

Abstract
We introduce a computational method to optimize the in vitro evolution of proteins. Simulating evolution with a simple model that statistically describes the fitness landscape, we find that beneficial mutations tend to occur at amino acid positions that are tolerant to substitutions, in the limit of small libraries and low mutation rates. We transform this observation into a design strategy by applying mean-field theory to a structure-based computational model to calculate each residue's structural tolerance. Thermostabilizing and activity-increasing mutations accumulated during the experimental directed evolution of subtilisin E and T4 lysozyme are strongly directed to sites identified by using this computational approach. This method can be used to predict positions where mutations are likely to lead to improvement of specific protein properties.
Keywords: in vitro directed evolution, computational protein design, combinatorial optimization, mean-field theory, protein tolerance
As techniques to alter the properties of proteins, directed evolution and computational design have matured separately. The aim of directed evolution is to accumulate stepwise improvements by iterations of random mutagenesis and screening (1, 2). As a fundamentally different approach, the objective of computational protein design (3) is to solve the inverse folding problem by using a force field paradigm that describes the interactions between amino acids and by then computing the globally optimal amino acid sequence (4, 5). Directed evolution has the benefit of improving any enzyme property that can be captured by a screen; however, the search is restricted by the number of mutants that can be experimentally screened at each generation (≈103–106). Conversely, computational design can effectively search a much larger number of sequences (>1026) (4) but is limited as to the size of the protein and is currently restricted to calculating the stabilization energy. This report introduces an approach to protein engineering in which computational design is used as a guide to focus an evolutionary search, thus combining the benefits of both design strategies.
An effective and widely used directed evolution strategy is to produce a library of mutants from a parent sequence through random point mutagenesis by using error-prone PCR (1, 2). The usual practice of mutagenizing the whole gene has several problems. The probability that any single random mutation improves a property is small, and the probability of improvement decreases rapidly when multiple simultaneous mutations are made. Therefore, the limited number of mutants that can be screened imposes a low upper limit on the mutation rate (6). Furthermore, the negligible probability that two or three mutations occur in a single codon and the significant biases of error-prone PCR severely restrict the possible amino acid substitutions. These effects can be overcome by intensely mutagenizing a limited number of positions (7–9). The challenge, however, is to identify the residues where such experiments are likely to be beneficial, as beneficial mutations often appear far from sites that would be predicted heuristically (e.g., catalytic sites) (1, 2). In this report, we first use a simple fitness model to demonstrate that positive mutations preferentially occur at residue positions that contribute independently to the fitness. Next, we use a detailed structural model to transform this observation into a design strategy.
Materials and Methods
Force Field and Rotamer Library.
The energy term consists of two contributions: rotamer/backbone e(ir) and rotamer/rotamer e(ir,js):
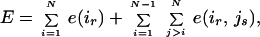 |
1 |
where N is the number of residues and ir is rotamer r at position i. Because the backbone remains fixed, its internal energy contribution is not relevant to the optimization procedure. Note that fitness is the negative of energy: F = −E. Potential functions and parameters for van der Waals interactions, hydrogen bonding, and electrostatics are described in previous work (10, 11). We use the DREIDING force field parameters for the atomic radii and internal coordinate parameters (12). The van der Waals energies are modeled by using a 6 − 12 Leonard–Jones potential with an additional 0.9 scale factor applied to the atomic radii to soften the lack of flexibility implied by the fixed backbone and the rotamer descriptions. A ceiling of 500 kcal/mol was set for the rotamer/rotamer energies to avoid unhindered van der Waals contributions and to expedite mean-field convergence. All rotamer/backbone and rotamer/rotamer energies are computed and stored before the mean-field calculation, requiring 165 (113) minutes for subtilisin E (T4 lysozyme) on 10 Silicon Graphics (Mountain View, CA) R10000 processors running at 195 MHz.
The rotamer library is backbone-dependent as described by Dunbrack and Karplus (13, 14). The following modifications were included, as previously described (15). The χ3 angles that were undetermined from the database statistics were assigned the values: Arg, −60°, 60°, and 180°; Gln, −120°, −60°, 0°, 60°, 120°, and 180°; Glu, 0°, 60°, and 120°; Lys, −60°, 60°, and 180°. The χ4 angles that were undetermined from the database statistics were assigned the following values: Arg, −120°, −60°, 60°, 120°, and 180°; Lys, −60°, 60°, and 180°. Rotamers with combination of χ3 and χ4 resulting in sequential g+/g− or g−/g+ angles were eliminated.
Rotamers that interact with the backbone with energies greater than 5 kcal/mol (subtilisin E) and 20 kcal/mol (T4 lysozyme) are eliminated from the calculation. The amino acids at residues 1–4 and 269–274 of subtilisin E are fixed in their wild-type conformations. For subtilisin E, an average of 121 rotamers per residue are considered, corresponding to 3.2 × 104 one-body energies, 5.1 × 108 two-body energies, and a rotamer space of 10497 combinations. For T4 lysozyme, an average of 176 rotamers per residue are considered, corresponding to 2.9 × 104 one-body energies, 4.1 × 108 two-body energies, and a rotamer space of 10384 combinations.
Mean-Field Theory.
The mean-field solution of Eq. 1 is
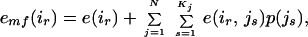 |
2 |
where emf(ir) is the mean-field energy felt by rotamer r at position i and Kj is the total number of rotamers at residue j (16–18). We can calculate the probability vector p(js) at some temperature T using the self-consistent equations
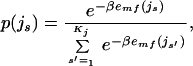 |
3 |
where β = 1/kBT, where kB is Boltzmann's constant. The probabilities are initially set to 1/Kj and the mean-field energies are calculated from Eq. 2 for each residue. The algorithm iterates between Eqs. 2 and 3 until self-consistency is achieved. Convergence is significantly improved if the probability vector p is updated with a memory of the previous step as described by Lee (16). An initially high temperature (50,000 K) is set, and the convergence algorithm is repeated as the temperature is lowered in increments of 100 K until the final temperature (600 K for subtilisin E and 300 K for T4 lysozyme) is reached. The final temperature corresponds with an estimated energy above which the structural stability is compromised. The sequence entropy at this temperature effectively counts the number of sequences that are stable in the fixed backbone. The mean-field solution of subtilisin E (T4 lysozyme) required 8,900 (6,402) minutes on a single Silicon Graphics R10000 Processor running at 195 MHz and 2.1 gigabytes of physical memory.
Results and Discussion
Simulations on a Generic Fitness Landscape.
The sequence space consists of all amino acid combinations for a fixed sequence length, connected through mutational moves (19). Each sequence has a corresponding fitness, representing the combination of properties (e.g., activity and stability) undergoing selection. The combination of sequence space and a fitness description constitutes the fitness landscape, the structure of which determines the difficulty of an evolutionary search (20, 21). Very rugged landscapes contain many local optima, creating a very difficult optimization problem. The underlying cause of ruggedness is coupling between residues. Coupled residues must be optimized simultaneously, whereas uncoupled ones could be optimized independently and combined. Coupling is experimentally observed as nonadditivity, in which the free energy contribution of multiple mutations does not equal the sum of the individual contributions from each mutation (22). Residues that are weakly coupled are tolerant to amino acid substitution (23, 24). The simplest description of the fitness landscape that captures the effect of coupling is to add a two-body term to an uncoupled fitness contribution (24),
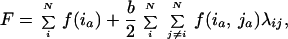 |
4 |
where N is the number of residues, ia is the amino acid identity at residue i, f(ia) is the contribution of ia to the fitness, and b determines the relative strength of coupled versus uncoupled interactions. If residues i and j are coupled, λij = 1; otherwise, λij = 0. Fitness approximations with one- and two-body terms have been used previously to model thermostability (4, 5, 24–26) and catalytic activity (27).
To investigate how coupling influences an evolutionary search, a hypothetical fitness landscape was generated by the random assignment of fitness contributions f(ia) from a Gaussian distribution and random placement of coupling interactions λij between residues. The directed evolution algorithm of mutagenesis and screening was then simulated at different fitness heights on the landscape. Mutations were made on the DNA level and then transcribed to the amino acid level. A mutation rate of three nucleotide substitutions (corresponding to an average of one amino acid substitution) per gene was applied to a N = 50-aa residue sequence. During each generation, 3,000 mutants were screened, and the coupling of the positions where mutations occurred on the most improved mutant was recorded.
We find that the probability of a positive mutation occurring at a highly coupled residue decreases significantly as the fitness of the parent increases (Fig. 1). The bias toward mutating uncoupled positions late in evolution is a result of the finite sampling size of the screening step. A highly coupled group of residues requires several simultaneous mutations to demonstrate improvement. When a mutation is made at a coupled residue, it is necessary to improve all of the coupled terms in addition to the uncoupled term, the probability of which rapidly decreases as the sequence becomes more highly optimized. This result is independent of the specific form of Eq. 4 and can be demonstrated by using any model that incorporates a variable degree of coupling between residues [such as Kauffman's NK-model (21), lattice proteins (26, 28), or RNA secondary structure models (29)].
Figure 1.

The probability distribution p(c) that a positive mutation occurs at a residue with c coupled interactions. The distribution is shown at two fitness values as the sequence ascends the fitness landscape, F = 0.0 (○) and F = 17.0 (▴). Data shown are for N = 50, b = 10.0, and 50 coupling interactions. The coupling is symmetric so two residues are affected for each interaction.
Calculating the Tolerance of Protein Structures.
As a strategy for directed evolution, concentrating mutagenesis on the regions of weak coupling reduces the search space to the positions that are most likely to show improvement. We can extend this result from the simple model to make experimentally relevant predictions by using a detailed protein design model that calculates the stabilizing energy of a sequence folded onto a fixed backbone (4, 5) to determine the coupling of each residue. The protein backbones of subtilisin E (274 amino acids) and T4 lysozyme (164 amino acids) were retrieved from high-resolution crystal structures (30, 31), and the interactions between residues were calculated by coarse-graining the flexibility of each amino acid into rotamers and constructing a force field to calculate the rotamer/backbone and rotamer/rotamer stabilizing energies (see Materials and Methods). An initial elimination of rotamers makes the problem computationally tractable; however, the combinatorial complexity remains enormous. The sequence space considered is hyperastronomically large: 10343-aa combinations for subtilisin E and 10214-aa combinations for T4 lysozyme. Searching the entire space for the global optimum is intractable both computationally and experimentally.
To circumvent the combinatorial difficulties, we apply statistical mechanics to determine the coupling of each position, using structural tolerance toward amino acid substitutions as a measure of the coupling. Structural tolerance is crucial for the success of directed evolution. Maintaining structure is required for the acquisition or fine-tuning of any other property, leading to the suggestion that properties such as stability and activity are correlated (32). The effect of structural tolerance is to increase the probability that a mutation is not destabilizing. Therefore, a structurally tolerant protein has a larger number of allowed mutations that can potentially improve a property, making it more likely that there is a connected path in sequence space of single mutations that leads to regions of higher fitness. By reducing the evolutionary search to regions of sequence space that are consistent with the structure, functional space can be more thoroughly explored.
Structural tolerance can be quantitated by counting the number of sequences (states) Ω compatible with a stabilization energy, defined as the sequence entropy, S(E) = kB ln Ω (24). As the energy is lowered, the number of compatible sequences decreases, thus decreasing the entropy. The site entropy is determined by the variability of the amino acid identity among the sequences consistent with an energy and is calculated from the probability p(ia) that an amino acid identity ia exists at site i,
 |
5 |
where A is total number of amino acids and kB is chosen to be 1. The amino acid probabilities are calculated as the sum of the amino acid's rotamer probabilities, as determined by mean-field theory (details of this computation are given in Materials and Methods). If the probabilities in Eq. 5 were based solely on the rotamers of the wild-type amino acid identity, then the site entropy would be a direct measure of the side chain flexibility. However, we are tabulating the probabilities of the existence of all amino acids at all positions and condensing this information into the site entropy. Therefore, the site entropy is a measure of the number of amino acid substitutions that can be made at each residue without disrupting the structure. A residue intolerant to mutations has a low entropy, whereas a tolerant residue has high entropy. A tabulation of the entropy at each position produces the entropy profile of subtilisin E shown in Fig. 2 and the distribution of site entropies of subtilisin E and T4 lysozyme shown in Fig. 3.
Figure 2.

The predicted sequence entropy profile (black line) and solvent accessibility (red line) for subtilisin E. If all amino acids are equally likely, then si = ln A ≈ 3.0. The solvent accessibility is the percent side chain surface area exposed, as calculated by the Lee and Richards method with a solvent radius of 1.4 Å (41).
Figure 3.

The probability distribution of site entropies p(si) for subtilisin E and T4 lysozyme. The bar indicates the mean and standard deviation of the distribution. The fraction of frozen residues are 0.078 and 0.039, as indicated by the arrows. The site entropies of positions where experimental directed evolution found positive mutations are indicated by the lines. (A) Mutations found from the in vitro evolution of subtilisin. (Top) Mutations made when the screen was to improve thermostability while retaining activity (6). From left to right, the positions (entropies) are 181 (0.36), 166 (0.96), 118 (2.37), 76 (2.45), 14 (2.50), 218 (2.54), 9 (2.55), 194 (2.59), and 161 (2.69). (Bottom) Mutations made when the screen was to improve activity toward s-AAPF-pNa in the organic solvent dimethyl formamide (33, 34). From left to right, the positions (entropies) are 181 (0.36), 107 (1.62), 182 (1.81), 206 (1.94), 156 (2.19), 131 (2.43), 188 (2.50), 218 (2.54), 255 (2.54). Note that residues 181 and 218 are common to both data sets (different amino acid substitutions were made at residue 181, whereas the same substitution was made at 218). In both studies, the mutations were found by screening 2,000–5,000 mutants generated with an average mutation rate of 2–3 nucleotide substitutions. (B) Mutations found during the evolution of T4 lysozyme (35). The red bars indicate mutations that improved stability, blue bars indicate mutations that improved activity, and purple bars indicate mutations that improved both properties. From left to right, the positions (entropies) are 153 (0.55), 26 (1.03), 151 (1.53), 22 (1.66), 41 (1.91), 16 (2.02), 147 (2.10), 119 (2.11), 163 (2.49), 116 (2.50), 93 (2.52), 113 (2.54), 40 (2.54), and 14 (2.59).
Correlation with Directed Evolution Experiments.
To test our prediction that beneficial mutations are made by directed evolution at structurally tolerant positions, we compared our calculations with mutations found from previous evolution experiments on subtilisin E (6, 33, 34) and T4 lysozyme (35) (see Materials and Methods). Seven of the nine mutations that improved the thermostability of subtilisin E occur at positions computed to be highly tolerant (Fig. 3A, red bars). The stabilizing mutations discovered by the evolution of T4 lysozyme also preferentially occur at the high-entropy positions (Fig. 3B). Thus, for both enzymes, the entropy predictions would aid an evolutionary search to improve thermostability, indicating that the computational method is valid independent of the specific protein or experimental protocol.
In directed evolution, improvement of properties other than stability is often desired. If the desired property is correlated with stability, then the structure-based entropy predictions will be more accurate. For instance, it has been suggested that improving thermostability is a good approach for enhancing activity at high temperatures (6, 36). When libraries of subtilisin E mutants were screened for improved thermostability while retaining activity, some mutations improved both properties. In addition, activity and stability are highly correlated in the screen used for T4 lysozyme; thus, activity-improving mutations also occur at highly tolerant positions. There is a weaker correlation with improving the activity of subtilisin E in organic solvent (Fig. 3A, blue bars), implying that retention of structure is less important. However, the mutations are still strongly biased toward the high entropy positions.
The site entropy profile is mapped onto the subtilisin E structure in Fig. 4. There is a trend toward the most variable sites being on the surface and the more conserved being in the core of the protein. However, the correlation between the entropy and solvent accessibility is poor (R2 = 0.55 for subtilisin E and 0.54 for T4 lysozyme; data in Fig. 2). The computed site entropies are derived from the fundamental physical features that lead to tolerance, whereas solvent accessibility is a secondary measure. The site entropy captures details of structural tolerance beyond solvent accessibility, including side chain packing, the coupling of backbone and side chain conformations, electrostatic interactions required by the backbone conformation, and a residue's local environment, and is therefore a better measure of tolerance.
Figure 4.

The structure of subtilisin E showing the entropy at each position. The yellow residues are the most variable sites (2.16 < s < 3.00, greater than one standard deviation above the mean), the red residues are moderately variable (1.31 < s < 2.16, between the mean and one standard deviation), and the gray residues have below average variability (s < 1.31). Site saturation experiments should be directed at yellow positions, whereas the contiguous yellow–red regions lend themselves to cassette mutagenesis. Figure generated by using molmol (40).
A comparison is made in Table 1 between the site entropies and solvent accessibilities of the positions where positive mutations were found. Site entropy predicts that certain positions with low solvent accessibility can have a high tolerance. Several specific residues have a high site entropy but a low solvent accessibility, which demonstrate the physical principles underlying our method. For example, residue 107 in subtilisin E has an above-average site entropy (1.62) but a very low solvent accessibility (1%). Residue 107 is on an α-helix, and the wild-type isoleucine side chain is oriented toward the center of the protein and is completely buried. However, the packing of the side chains of the surrounding residues is such that several other amino acids can be substituted without affecting the stabilization energy. After the mean-field calculation, the amino acids that are acceptable at this position (and their probabilities) are: Ile (0.42), Cys (0.23), Val (0.12), Met (0.09), Glu (0.09), Asp (0.03), Thr (0.01), Ser (0.01), and Ala (0.01). The result of the evolution experiment was an Ile→Val substitution, which increased the activity in organic solvent. A similar example exists in the T4 lysozyme data set. Residue 151 is on an α-helix near the surface and is partially blocked from the solvent by surrounding atoms. It has an above-average site entropy (1.53) and below-average solvent accessibility (17%). The mean-field calculation reveals that the amino acids possible at this position are: Met (0.37), Leu (0.34), Cys (0.11), Glu (0.09), Gln (0.05), Asp (0.03), Ser (0.01), and Thr (0.01). The evolution experiment generated a Thr→Ser substitution. Typically, the positions with high entropies (greater that one standard deviation above the mean) and below-average solvent accessibilities (<24% exposed) are close to the surface, and their side chains are partially buried. In the mean-field computation, we calculate the energies resulting from all amino acid substitutions, rather than using a measure based on the single wild-type amino acid identity, as in the solvent accessibility calculation. This leads to a more accurate assessment of the tolerance of a residue for amino acid substitutions.
Table 1.
Comparison of site entropies and solvent accessibility
| Residue | Site entropy | % exposed* | |
|---|---|---|---|
| Subtilisin E | 9 | 2.55 | 56 |
| 14 | 2.50 | 34 | |
| 76 | 2.45 | 46 | |
| 107 | 1.62 | 1 | |
| 118 | 2.37 | 79 | |
| 131 | 2.43 | 37 | |
| 156 | 2.19 | 53 | |
| 161 | 2.69 | 92 | |
| 166 | 0.96 | 8 | |
| 181 | 0.36 | 23 | |
| 182 | 1.81 | 52 | |
| 188 | 2.50 | 88 | |
| 194 | 2.59 | 71 | |
| 206 | 1.94 | 40 | |
| 218 | 2.54 | 50 | |
| 255 | 2.54 | 41 | |
| T4 lysozyme | 14 | 2.59 | 47 |
| 16 | 2.02 | 53 | |
| 22 | 1.66 | 19 | |
| 26 | 1.03 | 2 | |
| 40 | 2.54 | 80 | |
| 41 | 1.91 | 34 | |
| 93 | 2.52 | 81 | |
| 113 | 2.54 | 69 | |
| 116 | 2.50 | 51 | |
| 119 | 2.11 | 54 | |
| 147 | 2.10 | 50 | |
| 151 | 1.53 | 17 | |
| 153 | 0.55 | 0 | |
| 163 | 2.49 | 63 |
The percent surface area of the side chain accessible by solvent. The surface areas were calculated using the Lee and Richards definition of solvent accessible surface area using 1.4 Å as the radius of water (41). The average solvent accessibility is 24% and the standard deviation is 26%.
We also compared the calculated entropies with the diversity accumulated during natural evolution, calculated from a sequence alignment (data not shown). The sequence alignment entropy was determined from the sequences of subtilisins SSII, S41, S39, BPN′, E, Carlsberg, and thermitase (37). The amino acid probabilities pi(ia) are calculated as the fraction of aligned sequences where amino acid a exists at position i. We find that the calculated entropies correlate poorly with the natural amino acid variability (R2 = 0.27). Because the natural sequence variability among subtilisins is great, the correlation worsens as more sequences are compared.
That the site entropy can predict the positions where mutations occur in in vitro, but not in natural evolution, is interesting. This disparity is due to a combination of two effects, both related to the limited number of mutants that can be screened. First, the theory that we present relies on the assumption that the number of mutants screened is relatively small. The analog of this in nature is unclear; however, it is expected that many more mutants have been attempted in nature than can be currently analyzed in the laboratory. Second, long periods of neutral evolution have eroded the information in the sequence alignment. Multiple mutations can be made to achieve a punctuated fitness improvement over long time periods via the accumulation of neutral mutations, which eventually discover beneficial combinations (29). However, the probability of finding a good multiple mutant during in vitro evolution is small because of the sampling limitation of the experiment (analogous to a time limitation).
It is important to emphasize that our algorithm describes the positions where mutations will be discovered with the intention of optimizing directed evolution as a search algorithm. The probability that beneficial mutants are found increases when the high-entropy positions are targeted and low-entropy sites are neglected. Noncombinatorial experiments, such as rational design strategies, will not correlate with the entropy prediction. The requirement for a combinatorial component to the experiment is demonstrated by the example probabilities given above for residue 107 in subtilisin E and residue 151 in T4 lysozyme. In both examples, the amino acid substitution found by the evolution experiment does not correspond with the highest probability case determined by the computation. Once the algorithm determines the positions where substitutions do not disrupt the structure, evolutionary experiments can determine the specific mutations that generate the greatest fitness improvements.
Computationally Focused Mutagenesis.
The information from the structural entropy calculations can be incorporated in several experimental methods. First, site saturation mutagenesis can be applied at positions that are predicted to be the most tolerant. The positive mutants can then be recombined by using DNA shuffling (42) to compound the fitness improvement. As a second method, a portion of the gene that is determined to have an above-average total tolerance (such as residues 240 to 255 in subtilisin E) can be targeted by using regional combinatorial mutagenesis. The choice of experimental approach is determined by the accuracy of the entropy profile. If the correlation between the screened property and stability is high, then site saturation mutagenesis is appropriate. However, if the correlation is weaker, a combinatorial search of a region that is predicted to be able to withstand the additional diversity is better.
The experiment can also combine mutagenesis with recombination, a method conceptually similar to family shuffling, in which homologous genes are recombined (38, 39). In family shuffling, the sequences have previously survived natural selection; thus, the inherent diversity is less likely to have a deleterious effect on the structure and function. In our approach, the calculated entropy profile predicts the positions that are essential to maintain the structure, allowing the tolerant sites to be mutated en masse to produce a family of artificially divergent sequences. Recombining these sequences could generate a mutant library with large sets of mutations that are calculated to retain structural integrity.
Conclusions
Because positive mutations are found at high-entropy sites, we propose that mutagenesis should be preferentially applied to these regions. An alternative approach is to make specific mutations at a highly coupled set of residues, a strategy that has been successful in improving the stability of small proteins (4, 5). However, we are interested in improving properties such as activity, where the exact fitness contributions cannot be accurately computed. Experimentally incorporating a sufficiently high mutation rate to reliably discover highly coupled mutants requires a screening effort larger than is practically feasible. Our algorithm provides a methodology by which enzymes can be computationally prescreened, thus reducing the required experimental effort. By computationally calculating the entropy of each residue and by using this information to guide an experimental evolutionary search, the most powerful aspects of each technique are combined as an approach to protein design.
Acknowledgments
C.A.V. is supported by a National Science Foundation graduate research fellowship and by a California Institute of Technology Initiative in Computational Molecular Biology, a Burroughs Wellcome-funded program for science at the interface. Financial support was provided by the Howard Hughes Medical Institute (S.L.M.). We thank Hue Sun Chan, Peter Kollman, Alan Fersht, John Yin, and Walter Fontana for advance readings of this manuscript and critical comments.
References
- 1.Moore J C, Arnold F H. Nat Biotechnol. 1996;14:458–467. doi: 10.1038/nbt0496-458. [DOI] [PubMed] [Google Scholar]
- 2.Miyazaki K, Wintrode P, Grayling R, Rubingh D, Arnold F H. J Mol Biol. 2000;297:1015–1026. doi: 10.1006/jmbi.2000.3612. [DOI] [PubMed] [Google Scholar]
- 3.Street A G, Mayo S L. Structure (London) 1999;7:R105–R109. doi: 10.1016/s0969-2126(99)80062-8. [DOI] [PubMed] [Google Scholar]
- 4.Dahiyat B I, Mayo S L. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 5.Malakaukas S M, Mayo S L. Nat Struct Biol. 1998;5:470–475. doi: 10.1038/nsb0698-470. [DOI] [PubMed] [Google Scholar]
- 6.Zhao H, Arnold F H. Protein Eng. 1999;12:47–53. doi: 10.1093/protein/12.1.47. [DOI] [PubMed] [Google Scholar]
- 7.Skandalis A, Encell L P, Loeb L A. Chem Biol. 1997;4:889–898. doi: 10.1016/s1074-5521(97)90297-0. [DOI] [PubMed] [Google Scholar]
- 8.Nikolova P V, Henckel J, Lane D P, Fersht A R. Proc Natl Acad Sci USA. 1998;95:14675–14680. doi: 10.1073/pnas.95.25.14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyazaki K, Arnold F H. J Mol Evol. 1999;49:716–720. doi: 10.1007/pl00006593. [DOI] [PubMed] [Google Scholar]
- 10.Dahiyat B I, Mayo S L. Proc Natl Acad Sci USA. 1997;94:10172–10177. doi: 10.1073/pnas.94.19.10172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dahiyat B I, Mayo S L. Protein Sci. 1996;5:895–903. doi: 10.1002/pro.5560050511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayo S L, Olafson B D, Goddard W A., III J Phys Chem. 1990;94:8897–8909. [Google Scholar]
- 13.Dunbrack R L, Karplus M. J Mol Biol. 1993;230:543–574. doi: 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
- 14.Dunbrack R L, Karplus M. Nat Struct Biol. 1994;1:334–340. doi: 10.1038/nsb0594-334. [DOI] [PubMed] [Google Scholar]
- 15.Dahiyat B I, Gordon D B, Mayo S L. Protein Sci. 1997;6:1333–1337. doi: 10.1002/pro.5560060622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee C. J Mol Biol. 1994;236:918–939. doi: 10.1006/jmbi.1994.1198. [DOI] [PubMed] [Google Scholar]
- 17.Koehl P, Delarue M. J Mol Biol. 1994;239:249–275. doi: 10.1006/jmbi.1994.1366. [DOI] [PubMed] [Google Scholar]
- 18.Koehl P, Delarue M. Curr Opin Struct Biol. 1996;6:222–226. doi: 10.1016/s0959-440x(96)80078-9. [DOI] [PubMed] [Google Scholar]
- 19.Smith J M. Nature (London) 1970;225:563–564. doi: 10.1038/225563a0. [DOI] [PubMed] [Google Scholar]
- 20.Wright S. Proceedings of the Sixth International Congress on Genetics. Vol. 1. 1932. pp. 356–360. [Google Scholar]
- 21.Kauffman S. The Origins of Order. Oxford, U.K.: Oxford Univ. Press; 1993. [Google Scholar]
- 22.Wells J A. Biochemistry. 1990;29:8509–8517. doi: 10.1021/bi00489a001. [DOI] [PubMed] [Google Scholar]
- 23.Reidhaar-Olson J F, Sauer R T. Science. 1988;241:53–57. doi: 10.1126/science.3388019. [DOI] [PubMed] [Google Scholar]
- 24.Saven J G, Wolynes P G. J Phys Chem B. 1997;101:8375–8389. [Google Scholar]
- 25.Abkevich V I, Gutin A M, Shakhnovich E I. J Mol Biol. 1995;252:460–471. doi: 10.1006/jmbi.1995.0511. [DOI] [PubMed] [Google Scholar]
- 26.Li H, Helling R, Tang C, Wingreen N. Science. 1996;273:666–669. doi: 10.1126/science.273.5275.666. [DOI] [PubMed] [Google Scholar]
- 27.Matsuura T, Yomo T, Trakulnaleamsai S, Ohashi Y, Yamamoto K, Urabe I. Protein Eng. 1998;11:789–795. doi: 10.1093/protein/11.9.789. [DOI] [PubMed] [Google Scholar]
- 28.Shakhnovich E I. Phys Rev Lett. 1994;72:3907–3910. doi: 10.1103/PhysRevLett.72.3907. [DOI] [PubMed] [Google Scholar]
- 29.Fontana W, Shuster P. Science. 1998;280:1451–1455. doi: 10.1126/science.280.5368.1451. [DOI] [PubMed] [Google Scholar]
- 30.Matsumura M, Wozniak M, Dao-Pin S, Matthews B W. J Biol Chem. 1989;264:16059–16066. [PubMed] [Google Scholar]
- 31.Jain S C, Shinde U, Li Y, Inouye M, Berman H M. J Mol Biol. 1998;284:137–144. doi: 10.1006/jmbi.1998.2161. [DOI] [PubMed] [Google Scholar]
- 32.Shoichet B K, Baase W A, Kuroki R, Matthews B W. Proc Natl Acad Sci USA. 1995;92:452–456. doi: 10.1073/pnas.92.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen K, Arnold F H. Proc Natl Acad Sci USA. 1993;90:5618–5622. doi: 10.1073/pnas.90.12.5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.You L, Arnold F H. Protein Eng. 1996;9:77–83. doi: 10.1093/protein/9.1.77. and erratum (1996) 9, 719. [DOI] [PubMed] [Google Scholar]
- 35.Pjura P, Matsumura M, Baase W A, Matthews B W. Protein Sci. 1993;2:2217–2225. doi: 10.1002/pro.5560021221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giver L, Gershenson A, Freskgard P-O, Arnold F H. Proc Natl Acad Sci USA. 1998;95:12809–12813. doi: 10.1073/pnas.95.22.12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siezen R, Leunissen J A M. Protein Sci. 1997;6:501–523. doi: 10.1002/pro.5560060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crameri A, Raillard S-A, Bermudez E, Stemmer W P C. Nature (London) 1998;391:288–291. doi: 10.1038/34663. [DOI] [PubMed] [Google Scholar]
- 39.Altamirano M M, Blackburn J M, Aguayo C, Fersht A R. Nature (London) 2000;403:617–622. doi: 10.1038/35001001. [DOI] [PubMed] [Google Scholar]
- 40.Koradi R, Billeter M, Wuthrich K. J Mol Graphics. 1996;14:51–62. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 41.Lee B, Richards F M. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 42.Stemmer W P C. Nature (London) 1994;370:389–391. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]