.2%, n = 5; tACPD 5 min, 104.7 ± 3.7%, n = 5). The 2- and 5-min data have been pooled.
HEME OXYGENASE ACTIVITY IN HIPPOCAMPUS
These results are consistent with the possibility that during tetanic stimulation, NO synthase and heme oxygenase are activated in parallel by stimulation of NMDA and mGluRs, respectively. However, another possibility is that heme oxygenase activity is constitutive and provides a tonic, background level of stimulation that is necessary for potentiation by either tetanic stimulation or tACPD. To attempt to distinguish between these possibilities, we estimated hippocampal heme oxygenase activity by measuring the conversion of [14C]heme to [14C]bilirubin by the concerted activity of heme oxygenase, NADPH-cytochrome P-450 reductase and biliverdin reductase. In the CA1 region of the hippocampus, the mean basal heme oxygenase activity was 877 ± 57 pmoles/mg per hour (n = 25 slices) when 21.4 μm substrate concentration was used. Pretreating slices with ZnPP (10 μm) for at least 30 min at 28°C significantly decreased heme oxygenase activity ∼40% (Fig. 6B). In contrast, Nω-nitro-arginine (100 μm) did not affect heme oxygenase activity (data not shown). Strong tetanus or application of tACPD for either 2 or 5 min also did not affect heme oxygenase activity significantly.
The assay may not be sensitive enough to detect stimulation of heme oxygenase by tetanic stimulation if the tetanus releases endogenous substrates that compete with the exogenous heme in the assay. To test this possibility, we used a lower substrate concentration. However, the enzyme activity was also not altered 5 min after strong tetanic stimulation with 4.4 μm substrate concentration (111.0 ± 15.4%, n = 6). Basal heme oxygenase activity was significantly lower (227 ± 54 pmoles/mg per hour, n = 8) when assessed with 4.4 μm substrate concentration, as compared with the activity found with 21.4 μm heme (t = 6.09, P < 0.001), suggesting that substrate availability may be a rate-limiting step in hippocampal heme oxidation. This is somewhat surprising, as both known isozymes of heme oxygenase have a Km ∼0.5 μm for heme (Maines 1988). On the basis of a three-point Lineweaver-Burk plot, we estimated a Km of 37.2 μm and a vmax of 2250 pmoles/mg per hour for the basal hippocampal heme-degrading capacity. this result may suggest the presence of additional heme-degrading capacity within the hippocampal homogenate. heme-degrading capacity is not necessarily limited to the two known microsomal heme oxygenases, as there is some evidence for the presence of mitochondrial and cytosolic systems for heme degradation (maines 1988). the possible implication of these findings is that the present studies may have underestimated the actual capacity of hippocampal tissue to generate co.
The results of these assays favor the hypothesis that constitutive rather than stimulated heme oxygenase activity is critical for potentiation induced by strong tetanus or tacpd-paired training. However, our results do not exclude the possibility that heme oxygenase is activated in a restricted region or cellular compartment during ltp induction, and therefore might not be detected in our assays. similarly, because we applied tetanic stimulation or tacpd to slices and then measured heme oxygenase activity in homogenates, we may not have detected rapidly reversible stimulation of heme oxygenase.
Discussion
LTP in CA1 and dentate gyrus can be blocked by inhibitors, targeted mutation, or adenovirus-mediated inhibition of NO synthase, suggesting that NO is involved in potentiation (Böhme et al. 1991; O’Dell et al. 1991; Schuman and Madison 1991; Haley et al. 1992; Mizutani et al. 1993; Boulton et al. 1995; Doyle et al. 1996; Kantor et al. 1996; Son et al. 1996; Wu et al. 1997). However, NO synthase inhibitors failed to block LTP in some studies (Kato and Zorumski 1993; Bannerman et al. 1994; Cummings et al. 1994), and in other studies NO synthase inhibitors blocked LTP only under some experimental circumstances, but not under other circumstances (Gribkoff and Lum-Ragan 1992; Chetkovich et al. 1993; Haley et al. 1993, 1996; Williams et al. 1993; O’Dell et al. 1994; Malen and Chapman 1997). Williams et al. (1993) suggested that NO synthase inhibitors block LTP only in slices prepared from young animals and maintained at room temperature. However, we and others have found that NO synthase inhibitors can block LTP in slices maintained at 28–32°C (Böhme et al. 1991; O’Dell et al. 1991, 1994; Gribkoff and Lum-Ragan 1992; Chetkovich et al. 1993; Haley et al. 1993; Boulton et al. 1995; Son et al. 1996; Malen and Chapman 1997; Wu et al. 1997) and also in vivo (Mizutani et al. 1993; Doyle et al. 1996). We did not examine LTP in older rats, but several studies (Doyle et al. 1996; Haley et al. 1996; Malen and Chapman 1997) have reported that NO synthase inhibitors can block LTP in older animals. Therefore, we feel that age and temperature cannot explain most of the differences in the published results, and suggest that more of those differences might be explained by differences in the tetanic stimulation. We found that an NO synthase inhibitor blocked LTP produced by one train of tetanic stimulation and significantly reduced LTP by two trains of stimulation, but only slightly reduced LTP produced by four trains of stimulation. Similar results have been obtained in several other studies in which the strength of the tetanic stimulation was varied in different ways (Chetkovich et al. 1993; Haley et al. 1993; O’Dell et al. 1994; Malen and Chapman 1997; for review, see Gribkoff and Lum-Ragan 1992).
These results suggest that NO contributes importantly to LTP induced by relatively weak tetanic stimulation, but other messengers or enzymes contribute more to LTP induced by stronger tetanic stimulation. For example, previous studies have shown that cAMP-dependent protein kinase (PKA) makes little contribution to early-phase LTP induced by one train of tetanic stimulation, but makes more of a contribution to intermediate-phase LTP induced by two trains and makes a large contribution to late-phase LTP induced by four trains (Huang and Kandel 1994; Blitzer et al. 1995; Winder et al. 1998). Consistent with these results, Lu et al. (in prep.) have recently found that cGMP-dependent PKA contributes to late-phase LTP induced by three trains, but makes less of a contribution to LTP induced by four trains. Thus, the NO-cGMP-PKG signaling pathway and the cAMP-PKA pathway appear to play complementary roles in LTP. In addition to these pathways, other messengers and enzymes most likely also make contributions to LTP that are larger or smaller depending on the experimental conditions.
Another candidate retrograde messenger is CO. Inhibitors of heme oxygenase also block or significantly reduce the induction of LTP in the hippocampus (Stevens and Wang 1993; Zhuo et al. 1993; Ikegaya et al. 1994; Poss et al. 1995). However, there are concerns about the specificity of heme oxygenase inhibitors, one being that they may also inhibit NO production (Meffert et al. 1994; Okada, 1996). This seems unlikely in the present study, because LTP induced by either four trains of tetanic stimulation or tACPD paired with weak tetanus was only slightly reduced by an inhibitor of NO synthase (Nω nitro-arginine) but was significantly reduced by two inhibitors of heme oxygenase (ZnBG and ZnPP) at a 10-fold lower dosage. This pattern of results might occur if ZnBG and ZnPP were actually better inhibitors of NO synthase than Nω nitro-arginine. However, in biochemical assays of NO synthase activity in hippocampal tissue (East and Garthwaite 1991; Huang et al. 1993; Meffert et al. 1994) ZnBG did not inhibit NO synthase at all, and, although ZnPP did inhibit NO synthase, it was ∼1000-fold less potent than Nω nitro-arginine. Another concern is that inhibitors of heme oxygenase might also act by inhibiting soluble guanylyl cyclase (Ignarro et al. 1984; Luo and Vincent 1994). However, heme oxygenase inhibitors did not block potentiation induced by CO, which stimulates soluble guanylyl cyclase. These results are consistent with previous reports that heme oxygenase inhibitors do not inhibit soluble guanylyl cyclase in olfactory neuronal cultures (Verma et al. 1993), and that in endothelial and intestinal tissue heme oxygenase inhibitors are more selective for heme oxygenase than for either NO synthase or guanylyl cyclase (Zakhary et al. 1996, 1997). A third concern is that LTP appears to be normal in animals with a targeted mutation of heme oxygenase-2 (Poss et al. 1995). A possible explanation for this negative result is compensation by other isoforms of heme oxygenase or other messengers (Chen and Tonegawa 1996).
If both NO synthase and heme oxygenase are involved in LTP, it is not clear what their respective roles might be. One possibility suggested by our results is that they are activated by different stimulation patterns that might engage different receptors and second messengers. In many regions of the central nervous system, NO synthase activity is acutely controlled by glutamate NMDA receptors through a Ca2+-calmodulin-dependent mechanism. In the hippocampus, tetanic stimulation or NMDA application activates NO synthase, but the molecular mechanisms for activation of heme oxygenase are not known. Pharmacological studies with heme oxygenase inhibitors suggest that heme oxygenase may be activated in the brainstem or cerebellum by metabotropic glutamate receptor activation (Glaum and Miller 1993; Nathanson et al. 1995), suggesting the possibility that NO and CO could be activated in parallel by stimulation of NMDA and mGluRs, respectively. Consistent with this possibility, we found that inhibitors of heme oxygenase, but not NO synthase, block potentiation by the mGluR agonist tACPD.
Both NO and CO stimulate soluble guanylyl cyclase and increase the production of cGMP in hippocampus (East and Garthwaite 1991; Chetkovich et al. 1993; Verma et al. 1993). Furthermore, cGMP analogs can produce activity-dependent long-lasting potentiation and inhibitors of guanylyl cyclase or cGMP-dependent protein kinase can block LTP both in hippocampal slices (Haley et al. 1992; Zhuo et al. 1994; Boulton et al. 1995; Blitzer et al. 1995; Son et al. 1998) and in dissociated cultures of hippocampal neurons (Arancio et al. 1995, 1997). Other studies have failed to confirm some of these results (Schuman et al. 1994; Selig et al. 1996; Gage et al. 1997; Wu et al. 1998), but Son et al (1998) have recently identified experimental variables that may account in part for these discrepancies. We found that like LTP, tACPD-induced potentiation can be blocked by inhibitors of soluble guanylyl cyclase and cGMP-dependent protein kinase, and that tACPD stimulates guanylyl cyclase activity in hippocampal slices. These results suggest that mGluR activation might stimulate guanylyl cyclase through CO. However, in biochemical assays, we found that there is strong constitutive (basal) heme oxygenase activity in the CA1 region of hippocampus, and that this activity does not seem to be controlled by either tetanic stimulation or tACPD application. Although not conclusive, the assay results are most consistent with the possibility that heme oxygenase activity may not be acutely modulated by synaptic activation, as is NO synthase. Instead, constitutive heme oxygenase activity may be necessary for LTP and tACPD-induced potentiation, perhaps by producing tonic, background stimulation of guanylyl cyclase. If so, activation of mGluRs may produce acute stimulation of guanylyl cyclase through some other pathway, possibly arachidonic acid (Snider et al. 1984). Activation of mGluRs is thought to produce acute stimulation of guanylyl cyclase through NO in cerebellum (Okada 1995), but this seems unlikely in our experiments because potentiation by the mGluR agonist tACPD was blocked by inhibitors of guanylyl cyclase but not NO synthase.
The most likely conclusion from our results, therefore, is that NO synthase and heme oxygenase are not activated in parallel during the induction of LTP, but rather that heme oxygenase plays a more tonic role. This idea is also more consistent with the results shown in Figures 1 and 2, which indicate that the NO synthase- and heme oxygenase-dependent components of LTP are not additive. If heme oxygenase has a tonic function, it might act as a constitutive, housekeeping enzyme or it might have a more specific role as the source of a tonic retrograde messenger during LTP. Experiments on hippocampal neurons in culture support the idea that there may be both phasic and tonic retrograde messengers (Noel et al. 1996). One intriguing possibility is that although heme oxygenase may not play a phasic signaling role during the early phase of LTP, it could be induced by strong tetanic stimulation and thus play a more tonic role in the late, protein synthesis-dependent phase of LTP. Such a mechanism could contribute to a protein synthesis-dependent increase in presynaptic transmitter release during the late phase of LTP (Bolshakov et al. 1997; Sokolov et al. 1998).
Acknowledgments
We thank T. Abel, P. Nguyen, F. Rassendren, T. O’Dell, and S. Siegelbaum for their comments on an earlier draft, and A. Krawetz and H. Ayers for typing the manuscript. This work was supported in part by grants from the National Institute on Aging (AG08702), the National Institute of Mental Health (MH50733), and the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL rhawkins@nypimail.cpmc.columbia.edu; FAX (212) 543-5474.
References
- Arancio O, Kandel ER, Hawkins RD. Activity-dependent long-term enhancement of transmitter release by presynaptic 3′,5′-cyclic cGMP in cultured hippocampal neurons. Nature. 1995;376:74–80. doi: 10.1038/376074a0. [DOI] [PubMed] [Google Scholar]
- Arancio O, Kiebler M, Lee CJ, Lev-Ram V, Tsien RY, Kandel ER, Hawkins RD. Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell. 1996;87:1025–1035. doi: 10.1016/s0092-8674(00)81797-3. [DOI] [PubMed] [Google Scholar]
- Bannerman DM, Chapman PF, Kelly PAT, Morris RGM. Inhibition of nitric oxide synthase does not prevent induction of long-term potentiation in vivo. J Neurosci. 1994;14:7425–7425. doi: 10.1523/JNEUROSCI.14-12-07415.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskys A, Malenka RC. Agonists at metabotropic glutamate receptors presynaptically inhibit EPSCs in neonatal rat hippocampus. J Physiol. 1991;444:687–701. doi: 10.1113/jphysiol.1991.sp018901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- Böhme GA, Bon C, Stutzmann JM, Doble A, Blanchard JC. Possible involvement of nitric oxide in long-term potentiation. Eur J Pharmacol. 1991;199:379–381. doi: 10.1016/0014-2999(91)90505-k. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science. 1994;264:1148–1152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Golan H, Kandel ER, Siegelbaum SA. Recruitment of new sites of synaptic transmissin during the cAMP-dependent late phase of LTP at CA3-CA1 synapses in the hippocampus. Neuron. 1997;19:635–651. doi: 10.1016/s0896-6273(00)80377-3. [DOI] [PubMed] [Google Scholar]
- Bortolotto ZA, Bashir ZI, Davies CH, Collingridge GL. A molecular switch activated by metabotropic glutamate receptors regulates induction of long-term potentiation. Nature. 1994;368:740–743. doi: 10.1038/368740a0. [DOI] [PubMed] [Google Scholar]
- Boulton CL, Southam E, Garthwaite J. Nitric-oxide dependent long-term potentiation is blocked by a specific inhibitor of soluble guanylyl cyclase. Neuroscience. 1995;69:699–703. doi: 10.1016/0306-4522(95)00349-n. [DOI] [PubMed] [Google Scholar]
- Chen C, Tonegawa S. Molecular genetic analysis of synaptic plasticity, activity-dependent neural development, learning, and memory in the mammalian brain. Annu Rev Neurosci. 1996;20:157–184. doi: 10.1146/annurev.neuro.20.1.157. [DOI] [PubMed] [Google Scholar]
- Chetkovich DM, Klann E, Sweatt JD. Nitric oxide synthase-independent long-term potentiation in area CA1 of hippocampus. NeuroReport. 1993;4:919–922. doi: 10.1097/00001756-199307000-00020. [DOI] [PubMed] [Google Scholar]
- Clark GD, Happel LT, Zorumski CF, Bazan NG. Enhancement of hippocampal excitatory synaptic transmission by platelet-activating factor. Neuron. 1992;9:1211–1216. doi: 10.1016/0896-6273(92)90078-r. [DOI] [PubMed] [Google Scholar]
- Cummings JA, Nicola SM, Malenka RC. Induction in the rat hippocampus of long-term potentiation (LTP) and long-term depression (LTD) in the presence of a nitric oxide synthase inhibitor. Neurosci Lett. 1994;176:110–114. doi: 10.1016/0304-3940(94)90883-4. [DOI] [PubMed] [Google Scholar]
- Del Cerro S, Arai A, Lynch G. Inhibition of long-term potentiation by an antagonist of platelet-activating factor receptors. Behav Neurol Biol. 1990;54:213–217. doi: 10.1016/0163-1047(90)90595-w. [DOI] [PubMed] [Google Scholar]
- Doyle C, Holscher C, Rowan MJ, Anwyl R. The selective neuronal NO synthase inhibitor 7-nitro-indazole blocks both long-term potentiation and depotentiation of field EPSPs in rat hippocampal CA1 in vivo. J Neurosci. 1996;16:418–424. doi: 10.1523/JNEUROSCI.16-01-00418.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East SJ, Garthwaite J. NMDA receptor activation in rat hippocampus induces cyclic GMP formation through the L-arginine nitric oxide pathway. Neurosci Lett. 1991;123:17–19. doi: 10.1016/0304-3940(91)90147-l. [DOI] [PubMed] [Google Scholar]
- Gage AT, Reyes M, Stanton PK. Nitric-oxide-guanylyl-cyclase-dependent and independent components of multiple forms of long-term synaptic depression. Hippocampus. 1997;7:288–295. doi: 10.1002/(SICI)1098-1063(1997)7:3<286::AID-HIPO4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Glaum SR, Miller RJ. Zinc protoporphyrin-IX blocks the effects of metabotropic glutamate receptor activation in the rat nucleus tractus solitarii. Mol Pharmacol. 1993;43:965–969. [PubMed] [Google Scholar]
- Gribkoff VK, Lum-Ragan JT. Evidence for nitric oxide synthase inhibitor-sensitive and insensitive hippocampal synaptic potentiation. J Neurophysiol. 1992;68:639–642. doi: 10.1152/jn.1992.68.2.639. [DOI] [PubMed] [Google Scholar]
- Haley J E, Wilcox GL, Chapman PF. The role of nitric oxide in hippocampal long-term potentiation. Neuron. 1992;8:211–216. doi: 10.1016/0896-6273(92)90288-o. [DOI] [PubMed] [Google Scholar]
- Haley J E, Malen PL, Chapman PF. Nitric oxide synthase inhibitors block long-term potentaiton induced by weak but not strong tetanic stimulation at physiological brain temperatures in rat hippocampal slices. Neurosci Lett. 1993;160:85–88. doi: 10.1016/0304-3940(93)90919-c. [DOI] [PubMed] [Google Scholar]
- Haley J, Schaible E, Paulidis P, Murdock A, Madison DV. Basal and apical synapses of CA1 pyramidal cells employ different LTP induction mechanisms. Learn & Mem. 1996;3:289–295. doi: 10.1101/lm.3.4.289. [DOI] [PubMed] [Google Scholar]
- Hawkins RD, Kandel ER, Siegelbaum SA. Learning to modulate transmitter release: Themes and variations in synaptic plasticity. Annu Rev Neurosci. 1993;16:625–665. doi: 10.1146/annurev.ne.16.030193.003205. [DOI] [PubMed] [Google Scholar]
- Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC. Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 1993;75:1273–1286. doi: 10.1016/0092-8674(93)90615-w. [DOI] [PubMed] [Google Scholar]
- Huang Y-Y, Kandel ER. Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn & Mem. 1994;1:74–82. [PubMed] [Google Scholar]
- Ignarro LJ, Ballot B, Wood KS. Regulation of soluble guanylate cyclase activity by porphyrins and metalloporphyrins. J Biol Chem. 1984;259:6201–6207. [PubMed] [Google Scholar]
- Ikegaya Y, Saito H, Matsuki N. Involvement of carbon monoxide in long-term potentiation in the dentate gyrus of anesthetized rats. Jpn J Pharmacol. 1994;64:225–227. doi: 10.1254/jjp.64.225. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Kantor DB, Lanzrein M, Stary SJ, Sandoval GM, Smith WB, Sullivan BM, Davidson N, Schuman EM. A role for endothelial NO synthase in LTP revealed by adenovirus-mediated inhibition and rescue. Science. 1996;274:1744–1748. doi: 10.1126/science.274.5293.1744. [DOI] [PubMed] [Google Scholar]
- Kato K, Zorumski CF. Nitric oxide inhibitors facilitate the induction of hippocampal long-term potentiation by modulating NMDA responses. J Neurophysiol. 1993;70:1260–1263. doi: 10.1152/jn.1993.70.3.1260. [DOI] [PubMed] [Google Scholar]
- Korte M, Griesbeck O, Grand C, Carroll P, Staiger V, Thoenen H, Bonhoffer T. Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci. 1996;93:12547–12552. doi: 10.1073/pnas.93.22.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitinen JT, Juronen RO. A sensitive microassay reveals marked regional differences in the capacity of rat brain to generate carbon monoxide. Brain Res. 1995;694:246–252. doi: 10.1016/0006-8993(95)00761-e. [DOI] [PubMed] [Google Scholar]
- Linden D J, Narasimban K, Gurfel D. Protoprophyrins modulate voltage-gated Ca current in AtT-20 pituitary cells. J Neurophysiol. 1993;70:2673–2677. doi: 10.1152/jn.1993.70.6.2673. [DOI] [PubMed] [Google Scholar]
- Luo D, Vincent SR. Metalloporphyrins inhibit nitric oxide-dependent cGMP formation in vivo. Eur J Pharmacol. 1994;267:263–267. doi: 10.1016/0922-4106(94)90149-x. [DOI] [PubMed] [Google Scholar]
- Maines MD. Heme oxygenase: Function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2:2557–2568. [PubMed] [Google Scholar]
- ————— Carbon monoxide: An emerging regulator of cGMP in the brain. Mol Cell Neurosci. 1993;4:389–397. doi: 10.1006/mcne.1993.1049. [DOI] [PubMed] [Google Scholar]
- Malen P, Chapman PF. Nitric oxide facilitates long-term potentiaton, but not long-term depression. J Neurosci. 1997;17:2645–2651. doi: 10.1523/JNEUROSCI.17-07-02645.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin W, Villani GM, Jothianandan D, Furchgott RF. Selective blockade of endothelium-dependent and glycerol trinitrate-induced relaxation by hemoglobin and by methylene blue in the rabbit aorta. J Pharmacol Exp Ther. 1985;232:708–716. [PubMed] [Google Scholar]
- Meffert MK, Haley JE, Schuman EM, Schulman H, Madison DV. Inhibition of hippocampal heme oxygenase, nitric oxide synthase, and long-term potentiation by metalloporphyrins. Neuron. 1994;13:1225–1233. doi: 10.1016/0896-6273(94)90060-4. [DOI] [PubMed] [Google Scholar]
- Mizutani A, Saito H, Abe K. Involvement of nitric oxide in long-term potentiation in the dentate gyrus in vivo. Brain Res. 1993;605:309–311. doi: 10.1016/0006-8993(93)91756-i. [DOI] [PubMed] [Google Scholar]
- Nathanson JA, Scavone C, Scanlon C, McKee M. The cellular Na+ pump as a site of action for carbon monoxide and glutamate: A mechanism for long-term modulation of cellular activity. Neuron. 1995;14:781–794. doi: 10.1016/0896-6273(95)90222-8. [DOI] [PubMed] [Google Scholar]
- O’Dell TJ, Hawkins RD, Kandel ER, Arancio O. Tests of the roles of two diffusible substances in long-term potentiation: Evidence for nitric oxide as a possible early retrograde messenger. Proc Natl Acad Sci. 1991;88:11285–11289. doi: 10.1073/pnas.88.24.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dell TJ, Huang PL, Dawson TM, Dinerman JL, Snyder SH, Kandel ER, Fishman MC. Endothelial NOS and blockade of LTP by NOS inhibitors in mice lacking neuronal NOS. Science. 1994;265:542–546. doi: 10.1126/science.7518615. [DOI] [PubMed] [Google Scholar]
- Okada D. Protein kinase C modulates calcium sensitivity of nitric oxide synthase in cerebellar slices. J Neurochem. 1995;64:1298–1304. doi: 10.1046/j.1471-4159.1995.64031298.x. [DOI] [PubMed] [Google Scholar]
- ————— Zinc protoporphyrin IX suppresses nitric oxice production through a loss of L-arginine in rat cerebellar slices. Neurosci Res. 1996;25:353–358. doi: 10.1016/0168-0102(96)01057-7. [DOI] [PubMed] [Google Scholar]
- Otani S, Ben-Ari Y. Metabotropic receptor-mediated long-term potentiation in rat hippocampal slices. Eur J Pharmacol. 1991;205:325–326. doi: 10.1016/0014-2999(91)90920-l. [DOI] [PubMed] [Google Scholar]
- Poss KD, Thomas MJ, Ebralidze AK, O’Dell TJ, Tonegawa S. Hippocampal long-term potentiation is normal in heme oxygenase-2 mutant mice. Neuron. 1995;15:867–873. doi: 10.1016/0896-6273(95)90177-9. [DOI] [PubMed] [Google Scholar]
- Radpour S, Thomson AM. Synaptic enhancement induced by NMDA and Qp receptors and presynaptic activity. Neurosci Lett. 1992;138:119–122. doi: 10.1016/0304-3940(92)90486-q. [DOI] [PubMed] [Google Scholar]
- Schuman EM, Madison DV. A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science. 1991;254:1503–1506. doi: 10.1126/science.1720572. [DOI] [PubMed] [Google Scholar]
- Schuman EM, Meffert MK, Schulman H, Madison DV. An ADP-ribosyl transferase as a potential target for nitric oxide action in hippocampal long-term potentiation. Proc Natl Acad Sci. 1994;91:11958–11962. doi: 10.1073/pnas.91.25.11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selig DK, Segal MR, Liao D, Malenka RC, Malinow R, Nicoll RA, Lisman JE. Examination of the role of cGMP in long-term potentiation in the CA1 region of the hippocampus. Learn & Mem. 1996;3:42–48. doi: 10.1101/lm.3.1.42. [DOI] [PubMed] [Google Scholar]
- Snider RM, McKinney M, Forray C, Richelson E. Neurotransmitter receptors mediate cyclic GMP formation by involvement of arachidonic acid and lipoxygenase. Proc Natl Acad Sci. 1984;81:3905–3909. doi: 10.1073/pnas.81.12.3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov MV, Rossokhin AV, Bebnisch T, Reymann KG, Voronin LL. Interaction between paired-pulse facilitation and long-term potentiation of minimal excitatory postsynaptic potentials in rat hippocampal slices: A patch-clamp study. Neuroscience. 1998;85:1–13. doi: 10.1016/s0306-4522(97)00592-7. [DOI] [PubMed] [Google Scholar]
- Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, Kandel ER. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell. 1996;87:1015–1023. doi: 10.1016/s0092-8674(00)81796-1. [DOI] [PubMed] [Google Scholar]
- Son H, Lu Y-F, Zhuo M, Arancio O, Kandel ER, Hawkins RD. The specific role of cGMP in hippocampal LTP. Learn & Mem. 1998;5:231–245. [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Wang Y-Y. Reversal of long-term potentiation by inhibitors of heme oxygenase. Nature. 1993;364:147–149. doi: 10.1038/364147a0. [DOI] [PubMed] [Google Scholar]
- Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- Verma A, Hirsch DJ, Glatt CF, Ronnett GV, Snyder SH. Carbon monoxide: A putative neural messenger. Science. 1993;259:381–384. doi: 10.1126/science.7678352. [DOI] [PubMed] [Google Scholar]
- Wieraszko A, Li G, Kornecki E, Hogan MV, Ehrlich YH. Long-term potentiation in the hippocampus by platelet-activating factor. Neuron. 1993;10:553–557. doi: 10.1016/0896-6273(93)90342-o. [DOI] [PubMed] [Google Scholar]
- Williams JH, Errington ML, Lynch MA, Bliss TVP. Arachidonic acid induces a long-term activity-dependent enhancement of synaptic transmission in the hippocampus. Nature. 1989;341:739–742. doi: 10.1038/341739a0. [DOI] [PubMed] [Google Scholar]
- Williams JH, Li YG, Nayak A, Errington ML, Murphy K-PSJ, Bliss TVP. The suppression of long-term potentiation in rat hippocampus by inhibitors of nitric oxide synthase is temperature- and age-dependent. Neuron. 1993;11:877–884. doi: 10.1016/0896-6273(93)90117-a. [DOI] [PubMed] [Google Scholar]
- Winder DG, Mansuy IM, Osman M, Moallem TM, Kandel ER. Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell. 1998;92:25–37. doi: 10.1016/s0092-8674(00)80896-x. [DOI] [PubMed] [Google Scholar]
- Wu J, Wang Y, Rowan MJ, Anwyl R. Evidence for the involvement of the neuronal isoform of nitric oxide synthase during induction of long-term potentiation and long-term depression in the rat dentate gyrus in vitro. Neuroscience. 1997;78:393–398. doi: 10.1016/s0306-4522(97)84911-1. [DOI] [PubMed] [Google Scholar]
- ————— Evidence for involvement of the cGMP-protein kinase G signaling system in the induction of long-term depression, but not long-term potentiation, in the dentate gyrus in vitro. J Neurosci. 1998;18:3589–3596. doi: 10.1523/JNEUROSCI.18-10-03589.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakhary R, Gaine SP, Dinerman JL, Ruat M, Flavahan NA, Snyder SH. Heme oxygenase 2: Endothelial and neuronal localization and role in endothelium-dependent relaxation. Proc Natl Acad Sci. 1996;93:795–798. doi: 10.1073/pnas.93.2.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakhary R, Poss KD, Jaffrey SR, Ferris CD, Tonegawa S, Snyder S. Targeted gene deletion of heme oxygenase 2 reveals neural role for carbon dioxide. Proc Natl Acad Sci. 1997;94:14848–14853. doi: 10.1073/pnas.94.26.14848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M, Small SA, Kandel ER, Hawkins RD. Nitric oxide and carbon monoxide produce activity-dependent long-term synaptic enhancement in hippocampus. Science. 1993;260:1946–1950. doi: 10.1126/science.8100368. [DOI] [PubMed] [Google Scholar]
- Zhuo M, Hu Y, Schultz C, Kandel ER, Hawkins RD. Role of guanylyl cyclase and cGMP-dependent protein kinase in long-term potentiation. Nature. 1994;368:635–639. doi: 10.1038/368635a0. [DOI] [PubMed] [Google Scholar]