

Abstract
Allosteric effects in hemoglobin arise from the equilibrium between at least two energetic states of the molecule: a tense state, T, and a relaxed state, R. The two states differ from each other in the number and energy of the interactions between hemoglobin subunits. In the T state, constraints between subunits oppose the structural changes resulting from ligand binding. In the R state, these constraints are released, thus enhancing ligand-binding affinity. In the present work, we report the presence of four sites in hemoglobin that are structurally stabilized in the R relative to the T state. These sites are Hisα103(G10) and Hisα122(H5) in each α subunit of hemoglobin. They are located at the α1β1 and α2β2 interfaces of the hemoglobin tetramer, where the histidine side chains form hydrogen bonds with specific residues from the β chains. We have measured the solvent exchange rates of side chain protons of Hisα103(G10) and Hisα122(H5) in both deoxygenated and ligated hemoglobin by NMR spectroscopy. The exchange rates were found to be higher in the deoxygenated-T than in ligated-R state. Analysis of exchange rates in terms of the local unfolding model revealed that the structural stabilization free energy at each of these two histidines is larger by ≈1.5 kcal/(mol tetramer) in the R relative to the T state. The location of these histidines at the intradimeric α1β1 and α2β2 interfaces also suggests a role for these interfaces in the allosteric equilibrium of hemoglobin.
The Hb molecule has been a paradigm for understanding the molecular events underlying biological function. Cooperative oxygen-binding and allosteric effects have been rationalized by global thermodynamic models that postulate states of the molecule with specific energetic features (1–3). In the two-state model proposed by Monod, Wyman, and Changeux (2), the two energetic states are the tense (T) and the relaxed (R) states. The two states differ from each other in their affinity for ligand and in the interactions between subunits. Binding of oxygen to the deoxygenated (deoxy)-T state occurs with low affinity. However, the binding of the ligand destabilizes the T state and makes it more likely to switch to the high-affinity oxygenated (oxy)-R state. Thus, as ligation proceeds, the gradual enrichment in Hb molecules in the R state accounts for cooperativity in ligand binding. The success of this model in explaining the function of Hb has promoted its general application to a large variety of systems, including allosteric protein systems and nucleic acid-binding proteins (reviewed in refs. 4 and 5). However, the structural basis of this thermodynamic model is not yet fully understood.
The first insights into the molecular structures representing the T and R thermodynamic states of Hb have been provided by crystallographic results on deoxy and ligated Hb obtained by Perutz (6, 7). The deoxy-T and ligated-R structures maintain the same α1β1 and α2β2 interfaces within αβ dimers but differ greatly at the α1β2 and α2β1 interfaces between the two dimers. A network of intra- and intersubunit linkages is present at the amino and carboxy termini of α- and β-chains in the deoxy-T structure; these linkages break on ligand binding. Detailed analyses of these and other differences throughout the structures suggested a molecular mechanism by which binding of ligand at the heme groups promotes the breakage of intersubunit linkages and quaternary structure change (6). This mechanism has been a framework for analyzing oxygen-binding and allosteric effects in Hb in structural terms (7). Complete understanding of Hb function requires, however, the quantitative definition of the energetic changes involved in this structural mechanism. The structural pathways assumed by the molecule when binding ligand, once defined, must also be described by their free energy content and its changes.
Structural free energy changes in Hb have been measured by Ackers et al. by using oxygenation and tetramer–dimer equilibria (8) and by Englander et al. by using tritium–hydrogen exchange (9). Ackers et al. have defined the energetic contribution of individual sites to cooperativity by studying partially ligated intermediates of native Hb as well as of Hb molecules containing selected amino acid substitutions or modifications. The results revealed that the allosteric pathways assumed by Hb during ligand binding depend not only on the number of ligands bound but also on their distribution between the two αβ dimers (8, 10). The tritium–hydrogen exchange studies of Englander et al. have defined seven sets of allosterically sensitive peptide NH groups in Hb (11, 12). These sets contain 6–18 hydrogens each. In each set, the hydrogens exchange with solvent at similar rates. However, the exchange in each set is faster in the oxy than in the deoxy state by factors ranging from 15 to 10,000. Three of these sets of NH groups have been localized in the Hb structure near the amino termini of α-chains (13), at the carboxy termini of the β-chains (14), and at the F-FG helical segment in the β-chains (15). The enhancement in the exchange rates of these NH hydrogens on the transition from deoxy to oxy form reflects the local destabilization of the corresponding sites in the R relative to the T state (9, 14).
The present work extends these previous investigations in two directions. Firstly, by using NMR spectroscopy, we have extended the time scale of the proton exchange events observable in Hb. The tritium–hydrogen exchange method allows measurement of proton exchange rates lower than ≈0.02 s−1 (11, 12). The NMR method used in the present work measures exchange rates in the range from ≈5 to ≈300 s−1. Hence, these NMR proton exchange measurements probe a new window of structural fluctuations in Hb. Secondly, the resolution afforded by the NMR method allows us to monitor the exchange properties at four individual sites (two sites/αβ dimer). Although the number of sites that we observe is small, their exchange properties reveal a new pathway for the change in stabilization free energy on the T → R allosteric transition in Hb.
Materials and Methods
Hb Preparation.
All experiments reported in this work were carried out on human normal adult Hb (Hb A) that was purified from packed red blood cells by using the standard method (16) in the carbonmonoxy (CO) form. Several forms of ligated Hb A were investigated, i.e., HbCO A, oxy Hb A, CN-met Hb A, and azido-met Hb A. Oxy Hb A was prepared from HbCO A by photolysis of bound CO in the presence of oxygen. CN-met and azido-met Hb A were prepared as described (16). Samples in the deoxy form were obtained by flushing Hb solutions in the oxy form with nitrogen. All NMR samples were in 0.1 M Tris or Bis-Tris buffer containing 0.18 M chloride ions in 90%H2O/10%D2O. The Hb concentration ranged from 10 to 12%.
NMR Experiments.
The NMR experiments were performed at 37°C on a Varian INOVA 500 spectrometer operating at 11.75 T. Proton exchange rates were measured in experiments of transfer of magnetization from water. The water proton resonance was selectively inverted by using a Gaussian 180° pulse (6.4–6.8 ms), and the intensity of Hb proton resonances was measured as a function of the exchange delay, τ, after inversion. A weak gradient (0.21 G/cm) was applied during the delay τ to prevent the effects of radiation damping on the recovery of water magnetization to equilibrium. The observation was with a Jump-and-Return pulse sequence (17) modified as [90°(Φ)-t-90°(Φ)] to suppress the inverted water resonance. The dependence of the magnetization of an exchangeable proton on the delay τ is described by (18):
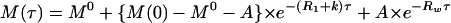 |
1 |
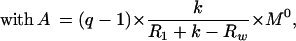 |
where M0 is the equilibrium magnetization,
R1 is the longitudinal relaxation
rate, and k is the exchange rate for the proton of interest;
Rw is the longitudinal relaxation rate
of water, and q =
Mw(0)/M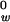 expresses the efficiency of inversion of the water magnetization (e.g.,
q = −1 for perfect inversion).
expresses the efficiency of inversion of the water magnetization (e.g.,
q = −1 for perfect inversion).
To minimize the effects of spin diffusion, the experiments were carried out for short exchange delays (τ ranging from 1 to 20 ms), and the initial-rate approximation of Eq. 1 was used:
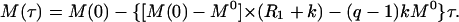 |
2 |
The magnetization of the exchangeable proton of interest was fitted as a function of the exchange delay τ to Eq. 2. M(0) was an independent parameter in the fit to allow for the small uncertainty in the definition of the exchange delay resulting from the length of the inversion pulse on water. Rw and q were measured in separated experiments. (R1 + k) values were measured in saturation–recovery experiments in which each exchangeable proton resonance of interest was selectively saturated and its recovery over delays τ ranging from 1 to 20 ms was monitored (18).
Analysis of Proton Exchange.
Proton exchange in proteins occurs by transient local unfolding of discrete segments in the structure. In the unfolding reaction, hydrogen bonds are transiently broken, and protons are moved into an open solvent accessible state. Exchange from this open state is catalyzed by H+ and/or OH− ions, water, or other catalysts present in the buffer (19, 20). The open state is short-lived and energetically unfavorable. Accordingly, the rate of exchange of a given proton is (19, 20):
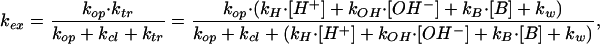 |
3 |
where kop is the rate constant for the opening reaction, and kcl is the rate constant for refolding into the native conformation. In the open state, kH and kOH are the rate constants for acid and base catalysis, respectively, kB is the rate constant for general buffer catalysis, and kw is the proton transfer rate for catalysis by water.
The experimental values for the exchange rates were fitted as a function of pH to Eq. 3 by using a nonlinear least-squares program. [H+] and [OH−] were expressed as a function of pH as: [H+] = 10−pH and [OH−] = 10(pH-pKw) with pKw = 13.622 at 37°C (21). The parameters independently determined from the fit were kop, kw/kOH and (kop + kcl)/kOH for base catalysis, and kop, kw/kH, kOH/kH and (kop+ kcl)/kH for acid/base catalysis. General base catalysis of the exchange by the buffer was checked in independent measurements at constant pH (pH 8.0) with Tris concentration increasing from 0.01 to 0.18 M. No significant changes in the exchange rates were observed (results not shown), indicating that the contribution of buffer catalysis to the exchange is negligible.
Results and Discussion
Proton NMR Resonances of Hisα103(G10) and Hisα122(H5).
The exchangeable proton resonances of Hb A that can be resolved in the NMR spectra downfield from ≈11 ppm are shown in Fig. 1. The two resonances of interest to the present work are those at ≈12.8 and ≈12.0 ppm. As Fig. 1 shows, these resonances are present in both deoxy and ligated Hb, and their spectral positions are practically constant on the deoxy-T → ligated-R transition (namely, their change in chemical shift is 0.2 ppm or less). Previous work has shown that the two resonances are not present in the NMR spectra of isolated α and β chains (22). Therefore, these resonances represent specific conformational markers for the tetrameric α2β2 Hb structure. The resonance at 12.0 ppm has been assigned to the side chain Nɛ2H group of Hisα103(G10), which is hydrogen-bonded to the side chain carbonyl group of Glnβ131(H9) [or to the peptide carbonyl of Asnβ108(G10)] at the α1β1 and α2β2 interfaces (22–24). The resonance at 12.8 ppm has been assigned to the side chain Nɛ2H group of Hisα122(H5), which forms a water-mediated intersubunit hydrogen bond with the side chain of Tyrβ35(C1) at the α1β1 and α2β2 interfaces (25).
Figure 1.

Exchangeable proton NMR resonances of Hb A in deoxy form (Lower) and CO form (Upper) in 0.1 M Bis-Tris buffer containing 0.18 M chloride ions at pH 7.1 and at 37°C.
Mechanism of Exchange of Nɛ2H protons of Hisα103(G10) and Hisα122(H5).
The rates of exchange with solvent for the Nɛ2H protons of Hisα103(G10) and Hisα122(H5) as a function of pH are shown in Figs. 2 and 3, respectively. As the data show, the exchange is acid and/or base catalyzed. In contrast, for free imidazole or for exposed histidine side chains in proteins, only base catalysis is observed, over the pH range above the imidazole's pKa (pKa≈6.5) (26). At pH values lower than the pKa, the exchange is limited by the off-rate of the NH protons and thus is pH-independent (26). Acid catalysis in exchange of histidine NH protons is observed in proteins when the histidine residue is buried in the protein structure in its neutral Nɛ2H tautomeric form and the protonation of the neutral histidine in the open state is the rate-limiting step in the exchange (27). The two histidine residues investigated here should be uncharged in the native Hb structure because of their location at the interface between α and β subunits in the αβ dimers. This suggestion is confirmed by the pH dependence of their Cɛ1- and Cδ2-proton resonances obtained from nuclear Overhauser effect (NOE) experiments. The Cɛ1 and Cδ2 protons of Hisα103(G10) and Hisα122(H5) are not directly observable in the one-dimensional NMR spectrum of Hb because of the severe overlap of their resonances with other aromatic proton resonances. However, as shown previously (22), these protons can be observed as NOEs from the histidine Nɛ2H protons (i.e., resonances at 12.0 and 12.8 ppm, respectively). Using this approach, in the present work we have monitored the Cɛ1 and Cδ2 protons of the two histidine residues of interest as a function of pH over the pH range from 5.6 to 9.0. We have found that the spectral positions of these Cɛ1 and Cδ2 protons are constant, indicating that in this pH range, Hisα103(G10) and Hisα122(H5) do not change their protonation state and, as expected, their pKa values in the native Hb structure are probably very low. This finding suggests that acid catalysis of the Nɛ2H exchange is observed for Hisα103(G10) (Fig. 2), because the protonation step in the open state is slow and rate limiting. In contrast, for Hisα122(H5), the open state may be such that the protonation of the neutral histidine is fast and, as for imidazole, the exchange rates are pH independent (Fig. 3). Another interesting feature of the pH dependence of the proton exchange observed in this work is that, for the Hisα103(G10) Nɛ2H proton, base catalysis of the exchange is greatly diminished (Fig. 2). In the deoxy-T state, the rate constant for base catalysis kOH is smaller than that for acid catalysis kH by a factor of ≈150 [kOH/kH = (6.0 ± 1.1) 10−3]. In the ligated-R state, base catalysis is practically absent (kOH/kH = 0). It is unlikely that this effect originates from a pH-induced conformational change in the Hb molecule for two reasons. First, as described above, the chemical shifts of the histidine Cɛ1 and Cδ2 protons are constant over the pH range of interest. Second, the longitudinal relaxation rates of the histidine Nɛ2H proton (measured as described in Materials and Methods) are also constant (i.e., R1 ≈22 s−1 in deoxy form, and R1 ≈25 s−1 in ligated form). Therefore, it is more likely that catalysis of the exchange by OH− is suppressed by effects of the negative overall charge of Hb and/or of the local electrostatic potential at Hisα103(G10) at pH values higher than ≈7.2 (28). Similar effects on base catalysis of exchange of peptide hydrogens in proteins have been previously observed (20, 29).
Figure 2.

Dependence of the exchange rate of Hisα103(G10) Nɛ2H proton on pH (A), and on the concentration of H+ ions in the pH range from 5.6 to 7.6 (B). (♦, ⋄) deoxy Hb; (●, ○) HbCO; (▾, ▿) oxy Hb; (□) CN-met Hb; (▵) azido-met Hb. Filled symbols: 0.1 M Bis-Tris with 0.18 M chloride ions. Open symbols: 0.1 M Tris with 0.18 M chloride ions. The solid lines correspond to nonlinear least-squares fits to Eq. 3 with the following fitted parameters: kop = (105 ± 16) s−1, kOH/kH = (6.0 ± 1.1) 10−3, kw/kH = (1.2 ± 0.4) 10−7 M and (kop + kcl)/kH = (6.2 ± 2.6) 10−7 M for deoxy Hb A, and kop = (76 ± 5) s−1, kOH/kH = 0, kw/kH = (9.4 ± 1.4) 10−8 M and (kop + kcl)/kH = (1.6 ± 0.2) 10−6 M for ligated Hb A.
Figure 3.

Dependence of the exchange rate of Hisα122(H5) Nɛ2H proton on pH (A) and on concentration of OH− ions (B). (♦, ⋄) deoxy Hb; (●, ○) HbCO; (▾, ▿) oxy Hb; (□) CN-met Hb; (▵) azido-met Hb. Filled symbols: 0.1 M Bis-Tris with 0.18 M chloride ions. Open symbols: 0.1 M Tris with 0.18 M chloride ions. Solid lines correspond to nonlinear least-squares fits to Eq. 3 with kH = 0 and the following fitted parameters: kop = (63 ± 2) s−1, kw/kOH = (5.7 ± 0.8) 10−7 M and (kop + kcl)/kOH = (7.2 ± 0.8) 10−6 M for deoxy Hb A, and kop = (19 ± 3) s−1, kw/kOH = (1.6 ± 0.6) 10−6 M and (kop+ kcl)/kOH = (6.6 ± 4.5) 10−6 M for ligated Hb A.
Allosteric Free Energy Changes at Hisα103(G10) and Hisα122(H5) in Hb.
The outstanding trend in the data presented in Figs. 2 and 3 is that the exchange rates of Hisα103(G10) and Hisα122(H5) are higher in deoxy than in ligated Hb. Moreover, in ligated Hb, the exchange rates are independent of the nature of the ligand and, for all ligands investigated, the exchange rates at a given pH are the same. Therefore, the exchange rates observed in ligated Hb are a general property of the R state of Hb.
In the framework of the local unfolding model, the differences in
exchange rates between deoxy-T and ligated-R states can be rationalized
in terms of changes in the structural stabilization free energies at
Hisα103(G10) and at Hisα122(H5). The structural stabilization free
energy is defined by the free energy change of the unfolding reaction,
ΔG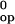 that, in turn, determines the equilibrium
constant for the reaction, Kop:
that, in turn, determines the equilibrium
constant for the reaction, Kop:
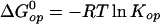 |
4 |
where Kop = kop/kcl. Accordingly, the gain in structural stabilization free energy at each site of interest on ligand binding to Hb is:
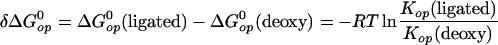 |
5 |
The exchange data allow unambiguous determination of the opening
rate, kop because, as shown in Figs.
2B and 3B, the exchange rates extend into
the EX1 regime where kex approaches
kop (Eq. 3). However,
determination of the closing rate,
kcl, requires the rate constants for
proton transfer in the open state, kH
and kOH. These rate constants can be
influenced by steric and electrostatic features of the open state,
which are not known (20, 30). Therefore, to estimate δ
ΔG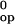 , we have made the following two assumptions:
(i) the rate constants kH
for Hisα103(G10) and kOH for
Hisα122(H5) are pH-independent, and their values are similar to those
in model compounds
(109–1010
M−1 s−1) (27, 31), and
(ii) these rate constants are the same in deoxy and in
ligated Hb A. With these assumptions, the changes in proton exchange
rates correspond to changes in stabilization free energy of ≈1.6
kcal/(mol tetramer) for Hisα103(G10) and ≈1.4 kcal/(mol
tetramer) for Hisα122(H5). In both cases, the stabilization free
energies are larger in the ligated-R than in the deoxy-T state.
, we have made the following two assumptions:
(i) the rate constants kH
for Hisα103(G10) and kOH for
Hisα122(H5) are pH-independent, and their values are similar to those
in model compounds
(109–1010
M−1 s−1) (27, 31), and
(ii) these rate constants are the same in deoxy and in
ligated Hb A. With these assumptions, the changes in proton exchange
rates correspond to changes in stabilization free energy of ≈1.6
kcal/(mol tetramer) for Hisα103(G10) and ≈1.4 kcal/(mol
tetramer) for Hisα122(H5). In both cases, the stabilization free
energies are larger in the ligated-R than in the deoxy-T state.
The molecular basis for the enhanced stabilization free energy at these two sites in the R state could be inferred from a comparison of the crystallographic structures of Hb in deoxy and ligated forms. For example, in deoxy Hb A, Hisα103(G10) makes a hydrogen bond with Glnβ131(H9), and five of its atoms are involved in contacts with this and other residues (32). In contrast, in horse met Hb, 16 atoms contribute to the contacts made by Hisα103(G10). These include contacts with Glnβ131(H9), Glnβ127(H5), Cysβ112(G14), Valβ111(G13), Asnβ108(G10), and a water molecule at the interface. The larger number of contact atoms for Hisα103(G10) in ligated as compared with deoxy Hb is expected to result in a larger stabilization free energy of this residue, in accordance with the present results.
The proton exchange rates and stabilization free energies obtained in this work further refine the energetic picture of Hb deduced previously by Englander et al. by using tritium–hydrogen exchange methods. According to these previous results, seven sets of allosterically sensitive hydrogens exist in Hb A (11, 12). These hydrogens represent approximately one-fourth of all peptide hydrogens in the molecule and, for all of them, the stabilization free energies are larger in deoxy-T than in ligated-R state. Our results reveal that, in addition to these sites, there are at least four other sites in the Hb A tetramer for which local stabilization energies are larger in the ligated-R than in the deoxy-T state. The structural fluctuations at these sites occur on a fast time scale (i.e., in the range of milliseconds to seconds; Figs. 2 and 3), explaining why they could not have been detected by tritium–hydrogen exchange measurements.
Role of α1β1/α2β2 Interfaces in the Allosteric Mechanism of Hb.
The crystallographic structures of deoxy and ligated Hb have shown that, in contrast to the interdimeric α1β2 and α2β1 interfaces, the intradimeric α1β1 and α2β2 interfaces remain essentially unchanged on the T → R transition (7, 33, 34). This structural equivalence has been regarded as evidence that the α1β1/α2β2 interfaces play no important role in the cooperative oxygen binding to Hb and in allosteric effects. A series of recent findings, however, suggest that, despite this structural equivalence, the α1β1 and α2β2 interfaces are not functionally inert. Ackers et al. have demonstrated that, when the second ligand binds to Hb, the microscopic species in which both ligands are on the same αβ dimer is energetically preferred over all other doubly ligated species (8, 35). This result implies the existence of favorable cooperativity between α and β hemes within the same αβ dimer, before the T → R transition. The intradimeric α1β1/α2β2 interfaces are most likely involved in this energetic coupling. Further evidence for a role of α1β1/α2β2 interfaces in the function of Hb has been provided by studies of Hb molecules containing amino acid substitutions at these interfaces. For example, mutating one of the residues in contact with Hisα103(G10), namely Cysβ112(G14) → Gly, increases the dimer–tetramer association constant 4-fold (36). This result suggests that structural changes at the α1β1/α2β2 interfaces can propagate to the α1β2/α2β1 interfaces and thus can affect the allosteric equilibrium. Support for this suggestion has been provided by Ho and coworkers, who found that Asnβ108(G10) → Lys and Asnβ108(G10) → Glu substitutions at the α1β1/α2β2 interfaces favor the T state relative to the R state, at low temperature and in the presence of allosteric effectors (24). Moreover, these two amino acid substitutions have been shown to have profound effects on the Bohr effect and on the linkage between oxygen and chloride ions (24). Therefore, it appears that the role of the α1β1/α2β2 interfaces in the Hb function is not negligible. Our present results suggest that this role could be associated with changes in the stabilization free energy of specific amino acid residues at these interfaces on the T → R transition. Correlation of these energetic changes with Hb function awaits characterization of the allosteric free energy changes in Hb molecules with single-site structural changes at the α1β1/α2β2 interfaces.
Acknowledgments
This work has been supported by a grant from the American Heart Association.
Abbreviations
- T
tense
- R
relaxed
- Hb A
human normal adult Hb
- deoxy
deoxygenated
- oxy
oxygenated
- CO
carbonmonoxy
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Koshland D E, Jr, Nemethy G, Filmer D. Biochemistry. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 2.Monod J, Wyman J, Changeux J-P. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 3.Wyman J, Gill S J. Binding and Linkage: Functional Chemistry of Biological Macromolecules. Mill Valley, CA: Univ. Sci. Books; 1990. [Google Scholar]
- 4.Perutz M F. Mechanisms of Cooperativity and Allosteric Regulation in Proteins. Cambridge, U.K.: Cambridge Univ. Press; 1990. [DOI] [PubMed] [Google Scholar]
- 5.Jardetzky O. Prog Biophys Mol Biol. 1996;65:171–219. doi: 10.1016/s0079-6107(96)00010-7. [DOI] [PubMed] [Google Scholar]
- 6.Perutz M F. Nature (London) 1970;228:726–739. doi: 10.1038/228726a0. [DOI] [PubMed] [Google Scholar]
- 7.Perutz M F. Annu Rev Physiol. 1990;52:1–25. doi: 10.1146/annurev.ph.52.030190.000245. [DOI] [PubMed] [Google Scholar]
- 8.Ackers G K. Adv Protein Chem. 1998;51:185–253. doi: 10.1016/s0065-3233(08)60653-1. [DOI] [PubMed] [Google Scholar]
- 9.Englander S W, Englander J J, McKinnie R E, Ackers G K, Turner G J, Westrick J A, Gill S J. Science. 1992;256:1684–1687. doi: 10.1126/science.256.5064.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LiCata V J, Dalessio P M, Ackers G K. Proteins Struct Funct Genet. 1993;17:279–296. doi: 10.1002/prot.340170306. [DOI] [PubMed] [Google Scholar]
- 11.Malin E L, Englander S W. J Biol Chem. 1980;255:10695–10701. [PubMed] [Google Scholar]
- 12.Rogero J R, Englander J J, Englander S W. Methods Enzymol. 1986;131:508–517. doi: 10.1016/0076-6879(86)31053-x. [DOI] [PubMed] [Google Scholar]
- 13.Ray J, Englander S W. Biochemistry. 1986;25:3000–3007. doi: 10.1021/bi00358a040. [DOI] [PubMed] [Google Scholar]
- 14.Louie G, Tran T, Englander J J, Englander S W. J Mol Biol. 1988;201:755–764. doi: 10.1016/0022-2836(88)90472-x. [DOI] [PubMed] [Google Scholar]
- 15.Englander J J, Rogero J R, Englander S W. J Mol Biol. 1983;169:325–344. doi: 10.1016/s0022-2836(83)80186-7. [DOI] [PubMed] [Google Scholar]
- 16.Antonini E, Brunori M. Hemoglobin and Myoglobin in Their Reactions with Ligands. Amsterdam: North–Holland; 1971. [Google Scholar]
- 17.Plateau P, Gueron M. J Am Chem Soc. 1982;104:7310–7311. [Google Scholar]
- 18.Ernst R R, Bodenhausen G, Wokaun A. Principles of Nuclear Magnetic Resonance in One and Two Dimensions. Oxford, U.K.: Clarendon; 1987. [Google Scholar]
- 19.Hvidt A, Nielsen S O. Adv Protein Chem. 1966;21:287–386. doi: 10.1016/s0065-3233(08)60129-1. [DOI] [PubMed] [Google Scholar]
- 20.Englander S W, Kallenbach N R. Quart Rev Biophys. 1984;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 21.Weast R C. CRC Handbook of Chemistry and Physics. Boca Raton, FL: CRC; 1986. [Google Scholar]
- 22.Russu I M, Ho N T, Ho C. Biochem Biophys Acta. 1987;914:40–48. doi: 10.1016/0167-4838(87)90159-2. [DOI] [PubMed] [Google Scholar]
- 23.Ho C. Adv Protein Chem. 1992;43:153–312. doi: 10.1016/s0065-3233(08)60555-0. [DOI] [PubMed] [Google Scholar]
- 24.Tsai C-H, Shen T-J, Ho N T, Ho C. Biochemistry. 1999;38:8751–8761. doi: 10.1021/bi990286o. [DOI] [PubMed] [Google Scholar]
- 25.Simplaceanu V, Lukin J A, Fang T-Y, Zou M, Ho N T, Ho C. Biophys J. 2000;79:1146–1154. doi: 10.1016/S0006-3495(00)76368-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eigen M. Angew Chem Int Ed Engl. 1964;3:1–19. [Google Scholar]
- 27.Connelly G P, McIntosh L P. Biochemistry. 1998;37:1810–1818. doi: 10.1021/bi972085v. [DOI] [PubMed] [Google Scholar]
- 28.Matthew J B, Hanania G I H, Gurd F R N. Biochemistry. 1979;18:1919–1928. doi: 10.1021/bi00577a011. [DOI] [PubMed] [Google Scholar]
- 29.Delepierre M, Dobson C M, Karplus M, Poulsen F M, States D J, Wedin R E. J Mol Biol. 1987;197:111–130. doi: 10.1016/0022-2836(87)90613-9. [DOI] [PubMed] [Google Scholar]
- 30.Pedersen T G, Thomsen N K, Andersen K V, Madsen J C, Poulsen F M. J Mol Biol. 1993;230:651–660. doi: 10.1006/jmbi.1993.1176. [DOI] [PubMed] [Google Scholar]
- 31.Bai Y, Milne J S, Mayne L, Englander S W. Proteins Struct Funct Genet. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fermi G, Perutz M F. Haemoglobin and Myoglobin. Oxford, U.K.: Clarendon; 1981. [Google Scholar]
- 33.Baldwin J, Chothia C. J Mol Biol. 1979;129:175–220. doi: 10.1016/0022-2836(79)90277-8. [DOI] [PubMed] [Google Scholar]
- 34.Perutz M F, Fermi G, Luisi B, Shaanan B, Liddington R C. Acc Chem Res. 1987;20:309–321. doi: 10.1101/sqb.1987.052.01.063. [DOI] [PubMed] [Google Scholar]
- 35.Ackers G K, Holt J M, Huang Y, Grinkova Y, Klinger A L, Denisov I. Proteins Struct Funct Genet. 2000;4:23–43. doi: 10.1002/1097-0134(2000)41:4+<23::aid-prot30>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 36.Fronticelli C, Gattoni M, Lu A-L, Brinigar W S, Bucci J L G, Chiancone E. Biophys Chem. 1994;51:53–57. doi: 10.1016/0301-4622(94)00028-x. [DOI] [PubMed] [Google Scholar]