

Abstract
In addition to their trophic functions, neurotrophins are also implicated in synaptic modulation and learning and memory. Although gene knockout techniques have been used widely in studying the roles of neurotrophins at molecular and cellular levels, behavioral studies using neurotrophin knockouts are limited by the early-onset lethality and various sensory deficits associated with the gene knockout mice. In the present study, we found that in a spontaneous mutant mouse, waggler, the expression of brain-derived neurotrophic factor (BDNF) was selectively absent in the cerebellar granule cells. The cytoarchitecture of the waggler cerebellum appeared to be normal at the light microscope level. The mutant mice exhibited no sensory deficits to auditory stimuli or heat-induced pain. However, they were massively impaired in classic eye-blink conditioning. These results suggest that BDNF may have a role in normal cerebellar neuronal function, which, in turn, is essential for classic eye-blink conditioning.
Neurotrophins are a class of molecules that regulate both developing and adult nervous systems. During early development, neurotrophins promote neuronal proliferation, differentiation, maturation, and survival. In the adult brain, neurotrophins and synaptic transmission exhibit mutual regulation. Acute application of neurotrophins enhances synaptic transmission (Lohof et al. 1993; Knipper et al. 1994; Lessmann et al. 1994; Kang and Schuman 1995; Levine et al. 1995; Moises et al. 1995) and long-term potentiation (Figurov et al. 1996), whereas induction of long-term potentiation increases brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT3) expression in vitro (Patterson et al. 1992; Dragunow et al. 1993) and in vivo (Springer et al. 1994). Furthermore, null mutation of the BDNF gene impairs both basal synaptic transmission and long-term potentiation in the hippocampus (Korte et al. 1995; Patterson et al. 1996), and some of the deficits can be rescued by culturing the slices with BDNF (Korte et al. 1996; Patterson et al. 1996). These results strongly suggest an acute function of neurotrophins in synaptic function in adult brain. Parallel with its roles in synaptic modulation, neurotrophins also affect behavioral learning. Although intraventricular nerve growth factor (NGF) infusion improves spatial memory in aged rats (Markowska et al. 1994, 1996), administrations of anti-NGF antibodies impair passive avoidance learning (Ricceri et al. 1994).
Various neurotrophin and tyrosine kinase (Trk)-receptor knockout mice provided animal models to study functions of neurotrophins in neuronal development and synaptic modulation. But they are not as useful in behavioral studies because mutant mice have severe sensory deficits and die during early postnatal development (Crowley et al. 1994; Enfors et al. 1994a,b; Jones et al. 1994; Klein et al. 1994; Smeyne et al. 1994). A recently discovered spontaneous mutant mouse stargazer, with localized BDNF deficit in the cerebellum (Qiao et al. 1996), provided a feasible model to study the function of BDNF in the cerebellum (Chen et al. 1996b). In the present study, we found that another mutant mouse allelic to stargazer (Sweet et al. 1991; Sweet 1993), waggler, also exhibited selective deficit in cerebellar granule cell BDNF expression without apparent abnormality in cerebellar cytoarchitecture. Unlike stargazer, the waggler mutant arises from C57BL/6 inbred strain, therefore allowing us to study potential roles of BDNF in cerebellar functions and learning with minimal interference from strain differences.
Materials and Methods
SUBJECTS
waggler mutant mice with C57BL6/J genetic background and wild-type littermates (+/+ or +/wag), originally from Jackson laboratory, were bred and maintained in a University of Southern California vivarium. The adult male animals used in the experiments were individually housed with ad lib access to food and water.
HISTOLOGY AND IN SITU HYBRIDIZATION
The adult mutant and wild-type mice were overdosed with sodium pentobarbital and transcardially perfused with 0.9% saline, followed by 10% buffered formalin. The brains were removed and postfixed for at least 24 hr at 4°C. The tissue was then sectioned at 40 μm and the slices were mounted onto subbed slides and stained with cresyl violet.
The procedures for in situ hybridization were the same as in Qiao et al. (1996). In brief, brains from both wild-type and mutant adult mice were excised after decapitation, frozen on dry ice, sliced on a cryostat at 16 μm, and mounted on subbed slides. The mutant and wild-type brain sections were mounted on the same slides and processed identically for optimal comparison. The sections were fixed with 4% p-formaldehyde in 0.1 m PBS for 30 min and pretreated with 0.25% acetic anhydride and 0.1 m triethanolamine for 10 min. Then, the tissue sections were incubated overnight at 50°C with 35S-labeled riboprobe (5 × 106 cpm/ml) prepared from a pGEM-4Z plasmid containing a 460-bp insert of rat BDNF cDNA clone (Phillips et al. 1990). The hybridization solution also contains 50% formamide, 10% dextran sulfate, 300 mm NaCl, 0.5 mg/ml yeast RNA, 10 μm dithiothreitol, 0.02% Ficoll, 0.02% polyvinyl pyrrolidone, 0.02% bovine serum albumin, 1 mm EDTA, and 10 mm Tris-HCl (pH 8.0). After hybridization, the sections were rinsed in 4× SSC, digested with 20 μg/ml RNase A at 37°C for 30 min, and washed through descending concentrations of 1× SSC to 0.1× SSC at 60–70°C. Finally, the slides were dehydrated in ethanol, dried, and exposed to X-ray film (Kodak X-OMAT, Rochester, NY) for 3–5 days.
SENSORY TEST
To record tone-induced neuronal responses, mice were anesthetized intraperitoneally (IP) with ketamine (80 mg/kg) and xylazine (20 mg/kg), and mounted onto a stereotaxic device. An epoxylite-coated electrode (50-μm tip exposure) was lowered to the primary auditory cortex (Franklin and Paxinos 1997), and extracellular multiple unit activity was amplified and sent to a window discriminator, from which spikes above the noise envelop were identified. Then, the spikes were counted in 4-msec bins and stored by a computer. The tone used in this test was the same as the one used in the behavioral training.
To test nociception, wagglers (n = 5) and wild types (n = 5) were restrained in a plastic cylinder (4-cm inner diam.), the tail was put on a hot plate (51°C), and the onset latency of tail flick was recorded three times with 1 min intervals.
TONE FEAR CONDITIONING
The procedure was modified from Kim et al. (1997). On day 1 of fear conditioning, each mouse was placed in the experimental chamber (11.5 × 11.5 × 11.5 in.; Coulbourn Instruments, Inc., PA) with the floor consisting of 26 stainless steel rods (3/16-in. diam.). After 3 min in the chamber, the mice received three tone and foot-shock pairings (tone: 20 sec, 85 dB, 2 kHz; foot shock: 1 sec, 0.5 mA; intertrial interval 1 min apart). One minute after the final foot shock, the mice were returned to their home cages. Each chamber was wiped with 5% ammonium hydroxide solution before training. On day 3, fear conditioning to the tone was assessed in a observation chamber that was completely different from the one used during conditioning. After 1 min in the new chamber, the same tone was presented for an 8-min test. Fear conditioning was measured as the percentage of freezing response. Freezing is defined as the absence of all visible movement of the body and vibrissae aside from movement necessitated by respiration. The freezing was scored every 2 sec, and the data were converted into percentage of freezing time for every minute of observation.
SURGERY
The surgical procedure was described previously (L. Chen et al. 1996a). Under anesthesia (ketamine, 80 mg/kg, and xylazine, 20 mg/kg, IP), animals were implanted with four electrodes (Teflon-coated stainless steel, 0.003 in. bare, 0.0045 in. coated, A-M Systems, Inc., Everett, WA) subcutaneously to the left upper eyelid to record differential electromyograph (EMG) from obicularis oculi, and to deliver periorbital shock. A four-pin strip connector to which the wires were soldered was cemented to the animal’s skull with dental acrylic.
DELAY EYE-BLINK CONDITIONING
The training procedure was a slight modification of that of Chen et al. (1996a). Training took place in a small Plexiglas cylinder (3.75-in. diam.) placed inside a sound- and light-attenuating chamber (Ralph Gerbrands Co., Arlington, MA). Background noise inside the chamber was ∼65 dB. The daily conditioning session consisted of 100 trials grouped into blocks of 10 trials [conditioned stimulus (CS), 4 CS-unconditioned stimuli (USs), US, 4 CS–USs, in sequence] with randomized intertrial intervals (ITIs) between 20 and 40 sec. The CS was a 352-msec tone (1000 Hz, 80 dB, 5 msec rise/fall time), and the US was a coterminating 100-msec shock (100 Hz biphasic square pulses delivered with a Grass SD9 stimulator). The initial US intensity was 20 V with 5-V increments every subsequent day to compensate for US habituation.
After 1 day of habituation, animals underwent 6 days of classic eye-blink conditioning, followed by 4 days of extinction (CS alone) training.
EMG ANALYSIS
For conditioned response (CR) analysis, the eyelid EMG signal was amplified, digitized, and stored by a computer for off-line analysis. The EMG sampling threshold was adjusted so that the baseline activity was ∼0.5 U/bin.
The CR period, the time window within which the digitized unit activity was used to score a CR, was defined as the period from 84 msec after the CS onset to the US onset (total of 168 msec) in paired CS–US trials, or to the termination of the CS (total of 268 msec) in CS-alone trials. The pre-CS period was defined as the 252-msec period before the onset of the CS.
The digitized EMG activity was analyzed trial by trial, and “bad” trials were excluded using the following criteria: (1) During the 252-msec pre-CS period, the average unit count per bin is higher than 2 (suggesting a high eyelid activity before the CS onset), or (2) the standard deviation (s.d.) of unit counts per bin is greater than the average unit counts per bin during the pre-CS period (suggesting an unstable EMG activity before the CS onset), or (3) the average unit count within 28 msec (7 bins) after the CS onset is 1 s.d. greater than the average of pre-CS activity (suggesting a short-latency startle response to the CS). The number of trials rejected was 10 ± 3 per session for each wild-type mouse and 6.4 ± 0.8 per session for waggler, with no statistically significant difference (P > 0.1).
The CR was defined as the average unit activity of any consecutive 7 bins during the CR period that was higher than “average + s.d. + 1” of the pre-CS unit activity. The CR onset latency was computed as the first time during the CR period, starting at which the average unit count of consecutive 7 bins (28 msec), was higher than average + s.d. + 1 of the pre-CS unit activity. The peak latency was calculated as the time during the CR period starting at which the average unit count of consecutive 7 bins (28 msec) was the highest and was higher than average + s.d. + 1 of the pre-CS unit activity. The unconditioned response (UR) amplitude was defined as the difference between average unit activity during the UR period (100-msec period 50 msec after the termination of the US) and averaged unit of the pre-CS period.
Results
BDNF mRNA EXPRESSION IS ABSENT IN CEREBELLAR GRANULE CELLS
Figure 1 presents the expression patterns of the BDNF mRNA. In normal adult brains, BDNF was highly expressed in the hippocampus, the neocortex, and the cerebellar cortex (mainly in the granule cells). In the mutant brain, the BDNF expression was normal in the hippocampus and the neocortex but virtually absent in cerebellar granule cells. The basal BDNF expression in the cerebellar molecular layer and deep nuclei was low in both mutant and wild-type mice and did not allow a comparison between them. BDNF is expressed transiently in the cerebellar deep nuclei around postnatal day 5 (Rocamora et al. 1993), when the phenotypes of the waggler mouse are not yet evident. We also examined the gross brain morphology by cresyl violet staining of the brain sections. Consistent with previous results from gene knockout studies (Jones et al. 1994), the mutant cerebellum appeared qualitatively normal (Fig. 2). In the cerebellar cortex, the laminar structure appeared well developed with normal molecular, Purkinje, and granule layers. In the cerebellar deep nuclei, cells appeared normal in their densities, shapes, and sizes. Several other brain regions involved in eye-blink conditioning were also examined (Fig. 3), including cerebellar nuclei, inferior olive, pontine gray, red nucleus, cochlear nuclei, and hippocampus. No noticeable difference was detected in any of these regions.
Figure 1.
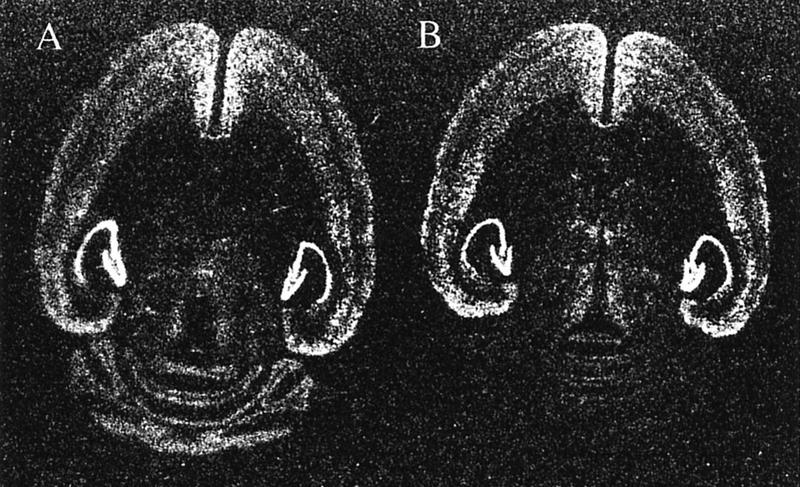
Expression patterns of BDNF mRNA in adult brains of wild-type (A) and waggler mice (B). The only noticeable difference is the absence of BDNF mRNA in the waggler cerebellar granule cell layer.
Figure 2.
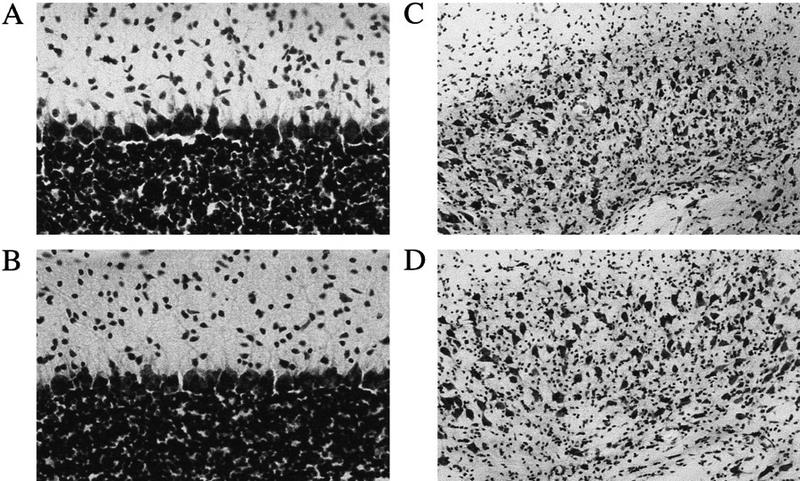
Sagittal sections of the cerebellar cortex (A, wild-type; B, waggler; 40× objectives) and coronal sections of the cerebellar interpositus nucleus (C, wild-type; D, waggler; 20×).
Figure 3.
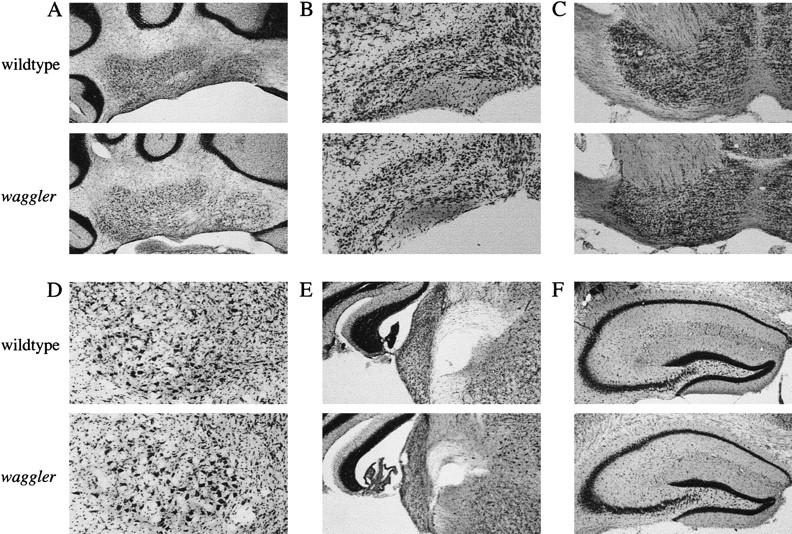
Coronal brain sections of the wild-type and waggler mice. (A) Cerebellar deep nuclei (5× objectives); (B) inferior olive (10×); (C) basal pontine gray (5×); (D) red nucleus (10×); (E) cochlear nuclei (5×); (F), hippocampus (5×).
CS AND US PERCEPTIONS ARE NORMAL IN WAGGLER
Because most neurotrophin knockout mice have sensory deficits, and some spontaneous ataxic mutant mice have inner ear defects (Lyon and Searle 1989), we tested the auditory and somatosensory responses in waggler mutant mice. When the tails of the mice were placed on a 51°C hot plate, both wild-type (n = 5) and mutant mice (n = 5) exhibited the characteristic tail flicks. The latency to tail flicks was measured three times with 1-min intervals. Both groups showed slight, but not significant, sensitization to the repeated measures. When the averaged latencies were compared (Fig. 4), no difference was found between the wild-type and the mutant mice [F(1, 8) = 0.003, P > 0.5]. Therefore, waggler mutant mice seem not to have the somatosensory deficits exhibited by the BDNF gene knockout mice. Furthermore, the unconditioned responses to the same US were not different between waggler and the wild-type mice (see Fig. 6, below), indicating that the specific US perception is normal in the mutant.
Figure 4.

Onset latencies of heat-induced tail flicks as an index of pain perception. It took a similar amount of time for both wild-type (solid bar) and waggler (open bar) mice to feel the 51°C hot plate-induced pain and to start tail flicks.
Figure 6.

Percent CR (top) and UR (bottom) amplitudes exhibited by wild-type (•) and waggler (█) mice. The percent CRs were calculated using CS–US paired trials.
Figure 5 shows a 10-trial summary of the extracellular neuronal responses recorded from the auditory cortex of the wild-type and mutant mice. The auditory stimulus was a 352-msec tone (1000 Hz, 80 dB, 5-msec rise/fall time), which was identical to the CS used in the behavioral training. Both the wild-type and the mutant mice showed responses with comparable patterns and amplitudes.
Figure 5.

(A,B) The 10-trial summary of tone-induced neuronal responses recorded from auditory cortex of wild-type (A) and waggler (B) mice. (C) Tone fear conditioning. Wild-type (•) and waggler (█) showed no difference in freezing percentage after three foot shocks or during the first minute of the tone test.
The normal hearing in the waggler mutant was also verified behaviorally with tone fear conditioning. Despite the apparent ataxia, waggler mice showed a similar amount of freezing to the wild-type after three foot shocks [F(1,13) = 1.31, P > 0.1] during the training session. Two days later, the tone test was conducted in a new context. During the first minutes without tone, no freezing was observed in either group. When the tone was presented, the mice in both groups exhibited profound freezing. The percent freezing times were similar between the mutant and the wild-type groups during the first minute after the tone onset [F(1, 13) = 0.42,P > 0.5], which indicates normal hearing of the mutant mice.
EYE-BLINK CONDITIONING IS SEVERELY IMPAIRED
Despite the normal sensory responses, the mutant mice were severely impaired in eye-blink conditioning. As evaluated using CR percentage of the paired trials (Fig. 6), wild-type mice exhibited a significant level of learning during the 6 days of paired training [linear trend analysis, F(1, 5) = 26.81, P < 0.005], whereas waggler mice only showed a marginal level of learning [F(1, 5) = 5.69, P =0.063]. The wild-type mice learned to a higher asymptotic level at a faster learning rate than the mutant mice [two-way ANOVA: group effect, F(1, 10) = 26.84, P < 0.001; group × day interaction, F(5, 50) = 4.78, P < 0.005]. The impairment did not appear to be caused by performance deficits, as the unconditioned response amplitudes (Fig. 6) were not statistically different between the two groups [two-way ANOVA: group effect, F(1, 10) = 3.62, P > 0.05; group × day interaction F(5, 50) = 1.28, P > 0.05].
When the learning was evaluated using CS-alone trials (Fig. 7), the impairment was also evident in waggler mice [F(1, 10) = 14.20, P < 0.005]. However, the mutant did show a mild increase in percent CRs [F(5, 25) = 2.88, P <0.05]. The CRs were extinguished to a similar level in both groups after repeated presentations of the tone CS [day effect: wild type, F(3, 15) = 8.84, P < 0.005; waggler, F(3, 15) = 4.33, P < 0.05]. No significant difference between the two groups was found during the extinction training, which might be attributable to the large variability.
Figure 7.

Percent CRs of the wild-type (•) and waggler (█) mice during acquisition and extinction phases. The percent CRs were calculated using CS-alone trials.
The histograms of the onset and peak CR latencies are presented in Figure 8. The onset latency of the wild-type mice exhibited a pattern similar to those observed before (Shibuki et al. 1996; Bao et al. 1998). A large number of CR onset latencies fell into the bin covering the 57- to 84-msec period (Fig 8A,C). The overall pattern resembled a normal distribution skewed toward the CS onset, indicating that the responses were triggered by the CS. The peaks of wild-type CRs had a bimodal pattern as seen before (Shibuki et al. 1996; also in C57BL6/J and BALB/c mice, S. Bao, L. Chen, and R.F. Thompson, unpubl.). For the mutant mice, however, both the onset and peak latencies were delayed. The distributions were also flatter in waggler than in wild-type mice. To rule out the effects caused by the large difference between the total numbers of CRs of the wild-type and the mutant groups, the frequencies of occurrence were also plotted in Figure 8 B and D. The differences are clear and statistically significant [two-way ANOVA on the group × latencies interaction: onset latency, F(8, 80) = 7.45, P < 0.000001; peak latency, F(8, 80) = 3.49, P < 0.005]. (The peak of eyelid EMG is not necessarily the peak of behavioral eye-blink. The peaks of behavioral eye-blink CRs in rabbits usually appear around the onset of the US).
Figure 8.

The onset (A,B) and peak (C,D) latencies exhibited by wild-type (solid bars) and waggler (open bars) mice. Both numbers of occurrence (A,C) and occurring frequencies (B,D) are presented.
Discussion
Our results demonstrated impaired eye-blink learning in mice with a localized BDNF deficiency. Unlike the BDNF gene knockout mice (Ernfors et al. 1994a; Jones et al. 1994), waggler mice do not exhibit sensory nucleus degeneration. They also do not have sensory or performance deficits pertaining to classic eye-blink conditioning. Although waggler mice show ataxia- and forebrain (but not cerebellar)-generalized spike-wave epilepsy, these features probably do not account for the learning impairments, as eye-blink conditioning is normal in tottering mice, another type of mutant mice with ataxia and similar epilepsy but without cerebellar BDNF deficiency (S. Bao, L. Chen, X. Qiao, and R.F. Thompson, unpubl.).
Neurotrophins and their receptors are expressed differentially in the central nervous system (Maisonpierre et al. 1990; Friedman et al. 1991; Ringstedt et al. 1993; Rocamora et al. 1993; Escandon et al. 1994). Generally, NT-3 expression decreases, whereas BDNF expression increases during development (Maisonpierre et al. 1990). In the rat cerebellum, NT-3 expression peaks ∼2 weeks postnatally and tapers down later, whereas BDNF expression peaks at postnatal day 20 and remains high throughout adulthood (Rocamora et al. 1993). A similar pattern is also observed in the mouse (Hofer et al. 1990; Qiao et al. 1996). In rats of postnatal day 20, granule cells have finished migration from the outer to the inner granule cell layer, and the maturation of the mossy fiber–granule cell synapses and the parallel fiber–Purkinje cell synapses is in progress (Altman 1972). Thus, BDNF may be more important in synaptic formation and/or maturation rather than in neuronal survival or differentiation. This notion is supported by the fact that waggler mice (also see Jones et al. 1994 for BDNF gene knockouts; Qiao et al. 1996 for stargazer mutant) develop qualitatively normal cerebellar laminar structure despite the lack of BDNF expression. The BDNF knockout mice show normal hippocampal morphology but deficient hippocampal basal synaptic transmission and long-term potentiation (Korte et al. 1995; Patterson et al. 1996). The similar deficits in basal synaptic transmission or synaptic plasticity may occur in the cerebellar cortical synapses in waggler mice, and consequently impair classic eye-blink conditioning.
Although it is enticing to attribute the motor learning deficits to the BDNF failure, further studies are necessary to test the hypothesis directly. The waggler mutant was first discovered in Jackson Laboratory (Sweet et al. 1991) and later was found to be allelic to stargazer mice (Sweet 1993), another ataxic mutant (Noebels et al. 1990). Although the mutation has been mapped on Chromosome 15 between D15Mit30 and the parvalbumin gene (Letts et al. 1997), it is not known yet how this mutation causes the failure of BDNF expression. It is possible that the absence of BDNF and the impaired eye-blink conditioning are separate results of the waggler mutation and independent of each other. A decisive experiment would be to rescue the learning by putting BDNF back into the cerebellum.
The timing of the classically conditioned eye-blink responses is altered in cerebellum-lesioned rabbits (Logan 1991; Perrett et al. 1993) and in some cerebellar mutant mice (Chen et al. 1996a). In waggler mutant mice, CR onset and peak latencies are also changed. Unlike early-onset CRs of Purkinje cell degeneration (pcd) mutant mice, the CRs exhibited by waggler mice are delayed. This is consistent with the fact that the Purkinje cells, the major inhibitory input to the cerebellar deep nuclei, are present in the waggler mutant but not in the pcd mutant.
The present results are in agreement with those of a developmental study (Stanton et al. 1992) showing that the learning capacity of rats in classic eye-blink conditioning increases dramatically during postnatal days 18–24, which is coincident with synapse maturation and BDNF expression of the cerebellar cortex (Altman 1972; Rocamora et al. 1993) but not with the transient BDNF expression in the cerebellar deep nuclei during postnatal days 5–15 (Rocamora et al. 1993). These results suggest that the cerebellar cortical synaptic transmission is important for eye-blink conditioning.
An alternative view is that the cerebellar deep nuclei are also affected by the cerebellar cortical BDNF deficiency, as the two regions have reciprocal projections. Although our light microscopic observations indicate that the morphology of the mutant cerebellar deep nuclei is normal in all aspects examined, further electrophysiological and electron microscopic examinations of the cerebellar deep nuclei of the waggler mutant are essential for a conclusion on this issue.
In the past two decades, evidence has accumulated pinpointing the cerebellum as the site of memory formation and storage for eye-blink conditioning (for review, see Thompson et al. 1997). The cerebellar cortex is necessary for optimal learning, although in most studies, cortical lesions only result in partial impairment (Yeo et al. 1985; Lavond et al. 1987; Lavond and Steinmetz 1989; Perrett et al. 1993; Chen et al. 1996a). Within the cerebellar cortex, it is hypothesized that the climbing fibers carry the US information and modify the parallel fiber–Purkinje cell synapses that convey the CS information from mossy fiber to cerebellar deep nuclei (Marr 1969; Albus 1971; Ito 1984; Thompson et al. 1997). Recent studies using gene knockout mice support this hypothesis. Compared to normal mice, gene knockout mice with multiple climbing fiber innervation learn faster (Chen et al. 1995), whereas mice with impaired parallel fiber–Purkinje cell long-term depression learn slower and to a lower asymptotic level in classic eye-blink conditioning (Aiba et al. 1994; Shibuki et al. 1996). However, a clear correlation between the cerebellar electrophysiology and learning has been hampered by the fact that the genetic deficiencies in conventional gene knockout mice are global, rather than localized. The present results indicate that the waggler mutant mouse may be a suitable model for studying the functions of BDNF in adult cerebellum.
In summary, we demonstrated that eye-blink conditioning is severely impaired in waggler, a mutant mouse with a localized cerebellar BDNF deficiency. The results support the hypothesis that the cerebellum is essential for classic eye-blink conditioning. The results also suggest that normal BDNF expression may be important for adult cerebellar functions and behavioral learning.
Acknowledgments
We thank Dr. Verity A. Letts (Jackson Laboratory) for kindly providing waggler mutant mice. This work was supported by National Science Foundation grant IBN-9215069, Office of Naval Research grant N00014-95-1152, National Institute of Mental Health grant 5P01-MH52194, and a grant from Sankyo Company to R.F.T.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
References
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- Albus JS. A theory of cerebellar function. Math Biosci. 1971;10:25–61. [Google Scholar]
- Altman J. Postnatal development of the cerebellar cortex in the rat. II. Phases in the maturation of Purkinje cells and of the molecular layers. J Comp Neurol. 1972;145:399–464. doi: 10.1002/cne.901450402. [DOI] [PubMed] [Google Scholar]
- Bao S, Chen L, Thompson RF. Classical eyeblink conditioning in two strain of mice: Conditioned responses, sensitization and spontaneous eyeblinks. Behav Neurosci. 1998;112:714–718. doi: 10.1037//0735-7044.112.3.714. [DOI] [PubMed] [Google Scholar]
- Chen C, Kano M, Abeliovich A, Chen L, Bao S, Kim JJ, Hashimoto K, Thompson RF, Tonegawa S. Impaired motor coordination correlates with persistent multiple climbing fiber innervation in PKC gamma mutant mice. Cell. 1995;83:1233–1242. doi: 10.1016/0092-8674(95)90148-5. [DOI] [PubMed] [Google Scholar]
- Chen L, Bao S, Lockard JM, Kim JK, Thompson RF. Impaired classical eyeblink conditioning in cerebellar-lesioned and Purkinje cell degeneration (pcd) mutant mice. J Neurosci. 1996a;16:2829–2838. doi: 10.1523/JNEUROSCI.16-08-02829.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Bao S, Qiao X, Knusel B, Thompson RF. Impaired classical eyeblink conditioning in mutant mice (stargazer) with localized deficiency in BDNF expression in the cerebellum. Soc Neurosci Abstracts. 1996b;22:1644. [Google Scholar]
- Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts Meek S, Armanini MP, Ling LH, MacMahon SB, Shelton DL, Levinson AD, Phillips HS. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 1994;76:1001–1011. doi: 10.1016/0092-8674(94)90378-6. [DOI] [PubMed] [Google Scholar]
- Dragunow M, Beilharz E, Mason B, Lawlor P, Abraham W, Gluckman P. Brain-derived neurotrophic factor expression after long-term potentiation. Neurosci Lett. 1993;160:232–236. doi: 10.1016/0304-3940(93)90420-p. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994a;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Kucera J, Jaenisch R. Lack of neurotrophin-3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell. 1994b;77:503–512. doi: 10.1016/0092-8674(94)90213-5. [DOI] [PubMed] [Google Scholar]
- Escandon E, Soppet D, Rosenthal A, Mendoza Ramirez JL, Szonyi E, Burton LE, Henderson CE, Parada LF, Nikolics K. Regulation of neurotrophin receptor expression during embryonic and postnatal development. J Neurosci. 1994;14:2054–2068. doi: 10.1523/JNEUROSCI.14-04-02054.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figurov A, Pozzo Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego, CA: Academic Press; 1997. [Google Scholar]
- Friedman WJ, Ernfors P, Persson H. Transient and persistent expression of NT-3/HDNF mRNA in the rat brain during postnatal development. J Neurosci. 1991;11:1577–1584. doi: 10.1523/JNEUROSCI.11-06-01577.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer M, Pagliusi SR, Hohn A, Leibrock J, Barde YA. Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. EMBO J. 1990;9:2459–2464. doi: 10.1002/j.1460-2075.1990.tb07423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M. The cerebellum and neural control. New York, NY: Raven; 1984. [Google Scholar]
- Jones KR, Farinas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Shih JC, Chen K, Chen L, Bao S, Maren S, Anagnostaras SG, Fanselow MS, Maeyerm ED, Serf I, Thompson RF. Selective enhancement of emotional, but not motor learning in monoamine oxidase A-deficient mice. Proc Natl Acad Sci. 1997;94:5929–5933. doi: 10.1073/pnas.94.11.5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Silos Santiago I, Smeyne RJ, Lira SA, Brambilla R, Bryant S, Zhang L, Snider WD, Barbacid M. Disruption of the neurotrophin-3 receptor gene trkC eliminates la muscle afferents and results in abnormal movements. Nature. 1994;368:249–251. doi: 10.1038/368249a0. [DOI] [PubMed] [Google Scholar]
- Knipper M, Leung LS, Zhao D, Rylett RJ. Short-term modulation of glutamatergic synapses in adult rat hippocampus by NGF. Neuroreport. 1994;5:2433–2436. doi: 10.1097/00001756-199412000-00007. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T. Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci. 1996;93:12547–12552. doi: 10.1073/pnas.93.22.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavond DG, Steinmetz JE. Acquisition of classical conditioning without cerebellar cortex. Behav Brain Res. 1989;33:113–164. doi: 10.1016/s0166-4328(89)80047-6. [DOI] [PubMed] [Google Scholar]
- Lavond DG, Steinmetz JE, Yokaitis MH, Thompson RF. Reacquisition of classical conditioning after removal of cerebellar cortex. Exp Brain Res. 1987;67:569–593. doi: 10.1007/BF00247289. [DOI] [PubMed] [Google Scholar]
- Lessmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. Neuroreport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- Letts VA, Valenzuela A, Kirley JP, Sweet HO, Davisson MT, Frankel WN. Genetic and physical maps of the stargazer locus on mouse chromosome 15. Genomics. 1997;43:62–68. doi: 10.1006/geno.1997.4780. [DOI] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan CG. Cerebellar cortical involvement in excitatory and inhibitory classical conditioning. Stanford, CA: Stanford University Press; 1991. [Google Scholar]
- Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- Lyon MF, Searle AG. Genetic variant and strains of the laboratory mouse. 2nd ed. Oxford, UK: Oxford University Press; 1989. [Google Scholar]
- Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: Parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- Markowska AL, Koliatsos VE, Breckler SJ, Price DL, Olton DS. Human nerve growth factor improves spatial memory in aged but not in young rats. J Neurosci. 1994;14:4815–4824. doi: 10.1523/JNEUROSCI.14-08-04815.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowska AL, Price D, Koliatsos VE. Selective effects of nerve growth factor on spatial recent memory as assessed by a delayed nonmatching-to-position task in the water maze. J Neurosci. 1996;16:3541–3548. doi: 10.1523/JNEUROSCI.16-10-03541.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr D. A theory of cerebellar cortex. J Physiol. 1969;202:437–470. doi: 10.1113/jphysiol.1969.sp008820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moises HC, Womble MD, Washburn MS, Williams LR. Nerve growth factor facilitates cholinergic neurotransmission between nucleus basalis and the amygdala in rat: An electrophysiological analysis. J Neurosci. 1995;15:8131–8142. doi: 10.1523/JNEUROSCI.15-12-08131.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels JL, Qiao X, Bronson RT, Spencer C, Davisson MT. Stargazer: A new neurological mutant on chromosome 15 in the mouse with prolonged cortical seizures. Epilepsy Res. 1990;7:129–135. doi: 10.1016/0920-1211(90)90098-g. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: A stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Perrett SP, Ruiz BP, Mauk MD. Cerebellar cortex lesions disrupt learning-dependent timing of conditioned eyelid responses. J Neurosci. 1993;13:1708–1718. doi: 10.1523/JNEUROSCI.13-04-01708.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HS, Hains JM, Laramee GR, Rosenthal A, Winslow JW. Widespread expression of BDNF but not NT3 by target areas of basal forebrain cholinergic neurons. Science. 1990;250:290–294. doi: 10.1126/science.1688328. [DOI] [PubMed] [Google Scholar]
- Qiao X, Hefti F, Knusel B, Noebels JL. Selective failure of brain-derived neurotrophic factor mRNA expression in the cerebellum of stargazer, a mutant mouse with ataxia. J Neurosci. 1996;16:640–648. doi: 10.1523/JNEUROSCI.16-02-00640.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricceri L, Alleva E, Calamandrei G. Impairment of passive avoidance learning following repeated administrations of antibodies against nerve growth factor in neonatal mice. NeuroReport. 1994;5:1401–1404. [PubMed] [Google Scholar]
- Ringstedt T, Lagercrantz H, Persson H. Expression of members of the trk family in the developing postnatal rat brain. Brain Res Dev Brain Res. 1993;72:119–131. doi: 10.1016/0165-3806(93)90165-7. [DOI] [PubMed] [Google Scholar]
- Rocamora N, Garcia Ladona FJ, Palacios JM, Mengod G. Differential expression of brain-derived neurotrophic factor, neurotrophin-3, and low-affinity nerve growth factor receptor during the postnatal development of the rat cerebellar system. Brain Res Mol Brain Res. 1993;17:1–8. doi: 10.1016/0169-328x(93)90065-w. [DOI] [PubMed] [Google Scholar]
- Shibuki K, Gomi H, Chen L, Bao S, Kim JJ, Wakatsuki H, Fujisaki T, Fujimoto K, Katoh A, Ikeda T, Chen C, Thompson RF, Itohara S. Deficient cerebellar long-term depression, impaired eyeblink conditioning, and normal motor coordination in GFAP mutant mice. Neuron. 1996;16:587–599. doi: 10.1016/s0896-6273(00)80078-1. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, Barbacid M. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature. 1994;368:246–249. doi: 10.1038/368246a0. [DOI] [PubMed] [Google Scholar]
- Springer JE, Gwag BJ, Sessler FM. Neurotrophic factor mRNA expression in dentate gyrus is increased following in vivo stimulation of the angular bundle. Brain Res Mol Brain Res. 1994;23:135–143. doi: 10.1016/0169-328x(94)90219-4. [DOI] [PubMed] [Google Scholar]
- Stanton ME, Freeman JH, Jr, Skelton RW. Eyeblink conditioning in the developing rat. Behav Neurosci. 1992;106:657–665. doi: 10.1037//0735-7044.106.4.657. [DOI] [PubMed] [Google Scholar]
- Sweet HO. Waggler and stargazer are allelic. Mouse Genome. 1993;91:312–313. [Google Scholar]
- Sweet HO, Bronson RT, Cook S, Spencer CA. Waggler (wag) Mouse Genome. 1991;89:552. [Google Scholar]
- Thompson RF, Bao S, Chen L, Cipriano B, Grethe JS, Kim JJ, Thompson JK, Tracy J, Weninger MS, Krupa DJ. Associative learning. In: Schmahmann JD, editor. The cerebellum and cognition. New York, NY: Academic Press; 1997. pp. 151–189. [DOI] [PubMed] [Google Scholar]
- Yeo CH, Hardiman MJ, Glickstein M. Classical conditioning of the nictitating membrane response of the rabbit. II. Lesions of the cerebellar cortex. Exp Brain Res. 1985;60:99–113. doi: 10.1007/BF00237023. [DOI] [PubMed] [Google Scholar]