

Abstract
Previous work has shown that mice missing the α-isoform of calcium–calmodulin-dependent protein kinase II (α-CaMKII) have a deficiency in CA1 hippocampal long-term potentiation (LTP). Follow-up studies on subsequent generations of these mutant mice in a novel inbred background by our laboratories have shown that whereas a deficiency in CA1 LTP is still present in α-CaMKII mutant mice, it is different both quantitatively and qualitatively from the deficiency first described. Mice of a mixed 129SvOla/SvJ;BALB/c;C57Bl/6 background derived from brother/sister mating of the α-CaMKII mutant line through multiple generations (>10) were produced by use of in vitro fertilization. Although LTP at 60 min post-tetanus was clearly deficient in these (−/−) α-CaMKII mice (42.6%, n = 33) compared with (+/+) α-CaMKII control animals (81.7%, n = 17), α-CaMKII mutant mice did show a significant level of LTP. The amount of LTP observed in α-CaMKII mutants was normally distributed, blocked by APV (2.7%, n = 8), and did not correlate with age. Although this supports a role for α-CaMKII in CA1 LTP, it also suggests that a form of α-CaMKII-independent LTP is present in mice that could be dependent on another kinase, such as the β-isoform of CaMKII. A significant difference in input/output curves was also observed between (−/−) α-CaMKII and (+/+) α-CaMKII animals, suggesting that differences in synaptic transmission may be contributing to the LTP deficit in mutant mice. However, tetani of increasing frequency (50, 100, and 200 Hz) did not reveal a higher threshold for potentiation in (−/−) α-CaMKII mice compared with (+/+) α-CaMKII controls.
Calcium–calmodulin-dependent protein kinase II (CaMKII) is a serine/threonine protein kinase that is highly expressed in the brain and is thought to be involved in such diverse mechanisms as neurotransmitter synthesis and release, microtubule assembly and disassembly, ion channel modulation and synaptic plasticity (Braun and Schulman 1995). The unique ability of CaMKII to autophosphorylate at Thr-286, producing a long-lasting calcium-independent form of the enzyme (Fong et al. 1989; Hanson et al. 1989; Waldmann et al. 1990; Waxham et al. 1990; Meyer et al. 1992), has led to proposals that CaMKII might serve as a memory molecule of activity at synapses and may underlie long-term changes in synaptic activity (Miller and Kennedy 1986; Lisman and Goldring 1988; Giese et al. 1998). Several lines of evidence indicate a role for CaMKII in long-term potentiation (LTP) (Lisman 1994), a well-characterized mechanism for increasing synaptic efficacy that has been observed at a number of excitatory synapses in the mammalian central nervous system, and most carefully studied at the CA3-CA1 hippocampal synapse (Bliss and Collingridge 1993; Roberson et al. 1996). CaMKII is highly expressed in the hippocampus, comprising 2% of total hippocampal protein (Erondu and Kennedy 1985), and is localized in both the pre- and post-sides of the synapse (Ouimet et al. 1984; Fukunaga et al. 1988). It is thought to be the major protein component of the postsynaptic density (PSD) (Kennedy et al. 1983; Goldenring et al. 1984; Kelly et al. 1984; Miller and Kennedy 1985), in which it is strategically located for the modification of proteins that might be involved in the expression and maintenance of LTP. Such proteins include glutamate receptors in the postsynaptic cell membrane and microtubule-associated protein 2 (MAP2), both of which are CaMKII substrates (Vallano et al. 1986; McGlade-McCulloh et al. 1993; Omkumar et al. 1996; Barria et al. 1997; Hayashi et al. 1997). CaMKII activation is dependent on the initial binding of calcium-clmodulin (Hanson and Schulman 1992), and it is known that calcium influx via the NMDA receptor is necessary for LTP induction (Bliss and Collingridge 1993). Correspondingly, it is known that LTP induction leads to an increase in CaMKII activity, an increase in autophosphorylated CaMKII, and an increase in the phosphorylation of the CaMKII substrates, MAP-2, synapsin I, and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptors (Fukunaga et al. 1993, 1995; Barria et al. 1997; Ouyang et al. 1997). CaMKII inhibitors, calmodulin inhibitors, and general kinase inhibitors have been shown to block LTP induction (Malenka et al. 1989; Malinow et al. 1989; Ito et al. 1991; Otmakhov et al. 1997), whereas constitutively active CaMKII has been shown to mimic and occlude LTP when introduced via a viral expression system (Pettit et al. 1994), as well as by direct injection into the postsynaptic cell (Lledo et al. 1995; A.M. Shirke and R. Malinow, unpubl), and to shift the LTP/LTD (long-term depression) modification threshold in transgenic animals (Mayford et al. 1995). In addition, mice mutant in the α-isoform of CaMKII are found to be deficient in hippocampal LTP, short-term potentiation (STP), and LTD (Silva et al. 1992a; Stevens et al. 1994), deficient in primary visual cortex (Gordon et al. 1996; A. Kirkwood, A. Silva, and M.F. Bear, unpubl.) and barrel cortex plasticity (Glazewski et al. 1996), to perform poorly in an assortment of learning (Silva et al. 1992b) and behavioral paradigms (Chen et al. 1994), and to exhibit hyperexcitability in multiple limbic structures (Butler et al. 1995). Recent recombinant mouse studies have further revealed that mutating the α-CaMKII autophosphorylation Thr-286 site alone (Thr-286-Ala) results in deficits in both CA1 LTP and learning (Giese et al. 1998).
We re-examined LTP in the CA1 region of the hippocampus of (−/−) α-CaMKII mice using field recordings. Preliminary studies indicated that LTP was diminished, but present, in hippocampal slices from young (−/−) α-CaMKII animals (12–28 days), ages that had not been examined in previous studies (Silva et al. 1992a; Stevens et al. 1994). Consequently, we extended our recordings to older adult animals (up to 108 days) to determine whether a developmentally dependent variable was affecting the probability of eliciting LTP in these animals. Basic characteristics of synaptic transmission were also examined to address whether differences in excitability may be contributing to the LTP deficits in these CaMKII mutant mice.
Materials and Methods
MICE
The mice used in the following electrophysiology experiments were of a mixed 129SvOla/SvJ;BALB/c;C57Bl/6 genetic background, inbred by brother/sister mating for over ten generations before the present experiments began. Because natural matings among heterozygous and homozygous α-CaMKII mutant animals were often found to be unproductive, animals were produced by use of in vitro fertilization. Hippocampal slices from mutant and wild-type animals were interleaved during experiments, by use of wild-type littermates and/or wild-type animals matched for background and age as controls. No significant differences were seen between those animals tested blindly and those tested with knowledge of phenotype, so data from these experiments were pooled. The genotypes of animals used were determined before, and confirmed after, the experiments by use of a PCR assay.
SLICE PREPARATION AND RECORDING
Transverse hippocampal slices (400 μm) were prepared from 12- to 108-day-old mice by the tissue chopper method (Malinow and Tsien 1990). CA3 and CA1 regions were surgically separated to prevent epileptiform activity. The slices were submerged and superfused with a bath solution containing 119 mm NaCl, 2.5 mm KCl, 2.5 mm CaCl2, 26.2 mm NaHCO3, 1.3 mm MgCl2, 1 mm NaH2PO4, and 11 mm glucose, equilibrated with 95% O2 and 5% CO2. Picrotoxin (100 μm) was added to the bath to block GABAA inhibitory postsynaptic currents. The temperature of the recording chamber was maintained at 30°C ± 0.5°C. Slices were allowed to recover at least 1 hr before recording was initiated.
By use of visual guidance, a glass-stimulating electrode filled with 2 m NaCl was placed in Schaffer collateral afferents to elicit field excitatory postsynaptic potentials (f-EPSPs) using a brief (<200 μsec) current pulse. An extracellular glass-recording electrode, filled with the above bath solution, was placed in the CA1 stratum radiatum ∼150–200 μm from the stimulating electrode. Visual guidance allowed specific and consistent placement of electrodes and rapid superficial assessment of the health of the slice. Stimulus intensity was adjusted to evoke base-line pretetanic responses of similar sizes for all of the slices (0.2–0.3 mV). Base-line stimulation frequency was 0.033 Hz and LTP was typically induced by use of a 100-Hz tetanus (five trains, 20 pulses per train, 10 sec between trains), unless otherwise stated. To block NMDA receptors in some experiments, 100 μm DL-aminophosphonovaleric acid (DL-APV) was added to the bath solution, as appropriate.
Individual f-EPSP peak amplitudes were determined by averaging the response size over a fixed 1- to 5-msec window encompassing the period of negative slope just preceding the peak amplitude of the signal, and subtracting a base-line estimate. For each experiment, the same windows were used on recorded sweeps in all experimental conditions. At the beginning of each experiment, the stimulating intensity (input) was varied and the amplitude of the f-EPSP response was measured (output). The intensity of input stimulation began at a level at which no responses were observed and increased until responses were maximal. These paired stimulation/response values were used to plot input/output curves of representative experiments (Fig. 5, below).
Figure 5.
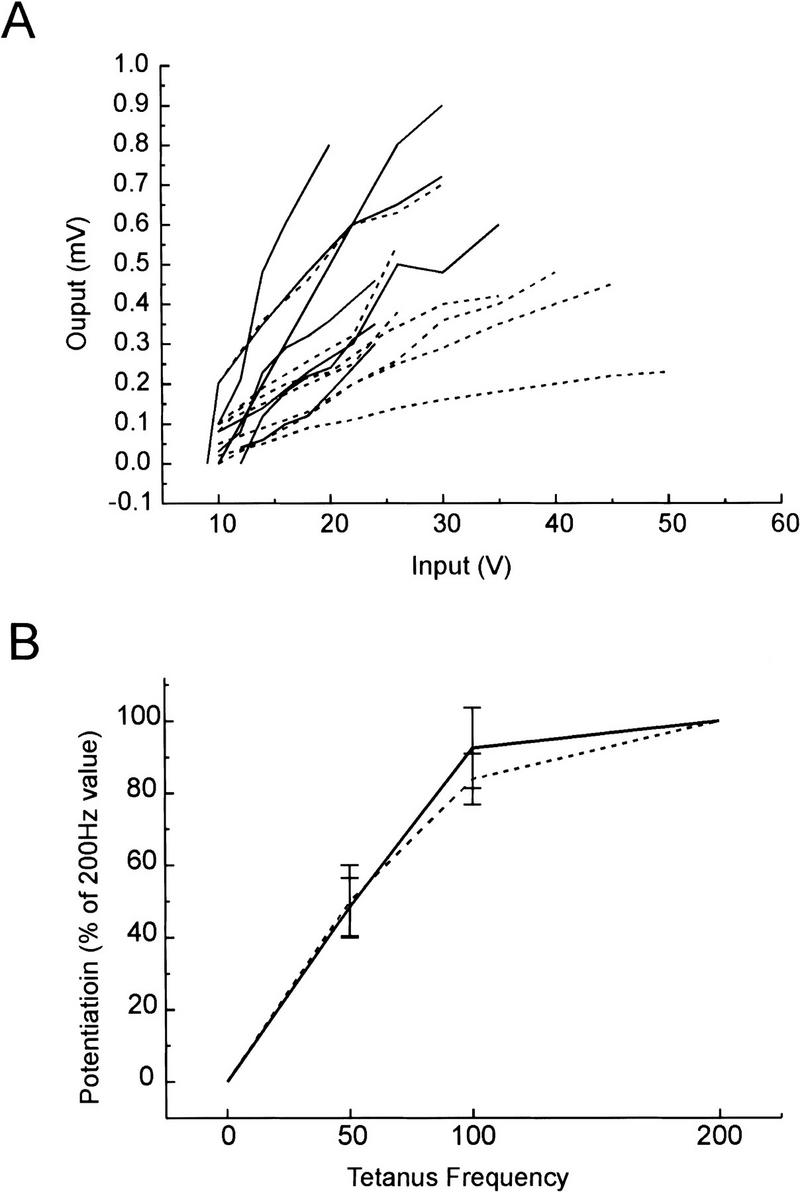
(A) Synaptic input–output curves are steeper in (+/+) α-CaMKII slices than in (−/−) α-CaMKII slices. Elicited f-EPSPs (mV) as a function of stimulation strength (V) are shown for a sampling of wild-type [α-CaMKII (+/+) broken lines, n = 7; 1–3 months old] and mutant [α-CaMKII (−/−) solid lines, n = 7; 1–2 months old] slices. For each curve, a slope was calculated by linear regression. The mean slope of the input–output curves is greater in wild-type slices (34 ± 7.2 μV/V) than in mutant slices (16 ± 2.7 μV/V) t test, P<0.05. (B) Successive tetani of increasing frequency do not reveal a higher threshold for potentiation in (−/−) α-CaMKII mice compared with (+/+) α-CaMKII mice. Following 30 min of stable recording of base-line f-EPSPs, three successive tetani of 50, 100, and 200 Hz (each five trains, 10–20 pulses per train, 10 sec between trains) were delivered to the slice, each separated by 30 min of recording. The amount of potentiation at 30 min following each tetanus was normalized to the potentiation following the 200-Hz tetanus and was plotted relative to the tetanus frequency for both wild-type (+/+, solid line, n = 4; 1–2 months old) and mutant (−/−, broken line, n = 5; 1–2 months old) slices.
For LTP experiments using the above tetanus paradigm, the magnitude of LTP was measured by comparing the average of responses over a 5-min interval 60 min after the tetanus with the average of responses over a 5-min interval preceding the tetanus. Consequently, the magnitude of LTP was expressed as a relative change (percent) in the response amplitude.
Results
We began by examining LTP in the CA1 region of hippocampal slices from young (−/−) α-CaMKII mice (12–28 days) from our mixed background inbred line. Synaptic transmission was monitored for stable base-line periods of up to 30 min before LTP was induced by use of a tetanus protocol (100 Hz, five trains, 20 pulses per train, 10 sec between trains). Surprisingly, LTP was observed in mutant slices, although it appeared to be smaller in magnitude than that observed in wild-type control experiments (Fig. 1A,B). This was in contrast to previously published analysis of the original (−/−) α-CaMKII line, in which 14 of 16 mutant slices showed no LTP (−1% ± 3 at 30 min post-tetanus), whereas the remaining 2 mutant slices showed significant LTP of a magnitude comparable with that observed in (+/+) α-CaMKII animals (68% ± 32 at 30 min post-tetanus), under given experimental conditions (Silva et al. 1992a). Subsequent studies also concluded that α-CaMKII mutants show significantly diminished LTP (Stevens et al. 1994), suggesting that it may be completely absent in these mice.
Figure 1.
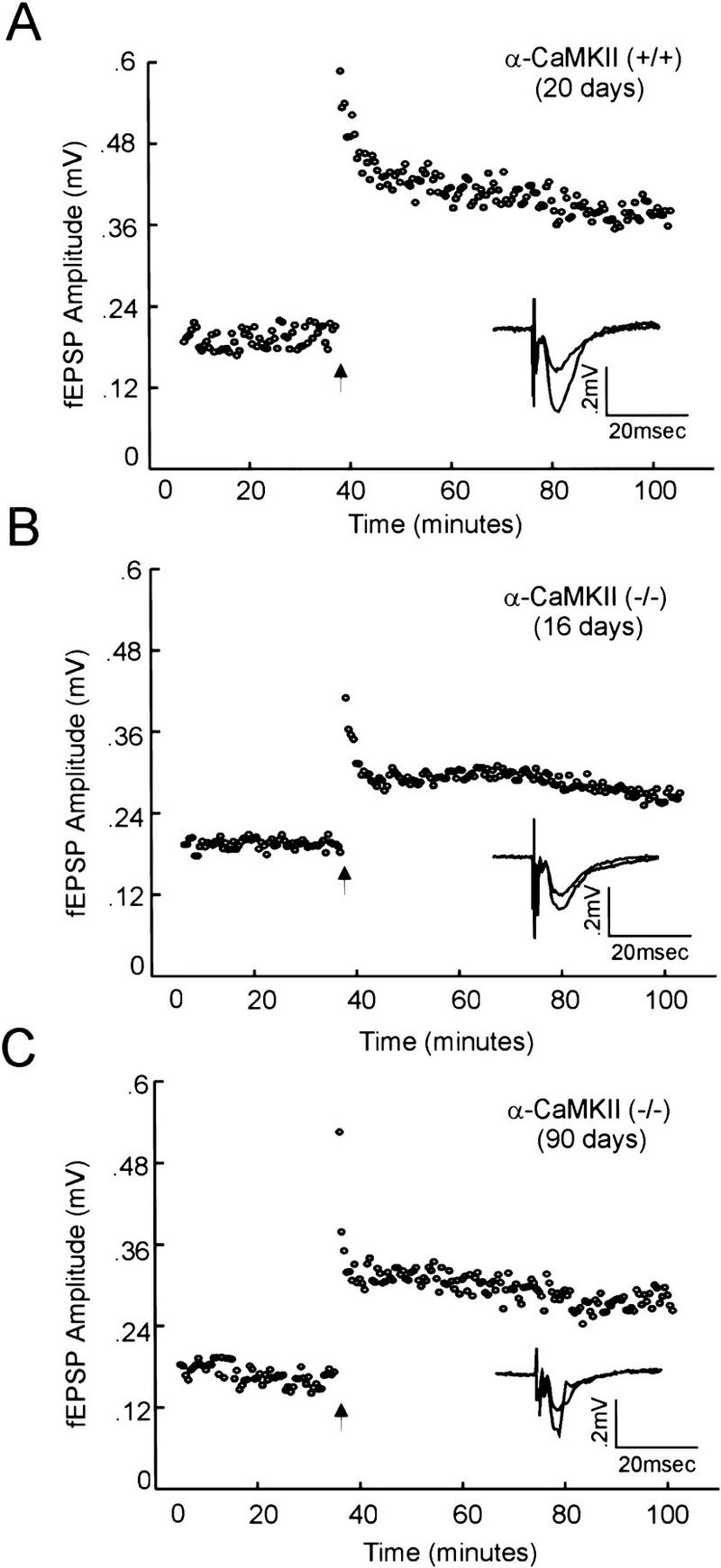
Tetanus-induced LTP of field EPSPs in the CA1 region of the hippocampus is present, but diminished, in (−/−) α-CaMKII mutant mice. Field EPSP amplitude (mV) is plotted versus time (minutes), with the arrow indicating the time of tetanus delivery (see Materials and Methods). Average field EPSP responses before (10 consecutive trials, 5 min prior) and after tetanus (10 consecutive trials 60 min after) are shown, overlapping, in the inset. Three representative experiments are shown, demonstrating CA1 LTP in slices from a wild-type mouse (A), a young (16 days) mutant mouse (B), and an adult (90 days) mutant mouse (C).
As the original analysis of (−/−) α-CaMKII mutant mice had been performed on animals primarily 1.5–3 months of age (Silva et al. 1992a), we considered that a developmentally dependent variable could be affecting the probability of eliciting LTP in young (−/−) α-CaMKII animals. To address this, we examined LTP in older animals (28–108 days) and found that it was also diminished, but present, in older (−/−) α-CaMKII mice compared with (+/+) α-CaMKII control animals (Fig. 1A,C), and there was no correlation between age and the amount of potentiation post-tetanus in either (−/−) α-CaMKII or (+/+) α-CaMKII mice (Fig. 2). Cumulative data across all age groups showed that whereas LTP at 60 min post-tetanus was clearly deficient in (−/−) α-CaMKII mice (42.6% ± 5.5, n = 33) compared with (+/+) α-CaMKII control animals (81.7% ± 14.1, n = 17), α-CaMKII mutant mice did show a significant level of LTP (Fig. 3). This α-CaMKII independent LTP was blocked when DL-APV (100 μm) was included in the bath (2.7% ± 10, n = 8) (Fig. 3), indicating that the potentiation is NMDA receptor dependent. The amount of potentiation observed in both (−/−) α-CaMKII and (+/+) α-CaMKII slices followed a normal distribution around a mean value (Fig. 4). The mean value for mutant slices is ∼50% of that observed in wild-type animals, with a comparable standard deviation/mean for both groups of animals [s.d./mean: (+/+) = 0.71; (−/−) = 0.75].
Figure 2.

No correlation between age and percent potentiation post-tetanus was observed in (+/+) α-CaMKII or (−/−) α-CaMKII mice. Potentiation of f-EPSP amplitude at 60 min post-tetanus is plotted vs. animal age (days). Synaptic potentiation is not correlated with age in either wild-type (n = 17, R = 0.28, P = 0.29, A) or mutant (n = 33, R = 0.27, P = 0.36, B) slices.
Figure 3.

α-CaMKII-independent CA1 LTP is ∼50% of that observed in (+/+) α-CaMKII mice and is blocked by APV. Cumulative data from tetanus induced CA1 LTP of f-EPSP responses in wild-type (α-CaMKII +/+, •, n = 17) and mutant (α-CaMKII −/−, ○, n = 33) slices, and in mutant slices in the presence of 100 μm DL-APV (α-CaMKII −/− APV, ▴, n = 8). The relative mean field EPSP amplitudes ± s.e.m. (mV) are plotted versus time (minutes), with the arrow indicating the time of tetanus delivery. The mean values are indicated for time points 30 min, 15 min, and 5 min before tetanus, and 1 min, 5 min, 30 min, and 60 min after tetanus and are calculated from f-EPSP amplitude values over a 5-min period, beginning at the time point indicated.
Figure 4.
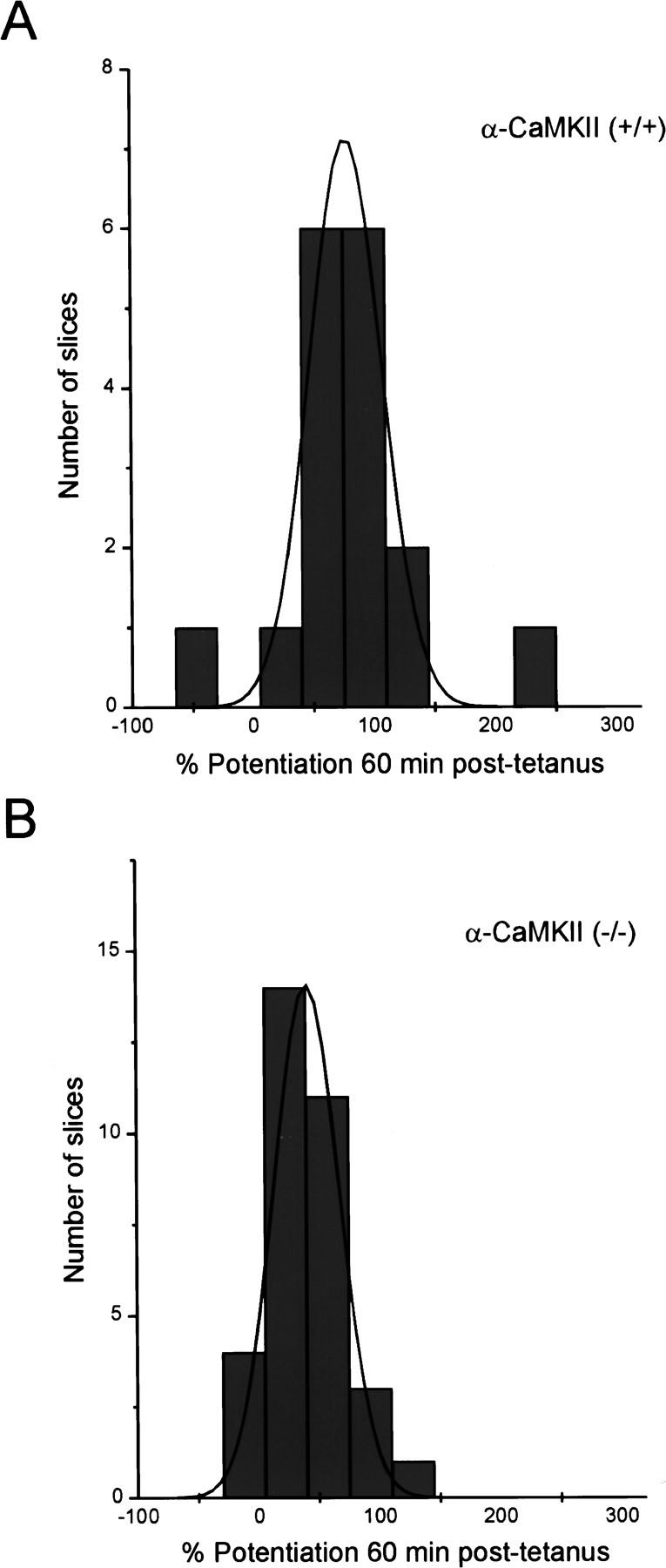
Potentiation in (−/−) α-CaMKII and (+/+) α-CaMKII slices 60 min post-tetanus follows a normal distribution. Histograms depicting the distribution of slices with the indicated amount of potentiation 60 min post-tetanus in mutant (A) and wild-type (B) slices.
It is possible that the LTP deficit observed in (−/−) α-CaMKII mutant mice may reflect differences in the basic properties of synaptic transmission between mutant and wild-type mice, which could consequently confound attempts to produce LTP efficiently in mutant animals. To examine this, we plotted input/output curves for data obtained from (−/−) α-CaMKII and (+/+) α-CaMKII animals (Fig. 5A). Synaptic input/output curves from age-matched mutant and wild-type mice (1–3 months old) are significantly different, with the mean slope, calculated by linear regression, of wild-type slices [n = 7; 34 ± 7.2 μV/V (microvolt response per stimulus volt)] being over twice that observed in mutant slices (n = 7; 16 ± 2.7 μV/V).
If differences in some aspect of depolarization and/or cell excitability are present in the (−/−) α-CaMKII mutant mice, it might be possible to overcome these deficits when trying to induce LTP by use of stimuli of increasing strength. To address this, stable base-line field, EPSP responses were recorded for 30 min in (−/−) α-CaMKII (n = 5) and (+/+) α-CaMKII slices (n = 4), followed by successive tetani of 50, 100, and 200 Hz (each five trains, 10–20 pulses per train, 10 sec between trains), with each tetanus separated by 30 min of recording (Fig. 5). The amount of potentiation at 30 min following each tetanus was normalized to the maximum level of potentiation obtained following the 200 Hz tetanus. The resulting frequency—potentiation curves are shown, and they do not reveal a higher threshold for potentiation in (−/−) α-CaMKII mice compared with (+/+) α-CaMKII mice. Although mutant mice show decreased responses following each tetanus when compared with wild-type, these responses appear to be saturated with tetani of similar strengths in both mutant and wild-type slices.
Discussion
We have examined LTP in the CA1 region of hippocampal slices from mice lacking the α-isoform of CaMKII and find that LTP is deficient in these mutant mice. However, the deficiency is different from that first described (Silva et al. 1992b). We find that there is a significant amount of potentiation in (−/−) α-CaMKII mutant mice, although it is reduced to ∼50% of that observed in wild-type animals. This α-CaMKII independent potentiation is blocked by APV, suggesting that it is traditional NMDA receptor-dependent LTP, and is consistent in quantity and distribution (normal) across all ages examined. α-CaMKII is therefore not absolutely required for CA1 LTP, although it clearly may contribute and/or play a modifying role. This result is not in opposition to the idea that the CaMKII holoenzyme does play a critical role in the induction and maintenance of LTP, or that perhaps the α-CaMKII isoform, specifically, is the dominant contributing molecule in wild-type mice. The residual LTP observed in α-CaMKII mutant mice does suggest that some other molecule(s), such as other isoforms of CaMKII or other implicated kinases, are able to complement the CaMKII deficiency. In addition, it is possible that in the wild-type animal, it is the α-CaMKII isoform alone that contributes, because of its high expression levels and optimal localization, and that only in its absence are the potential contributing functions of other molecules revealed.
The α isoform of CaMKII is the most abundant subunit expressed in the adult mammalian hippocampus, in which it is found at a concentration three times higher than the β subunit, the next most highly expressed brain isoform (McGuinness et al. 1985; Miller and Kennedy 1985; Burgin et al. 1990). Previous results have shown that total CaMKII activity in forebrain from (−/−) α-CaMKII mutant mice is decreased to 45% ± 4, when measured by an in vitro assay, indicating that substantial CaMKII activity still remains in these mutant mice (Silva et al. 1992a). Recent studies have also shown that the β isoform is able to target to synapses in the absence of the α isoform, as it is observed to be present at normal levels in the postsynaptic densities of α-CaMKII mutant mice (M. Kennedy, in prep.). These levels are low compared with the wild-type levels of the α isoform, as previous studies have also indicated that mRNA for the α but not the β isoform is localized in dendrites (Burgin et al. 1990) and that the 3′-untranslated region of the α subunit mRNA is critical for dendritic targeting (Mayford et al. 1996). Consequently, the β isoform may be positioned appropriately to receive an incoming calcium transient in α-CaMKII mutant animals, although its relative lower abundance and perhaps lack of efficient mRNA targeting would still lead to a less efficient subsequent signaling response and decreased LTP.
The α isoform of CaMKII is expressed postnatally in the hippocampus, initially at very low levels, increasing fivefold to high adult levels by 3 weeks of age (Kelly and Vernon 1985; Burgin et al. 1990). It contrast, the β-isoform is expressed at moderate levels at birth and increases just slightly during development. During the first week of postnatal development, tetanus-induced LTP is diminished, but present (Harris and Teyler 1984; Muller et al. 1989; Dudek and Bear 1993; Bolshakov and Siegelbaum 1995; Liao and Malinow 1996), and increases in a fashion that parallels the expression of α-CaMKII. Interestingly, in one study, the amount of tetanus-induced LTP observed in the youngest animals tested (postnatal day 4–9) was ∼50% of that observed in older animals (postnatal day 10–14) (Liao and Malinow 1996), comparable with the difference we observe between α-CaMKII mutant and wild-type mice (Fig. 3). Because LTP, although reduced, is still attainable in young wild-type animals when the α-isoform is known to be poorly expressed, it suggests that the β isoform of CaMKII (or other mediators) can partially complement the absence of α-CaMKII.
Whereas tetanus-induced LTP is reduced in very young rats, LTP induced by low frequency stimulation paired with depolarization of the postsynaptic cell during whole cell recording produces a robust potentiation similar to that observed in older animals (Liao and Malinow 1996; Durand et al. 1996; J.A. Cummings, J.T.R. Isaac, R.A. Nicoll, and R.C. Malenka, unpubl.). This result, combined with the observation that the majority of CA1 transmission in young animals acts through NMDA receptors, making them effectively silent at hyperpolarized potentials, led to the hypothesis that an insufficient depolarization during LTP induction is responsible for the diminished LTP observed in immature animals. To address whether our deficiency in LTP is attributable to similar problems with induction or with subsequent expression mechanisms, we examined other parameters of synaptic transmission. Basal transmission, as indicated by the slope of input-output curves, is diminished in our mutant animals, compared with wild-type animals. This also parallels what was observed in young wild-type rats, which show reduced input-output slopes when compared with older animals (Liao and Malinow 1996). These similarities suggest that ineffective depolarization could be contributing to LTP induction deficits in α-CaMKII mutant mice. It will be interesting to examine in future studies whether α-CaMKII mutant mice have increased numbers of silent synapses. Although a high proportion of pure NMDA silent synapses could be a barrier to eliciting LTP, it could also be a reflection of deficient LTP during the development of α-CaMKII mutant mice, as it is thought that an LTP-like process is necessary to convert pure NMDA-receptor synapses to synapses with mixed AMPA and NMDA receptor function (Isaac et al. 1995; Liao et al. 1995; Wu et al. 1996).
If α-CaMKII mutant mice have a deficit in LTP-induction mechanisms, then one may be able to overcome this deficit by delivering stronger tetani. Our results, however, indicate that increasing tetanic strength or number does not overcome the mutant phenotype. Mutant mice appear to have a similar threshold for LTP induction and their LTP saturates with similar tetanic strengths as wild-type mice. Although this implicates deficient expression mechanisms in α-CaMKII mutant mice, it is possible that tetanus-delivered stimulation does not produce effective maximal depolarization. Whole cell experiments using a pairing protocol for LTP induction would be useful to determine if such a limit to effective postsynaptic membrane depolarization exists in these animals.
The complete lack of LTP and/or bimodal distribution of LTP values observed in previous studies (Silva et al. 1992; Stevens et al. 1994) is in contrast to that observed in our present studies, in which LTP was typically present in a normal distribution across mutant slices, but reduced compared with wild-type values. The bimodal distribution suggests that a variable at the level of the entire slice or the entire animal may be working to affect the probability of eliciting LTP in these experiments. A normal distribution of LTP would be expected if there is a large number of synapses acting independently, some producing LTP and some not, with the absence of α-CaMKII decreasing the likelihood or amount of LTP that can be obtained at any synapse.
There are several factors that could be contributing to the contrasting results observed in our present and previously reported CA1 LTP experiments. Two major variables are the precise experimental conditions used and the specific mutant animals examined. First, it is possible that minor differences in slice preparation and maintenance, stimulating and recording electrode type and placement, and specific recording conditions could have various effects on either slice health, effective stimulation, or postsynaptic cell depolarization, and subsequent LTP induction. It is also possible that the differences between present and past analyses of α-CaMKII mutant mice are caused by the differences in the genetic background of the mice used (Gerlai 1996a,b; Crawley 1996; Crusio 1996; Lathe 1996; Banbury Conference 1997). The earlier study was carried out with F2 and F3 mice produced between the 129Sv and BALB/c strains, whereas the present study utilized progeny generated by repeated sister/brother mating of litters that included contributions from the C57Bl/6 strain. It is quite possible that each of these mouse strains contains a unique assortment of alleles at multiple loci, and that these alleles could play roles directly in the LTP mechanism, peripherally as modifiers of LTP, or indirectly by changing other aspects of neurologic function that could affect plasticity. In fact, it has been shown recently that the amount of hippocampal LTP produced following υ-burst stimulation varies among different inbred strains of wild-type mice (T. Abel, P.V. Nguyen, R. Bourtchouladz, M.E. Bach, I. Gaprindashvili, P. Jain, and E.R. Kandel, unpubl.). For example, a 129Sv inbred line has been shown to exhibit reduced levels of hippocampal LTP following υ-burst stimulation (T. Abel, P.V. Nguyen, R. Bourtchouladz, M.E. Bach, I. Gaprindashvili, P. Jain, and E.R. Kandel, unpubl.). Because a 129Sv embryonic stem (ES) cell line was used to create the original α-CaMKII mutant mice, it is possible that 129Sv gees that account for this reduction in LTP could be closely linked to the mutated α-CaMKII locus, and therefore more readily retained in early generations of animals. For this reason, as well as the numerous behavioral deficits that are known to be associated with the 129Sv background (Gerlai 1996a), ES cells from the C57Bl/6J line are being used in our laboratory for creating future mutant mice.
It is possible that in the mice used in the earlier LTP study, the combinations of the alleles of the relevant genes fell into two classes: one in which α-CaMKII is essential for LTP and another in which α-CaMKII is entirely dispensable, explaining the bimodal distribution of LTP values. As the C57Bl/6 background was introduced and brother–sister mating was repeated, a combination of alleles that permits partial dependency on α-CaMKII may have been established in the pedigree. Recently, a bimodal distribution of other phenotypes was also observed among the progeny of early crosses of the original α-CaMKII mutant line that was repeatedly crossed into C57Bl/6 (Gordon et al. 1996). Visual cortical plasticity experiments revealed a severe deficit following monocular occlusion in only about one half of the α-CaMKII mutants of this line, with a similar fraction of animals showing a deficit in spatial learning in the Morris water maze following a period of intensive training. Mapping the genetic loci responsible for phenotypic differences both within, and between, these strains could be a valuable tool for identifying other genes that influence hippocampal LTP, and other α-CaMKII-associated phenotypes.
In conclusion, we find that NMDA receptor-dependent CA1 LTP is present, but reduced in mice lacking the α subunit of CaMKII in the present background examined. Therefore, α-CaMKII is not absolutely required for LTP. However, α-CaMKII appears to play a role, and these results do not preclude an absolute requirement for CaMKII in LTP, as other isoforms of CaMKII could be complementing the deficiency in the α-isoform. Although we cannot conclude whether the deficit is in the induction or expression of LTP, future whole cell recording experiments looking at silent synapses and postsynaptic depolarization in α-CaMKII mutant mice could be enlightening.
Acknowledgments
We are grateful to members of the Malinow and Tonegawa laboratories for helpful discussions, with special thanks to A. Aiba, T. Iwasato, K. Poss, and C. Lovett for their invaluable assistance with the technique of in vitro fertilization, and to Charles Steinberg for reading the manuscript. H.L.H. is supported by the National Institutes of Health Medical Science Training Program (NIH MSTP) (Harvard) and a Sackler Fellowship. S.T. is supported by NIH grant NS32925. R.M. is supported by the Mathers Charitable Foundation and the NIH.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
References
- Banbury conference. Neuron. 1997;19:755–759. doi: 10.1016/s0896-6273(00)80958-7. [DOI] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach R, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Regulation of hippocampal transmitter release during development and long-term potentiation. Science. 1995;269:1730–1734. doi: 10.1126/science.7569903. [DOI] [PubMed] [Google Scholar]
- Braun AP, Schulman H. The multifunctional calcium/calmodulin-dependent protein kinase: From form to function. Annu Rev Physiol. 1995;57:417–445. doi: 10.1146/annurev.ph.57.030195.002221. [DOI] [PubMed] [Google Scholar]
- Burgin KE, Waxham MN, Rickling S, Westgate SA, Mobley WC, Kelly PT. In situ hybridization histochemistry of Ca2+/calmodulin-dependent protein kinase in developing rat brain. J Neurosci. 1990;10:1788–1798. doi: 10.1523/JNEUROSCI.10-06-01788.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler LS, Silva AJ, Abelovich A, Watanabe Y, Tonegawa S, McNamara JO. Limbic epilepsy in α–calcium-calmodulin kinase II mutant mice. Proc Natl Acad Sci. 1995;92:6852–6855. doi: 10.1073/pnas.92.15.6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Rainnie DG, Greene RW, Tonegawa S. Abnormal fear response and aggressive behavior in mutant mice deficient for α–calcium-calmodulin kinase II. Science. 1994;265:291–294. doi: 10.1126/science.7939668. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Unusual behavioral phenotypes of inbred mouse strains. Trends Neurosci. 1996;19:181–182. doi: 10.1016/s0166-2236(96)20021-9. [DOI] [PubMed] [Google Scholar]
- Crusio WE. Gene-targeting studies: New methods, old problems. Trends Neurosci. 1996;19:186–187. doi: 10.1016/s0166-2236(96)20023-2. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J Neurosci. 1993;13:2910–2918. doi: 10.1523/JNEUROSCI.13-07-02910.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand GM, Kovalchuk Y, Konnerth A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature. 1996;381:71–75. doi: 10.1038/381071a0. [DOI] [PubMed] [Google Scholar]
- Erondu NE, Kennedy MB. Regional distribution of type II Ca2+/calmodulin-dependent protein kinase in rat brain. J Neurosci. 1985;5:3270–3277. doi: 10.1523/JNEUROSCI.05-12-03270.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong Y-L, Taylor WL, Means AR, Soderling TR. Studies of the regulatory mechanism of Ca2+/calmodulin-dependent protein kinase II. Mutation of threonine 286 to alanine and apartate. J Biol Chem. 1989;264:16759–16763. [PubMed] [Google Scholar]
- Fukunaga K, Goto S, Miyamoto E. Immunohistochemical localization of Ca2+/calmodulin-dependent protein kinase II in rat brain and various tissues. J Neurochem. 1988;51:1070–1078. doi: 10.1111/j.1471-4159.1988.tb03070.x. [DOI] [PubMed] [Google Scholar]
- Fukunaga K, Stoppini L, Miyamoto E, Muller D. Long-term potentiation is associated with an increased activity of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1993;268:7863–7867. [PubMed] [Google Scholar]
- Fukunaga K, Muller D, Miyamoto E. Increased phosphorylation of Ca2+/calmodulin-dependent protein kinase II and its endogenous substrates in the induction of long term potentiation. J Biol Chem. 1995;270:6119–6124. doi: 10.1074/jbc.270.11.6119. [DOI] [PubMed] [Google Scholar]
- Gerlai R. Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci. 1996a;19:177–181. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]
- ————— Gene-targeting in neuroscience: The systemic approach. Trends Neurosci. 1996b;19:188–189. [Google Scholar]
- Giese KP, Fedorov NB, Filipkowshki RK, Silva AJ. Autophosphorylation at Thr286 of the α-calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- Glazewski S, Chen C-M, Silva A, Fox K. Requirement for α CaMKII in experience-dependent plasticity of the barrel cortex. Science. 1996;272:421–423. doi: 10.1126/science.272.5260.421. [DOI] [PubMed] [Google Scholar]
- Goldenring JR, McGuire JS, Jr, DeLorenzo RJ. Identification of the major postsynaptic density protein as homologous with the major calmodulin-binding subunit of a calmodulin-dependent protein kinase. J Neurochem. 1984;42:1077–1084. doi: 10.1111/j.1471-4159.1984.tb12713.x. [DOI] [PubMed] [Google Scholar]
- Gordon JA, Cioffi D, Silva AJ, Stryker M. Deficient plasticity in the primary visual cortex of α-calcium/calmodulin-dependent protein kinase II mutant mice. Neuron. 1996;17:491–499. doi: 10.1016/s0896-6273(00)80181-6. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Schulman H. Neuronal Ca2+/calmodulin-dependent protein kinases. Annu Rev Biochem. 1992;61:559–601. doi: 10.1146/annurev.bi.61.070192.003015. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Kapiloff MS, Lou LL, Rosenfeld MG, Schulman H. Expression of a multifunctional Ca2+/calmodulin-dependent protein kinase and mutational analysis of its autoregulation. Neuron. 1989;3:59–70. doi: 10.1016/0896-6273(89)90115-3. [DOI] [PubMed] [Google Scholar]
- Harris KM, Teyler TJ. Development onset of long-term potentiation in area CA1 of the rat hippocampus. J Physiol. 1984;346:27–48. doi: 10.1113/jphysiol.1984.sp015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Ishida A, Katagiri H, Mishina M, Fujisawa H, Manabe T, Takahashi T. Calcium- and calmodulin-dependent phosphorylation of AMPA type glutamate receptor subunits by endogenous protein kinases in the post-synaptic density. Brain Res Mol Brain Res. 1997;46:338–342. doi: 10.1016/s0169-328x(97)00073-9. [DOI] [PubMed] [Google Scholar]
- Isaac JTR, Nicoll RA, Malenka RC. Evidence for silent synapses: Implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Ito I, Hidaka H, Sugiyama H. Effects of KN-62, a specific inhibitor of calcium/calmodulin-dependent protein kinase II, on long-term potentiation in the rat hippocampus. Neurosci Lett. 1991;121:119–121. doi: 10.1016/0304-3940(91)90663-e. [DOI] [PubMed] [Google Scholar]
- Kelly PT, Vernon P. Changes in the subcellular distribution of calmodulin-kinase II during brain development. Dev Brain Res. 1985;18:211–224. doi: 10.1016/0165-3806(85)90265-2. [DOI] [PubMed] [Google Scholar]
- Kelly PT, McGuinness TL, Greengard P. Evidence that the major PSD protein is a component of a calcium/calmodulin-dependent protein kinase. Proc Natl Acad Sci. 1984;81:945–949. doi: 10.1073/pnas.81.3.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MB, Bennett MK, Erondu NE. Biochemical and immunochemical evidence that the “major postsynaptic density protein” is a subunit of a calmodulin-dependent protein kinase. Proc Natl Acad Sci. 1983;80:7357–7361. doi: 10.1073/pnas.80.23.7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathe R. Mice, gene targeting and behaviour: More than just genetic background. Trends Neurosci. 1996;19:183–186. doi: 10.1016/s0166-2236(96)20022-0. [DOI] [PubMed] [Google Scholar]
- Liao D, Malinow R. Deficiency in induction but not expression of LTP in hippocampal slices from young rats. Learn & Mem. 1996;3:138–149. doi: 10.1101/lm.3.2-3.138. [DOI] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–414. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Lisman JE. The CaMKII hypothesis for storage of synaptic memory. Trends Neurosci. 1994;17:406–412. doi: 10.1016/0166-2236(94)90014-0. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Goldring MA. Feasibility of long-term storage of graded information by the Ca2+/calmoudlin-depedent protein kinase molecules of the postsynaptic density. Proc Natl Acad Sci. 1988;85:5320–5324. doi: 10.1073/pnas.85.14.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lledo PM, Hjelmstad GO, Mukherfi S, Soderling TR, Malenka RC, Nicoll RA. Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci. 1995;92:11175–11179. doi: 10.1073/pnas.92.24.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka R, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA, Waxham MN. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 1989;340:554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recording of long-term potentiation in hippocampal slices. Nature. 1990;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Mayford M, Wang J, Kandel ER, O’Dell TJ. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell. 1995;81:891–904. doi: 10.1016/0092-8674(95)90009-8. [DOI] [PubMed] [Google Scholar]
- Mayford M, Baranes D, Podsypanina K, Kandel ER. The 3′-untranslated region of CaMKIIα is a cis-acting signal for the localization and translation of mRNA in dendrites. Proc Natl Acad Sci. 1996;93:13250–13255. doi: 10.1073/pnas.93.23.13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlade-McCulloh E, Yamamoto H, Tan S-E, Brickey DA, Soderling TR. Phosphorylation and regulation of glutamate receptors by calcium/calmodulin-dependent protein kinase II. Nature. 1993;362:640–642. doi: 10.1038/362640a0. [DOI] [PubMed] [Google Scholar]
- McGuinness TL, Lai Y, Greengard P. Ca2+/calmodulin-dependent protein kinase II. Isozymic forms from rat forebrain and cerebellum. J Biol Chem. 1985;260:1696–1704. [PubMed] [Google Scholar]
- Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 1992;256:1199–1201. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- Miller SG, Kennedy MB. Distinct forebrain and cerebellar isozymes of type II Ca2+/calmodulin-dependent protein kinase associate differently with the postsynaptic density fraction. J Biol Chem. 1985;260:9039–9046. [PubMed] [Google Scholar]
- ————— Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation: A Ca2+-triggered molecular switch. Cell. 1986;44:861–870. doi: 10.1016/0092-8674(86)90008-5. [DOI] [PubMed] [Google Scholar]
- Muller D, Oliver M, Lynch G. Developmental changes in synaptic properties in hippocampal neonatal rats. Dev Brain Res. 1989;49:105–114. doi: 10.1016/0165-3806(89)90063-1. [DOI] [PubMed] [Google Scholar]
- Omkumar RV, Kiely MJ, Rosenstein AM, Min KT, Kennedy MB. Identification of a phosphorylation site for calcium/calmodulin-dependent protein kinase II in the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 1996;270:31670–31678. doi: 10.1074/jbc.271.49.31670. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Griffith LC, Lisman JE. Postsynaptic inhibitors of calcium/calmodulin-dependent protein kinase type II block induction but not maintenance of pairing-induced long-term potentiation. J Neurosci. 1997;17:5357–5365. doi: 10.1523/JNEUROSCI.17-14-05357.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet CC, McGuinness TL, Greengard P. Immunocytochemical localization of calcium/calmodulin-dep endent protein kinase II in rat brain. Proc Natl Acad Sci. 1984;81:5604–5608. doi: 10.1073/pnas.81.17.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Y, Kantor D, Harris KM, Schuman EM, Kennedy MB. Visualization of the distribution of autophosphorylated Calcium/calmodulin-dependent protein kinase II after tetanic stimulation in the CA1 area of the hippocampus. J Neurosci. 1997;17:5416–5427. doi: 10.1523/JNEUROSCI.17-14-05416.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit DL, Perlman S, Malinow R. Potentiated transmission and prevention of further LTP by increased CaMKII activity in postsynaptic hippocampal slice neurons. Science. 1994;266:1881–1885. doi: 10.1126/science.7997883. [DOI] [PubMed] [Google Scholar]
- Roberson ED, English JD, Sweatt JD. A biochemist’s view of long-term potentiation. Learn & Mem. 1996;3:1–24. doi: 10.1101/lm.3.1.1. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in α–calcium-calmodulin kinase II mutant mice. Science. 1992a;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S. Impaired spatial learning in α-calcium-calmodulin kinase II mutant mice. Science. 1992b;257:206–211. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Tonegawa S, Wang Y. The role of calcium-calmodulin kinase II in three forms of synaptic plasticity. Curr Biol. 1994;4:687–693. doi: 10.1016/s0960-9822(00)00153-6. [DOI] [PubMed] [Google Scholar]
- Vallano ML, Goldenring JR, Lasher RS, Delorenzo RJ. Association of calcium/calmodulin-dependent kinase with cytoskeletal preparations: Phosphorylation of tubulin, neurofilament, and microtubule associated proteins. Ann NY Acad Sci. 1986;466:357–374. doi: 10.1111/j.1749-6632.1986.tb38406.x. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Hanson PI, Schulman H. Multifunctional Ca2+/calmodulin-dependent protein kinase made Ca2+-independent for functional studies. Biochemistry. 1990;29:1679–1684. doi: 10.1021/bi00459a002. [DOI] [PubMed] [Google Scholar]
- Waxham MN, Aronowski J, Westgate SA, Kelly PT. Mutagenesis of Thr286 in monomeric Ca2+/calmodulin-dependent protein kinase II eliminates Ca2+/calmodulin-independent activity. Proc Natl Acad Sci. 1990;87:1273–1277. doi: 10.1073/pnas.87.4.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Malinow R, Cline HT. Maturation of a central glutamatergic synapse. Science. 1996;274:972–976. doi: 10.1126/science.274.5289.972. [DOI] [PubMed] [Google Scholar]