

Abstract
Nerve injury, tissue damage, and inflammation all cause hyperalgesia. A factor contributing to this increased sensitivity is a long-term (>24 hr) hyperexcitability (LTH) in the sensory neurons that mediate the responses. Using the cluster of nociceptive sensory neurons in Aplysia californica as a model, we are examining how inflammation induces LTH. A general inflammatory response was induced by inserting a gauze pad into the animal. Within 4 days, the gauze is enmeshed in an amorphous material that contains hemocytes, which comprise a cellular immune system. Concurrently, LTH appears in both ipsilateral and contralateral sensory neurons. The LTH is manifest as increased action potential discharge to a normalized stimulus. Immunocytochemistry revealed that hemocytes have antigens recognized by antibodies to TGFβ1, IL-6, and 5HT. When a localized inflammation was elicited on a nerve, hemocytes containing the TGFβ1 antigen were present near axons within the nerve and those containing the IL-6 were on the surface. Western blots of hemocytes, or of gauze that had induced a foreign body response, contained a 28-kD polypeptide recognized by the anti-TGFβ1 antibody. Exposure of the nervous system to recombinant human TGFβ1 elicited increased firing of the nociceptive neurons and a decrease in threshold. The TGFβ1 also caused an activation of protein kinase C (PKC) in axons but did not affect a kinase that is activated in axons after injury. Our findings, in conjunction with previous results, indicate that a TGFβ1-homolog can modulate the activity of neurons that respond to noxious stimuli. This system could also contribute to interactions between the immune and nervous systems via regulation of PKC.
Animals have developed sophisticated mechanisms to respond to noxious stimuli. Events that damage the skin or injure a nerve activate nociceptive neurons whose cell bodies reside in dorsal root ganglia. The information is then relayed to neurons in the spinal cord, which subsequently relay it to centers in the brain. One consequence of injury is an increase in the excitability of the nociceptors whose peripheral fields have been damaged (Lamotte et al. 1996). These changes depend on transcription and translation and therefore constitute a memory of injury (Zimmermann and Herdegen 1996). The increase in excitability contributes to hyperalgesia at the site of injury, making the area more sensitive and serving as a protective mechanism (Walters 1994). A similar response occurs when an animal has been invaded by a foreign substance. In these situations, however, the immune system is an intermediary, releasing factors that induce the hyperexcitability in the sensory neurons (Bennett and Xie 1988; Cesare and McNaughton 1997). The hyperexcitability caused by injury or inflammation can last for days but generally diminishes as the injury is repaired and the inflammation subsides. In some unfortunate cases, the hyperexcitability persists, leading to chronic neuropathic pain.
Chronic pain is a significant clinical problem that might be alleviated if we had a more complete understanding of the molecular events that cause long-term hyperexcitability (LTH). The pathways responsible for inducing LTH after injury are gradually being identified (e.g., Ambron and Walters 1996; Zimmermann and Herdegen 1996), but defining interactions between sensory neurons and the immune system has proven more difficult. Given that both injury and inflammation induce a similar LTH, there should be points of convergence in the pathways activated under each condition. We are attempting to identify these points by using the bilateral cluster of wide-dynamic-range sensory neurons in the marine mollusc Aplysia californica as a model. Because these neurons are the primary responders to noxious stimuli, their functions overlap those of the nociceptive neurons of vertebrates (Illich and Walters 1997).
Injury to the axon of a sensory cell causes the soma to become hyperexcitable and produces a site-specific sensitization of the skin in the receptive field of the injured neuron (Walters 1987a,b). The sensitization, which can last for weeks, is analogous to hyperalgesia in higher animals and is the behavioral manifestation of the memory of injury (Walters 1994). The sensory neurons also become hyperexcitable when an inflammatory response is elicited by tying a nondamaging ligation around a nerve containing their axons (Clatworthy et al. 1994). The site of the inflammation is marked by an accumulation of hemocytes, which comprise an invertebrate cellular immune system (Bayne 1983; Ottaviani and Franceschi 1996). Hemocytes contain homologs of cytokines, bioactive peptides (Ottaviani and Cossarizza 1990; Ottaviani et al.1992), and proteins, such as transforming growth factor-β1(TGFβ1) (Franchini et al. 1996), which when released could contribute to the induction of LTH. It was therefore significant that Zhang et al. (1997) recently showed that recombinant TGFβ1 could induce changes in the electrical properties of Aplysia nociceptive neurons. In this paper we show that Aplysia hemoctyes contain a TGFβ1-like factor and that these cells appear at sites of inflammation. In addition, recombinant vertebrate TGFβ1 causes sensory neurons to become hyperexcitable. These findings indicate that Aplysia will be useful in determining how different noxious stimuli can cause LTH in a well-defined population of nociceptive neurons.
Materials and Methods
A total of 100–200 grams of A. californica were obtained from the University of Miami (FL) animal facility and were maintained in artificial seawater (ASW) at 15°C. Animals were anesthetized with isotonic MgCl2, a small incision was made in the dorsal body wall, and either a 0.5 × 0.5-cm piece of nonsterile gauze was inserted into the hemocoel or a string of gauze was tied loosely around a peripheral nerve. The animal was then sutured with 5-0 monofilament nylon and returned to the tank.
ELECTROPHYSIOLOGY
Animals were anesthetized, incised along their ventral surface, and pinned to a dish. The entire CNS was removed and placed in ASW and the pleural ganglia were desheathed. The nociceptive sensory neurons were impaled and the threshold and the number of action potentials in response to a stimulus 2.5× threshold were determined by standard procedures (Walters et al. 1991; Ambron et al. 1996). All data are expressed as means ±s.e.m.. Experiments in which one sensory cluster was the experimental and the other the control, the person recording from the cells did not know which preparation was being examined. The mean excitability from 5–10 cells in each cluster—one cell of the pair in the right cluster and the other in a comparable position in the left cluster—was compared using a paired two-tailed t-test. Comparison of the mean excitability of the sensory neurons between animals was carried out using an unpaired t-test.
IMMUNOHISTOCHEMISTRY
Hemocytes and pedal nerves were fixed with 5% paraformaldehyde in Tris-HCl (pH 7.6) for 3 hr at room temperature and permeabilized with 0.5% Triton X-100 in buffer. The nerves were frozen, and 10- 20-μm sections were obtained with a cryostat. Nerve sections and permeabilized hemocytes were exposed to antibodies against TGFβ1, interleukin-6 (IL-6), and IL-1 (1:200) (Santa Cruz Biotechnology) and antigen/antibody complexes were detected using an HRP-conjugated secondary antibody (1:500). Controls were exposed to secondary antibody only, and there was no staining.
TGFβ1
Human recombinant TGFβ1 was obtained as the active 25-kD disulfide-linked homodimer (R&D Systems). Samples of the stock solution were subjected to SDS-PAGE, and silver staining showed that ∼90% of the protein was in a single 13-kD band. The nervous system was excised from the animal in isotonic MgCl2/ASW (1:1) leaving nerve p9 as long as possible. The animal was bisected and the two halves incubated separately. The bath on one side was replaced with medium containing TGFβ1 (100 ng/ml). After 24 hr at 15°C, the sheath over the sensory cluster was removed and the neurons in the p9 region were impaled for recording. In experiments to assay the effects of TGFβ1 on PKC, we employed a three-chambered dish in which the distal portions of the pedal nerves on each side were placed in chambers separated from each other and from the attached pleuropedal ganglia. The chambers were sealed from one another using vacuum grease. The factor (100 ng/ml) was then added to one of the chambers containing the distal portions of the nerves. After 30 min at 15°C, the nerves on both sides were removed and assayed.
CELL CULTURE
Juvenile animals were anesthetized, and the abdominal ganglion was removed. Neurons were individually placed onto culture dishes coated with polylysine as described previously (Ambron et al. 1996). After 1 day, hemolymph-containing hemocytes was added, and the interaction between the hemocytes and neurons was monitored by phase-contrast microscopy. Images were captured using an Argus 20 System linked to Adobe Photoshop.
PKC ASSAYS
We adapted previously published procedures (Kruger et al. 1991; Sossin and Schwartz 1992). Tissues were homogenized in lysis buffer containing EDTA, EGTA, sodium pyrophosphate, β-glycerol, sodium vanadate, and leupeptin, in Tris-HCl (pH 7.4). Extracts were clarified by centrifugation, and samples containing 25 ng of protein were removed and incubated in 10 mm MgCl2, 10 mm EGTA, and 500 nm synthetic peptide substrate (Sossin and Schwartz 1992). A preparation of phorbol myristic acid (PMA) and phosphatidyl serine (PS) (Life Technologies) was added to an identical tube to maximally activate the protein kinase C (PKC) and chelerythrine (CalBiochem) was used as a specific inhibitor. The reactions were initiated with [γ-32P]ATP (Du Pont–New England Nuclear). After 30 min at room temperature, the reaction mix was spotted onto a phosphocellulose disk (Whatman). The disk was quenched with 1% ATP followed by washes with 5% phosphoric acid and water. Incorporation into the substrate was determined by liquid scintillation. To assess PKC activity, background phosphorylation, which occurs in the absence of substrate, was subtracted in each sample. The specific activity of PKC in each sample was then calculated and expressed as pmoles of 32P incorporated into substrate per minute per microgram of protein. The phosphorylation of the substrate, which ranged from 8,000 to 10,000 cpm, was prevented by chelerythrine.
Results
AN INFLAMMATORY RESPONSE INDUCES LONG-TERM HYPEREXCITABILITY IN THE NOCICEPTIVE NEURONS
The sensory neurons are found in bilateral clusters containing ∼200 neurons each (Walters et al. 1983). Each neuron extends an axon into a single pedal nerve to the body wall (Fig. 1; Walters et al. 1983). Within the cluster, neurons with axons in the same nerve tend to be grouped. Because each nerve innervates a discrete area of skin, the groups collectively form a rough somatotopic map of the body wall (Billy and Walters 1989). To test the hypothesis that LTH is caused by the release of a factor from the hemocytes into the hemolymph, we induced a general inflammation by inserting a piece of gauze into the hemocoel through a small incision in the body wall. The incision was sutured and the animal was returned to its tank. The 70% of the animals that recovered regained mobility and found and consumed seaweed as usual but were more sensitive to touch and tended to release ink more readily than nontreated animals. By the fourth day the gauze was enveloped in a viscous, brown amorphous material that contained hemocytes and was strongly attached to either the nerves or the head ganglia on the side of the incision. In some cases, the gauze was also fused to muscles of the body wall. We assessed the excitability of sensory neurons in each cluster from 11 animals and found that neurons on both sides fired significantly more action potentials in response to a normalized stimulus than did cells from five animals tested on day 0 (P < 0.001, Table 1). The inflammation did not affect threshold, however. Incising the body wall also increased the excitability of the sensory neurons 4 days later, although not to the level seen on the side containing the gauze. Thus, comparison of the mean activity in the sensory neurons in the 11 animals treated with the gauze to that recorded from the cells in clusters from 5 animals that were operated on, but not exposed to the gauze, showed a significant difference (P < 0.002, Table 1).
Figure 1.

Diagram of the ventral surface of the left pleural and pedal ganglion showing pedal (p) nerves p9 and p8. p9, which is the longest nerve in the animal, extends caudally to innervate the tail. The other pedal nerves have been severed close to the ganglion.
Table 1.
Induction of LTH in the nociceptive neurons
| Spikesa
|
Threshold
|
|||||
|---|---|---|---|---|---|---|
| Day
|
left
|
right
|
left
|
right
|
N
|
SNb
|
| Operated | ||||||
| 0 | 3.7 ± 0.3 | 4.0 ± 0.3 | 1.4 ± 0.1 | 1.4 ± 0.1 | 5 | 34 |
| 1 | 2.7 ± 0.2 | 3.7 ± 0.6 | 1.2 ± 0.1 | 1.4 ± 0.2 | 4 | 15 |
| 4 | 5.6 ± 0.6 | 5.1 ± 0.5 | 1.0 ± 0.1 | 1.2 ± 0.1 | 5 | 30 |
| Gauze | ||||||
| 4 | 8.4 ± 0.6 | 6.1 ± 0.4 | 1.0 ± 0.1 | 1.0 ± 0.6 | 11 | 65 |
| TGFβ1 | ||||||
| 1 | 8.1 ± 0.6 | 4.5 ± 0.4 | 0.9 ± 0.04 | 1.1 ± 0.03 | 4 | 40 |
The number of action potentials elicited by a stimulus normalized to threshold, and the cell threshold (nA) of sensory neurons in the right and left pleural ganglion after an incision (operated), insertion of gauze, or exposure to TGFβ1.
The side exposed to TGFβ1. Each value is the mean (±s.e.m.) of the average scores from N animals. Each animal contributed an average score from 5–10 cell pairs—one cell in each pair from the right sensory cluster and the other from the left.
(SN) The total number of sensory neuron pairs.
HEMOCYTES CONTAIN BIOACTIVE FACTORS
We used indirect immunocytochemistry to screen the hemocytes for factors that might cause hyperexcitability but limited our search to constituents that have been identified in invertebrates (Franchini et al. 1996). Hemolymph was added to polylysine-coated plastic dishes and when the round, phase-bright hemocytes settled onto the substratum and become phase dark and motile, the cells were fixed, permeabilized, and exposed to antibodies. Approximately 15% of the hemocytes stained with an antibody to vertebrate TGFβ1 (Fig. 2). The staining was concentrated in large vesicular structures, a distribution that has been observed in hemocytes from other invertebrates (Franchini et al. 1996). We found that the stained cells were large and contained many broad lamellipodia and filamentous extensions. Approximately 60% of the cells were stained with an antibody to IL-6 but none with an antibody to IL-1 (Fig. 2). The cells containing the putative IL-6 homolog were smaller than those that were positive for TGFβ1, suggesting that the two factors are present in distinct populations of cells. Approximately 10% of the cells were reactive to an antibody to serotonin (5-HT; Fig. 2).
Figure 2.

Immunocytochemistry of hemocytes migrating on polylysine-coated plastic dishes. The cells were fixed, permeabilized, and exposed to the primary antibody indicated. Antigen-antibody complexes were subsequently detected with HRP-coupled secondary antibody. Hemocytes were stained by antibodies to vertebrate TGFβ1 and IL-6, but not to IL-1. Positive staining was also obtained with an antibody to 5-HT. The staining was most intense in large granules (arrows). Hemocytes that were recognized by the anti-TGFβ1 antibody were larger than most of the other cells and contained especially prominent lamellipodia (Lam). Bars, 2 μm.
CELLS CONTAINING THE PUTATIVE TGFβ1 HOMOLOG ARE PRESENT AT SITES OF INFLAMMATION
We next examined the site of an inflammation. A loose ligation was tied around nerve p9, and 4 days later, when the gauze is known to elicit a foreign body response (Table 1), the animal was sacrificed and a nerve segment containing the inflammation site and an equal-sized segment of control nerve were removed, fixed, frozen, and sectioned. Clusters of cells at the ligation site were detected with the anti-TGFβ1 antibody (Fig. 3). Significantly, the stained cells were within the nerve near the axon bundles. In contrast, cells that stained with the antibody to IL-6 were found in groups on the surface of the inflamed nerve (Fig. 3). Preliminary experiments indicate that IL-6 has no effect on the activity of the sensory neurons.
Figure 3.
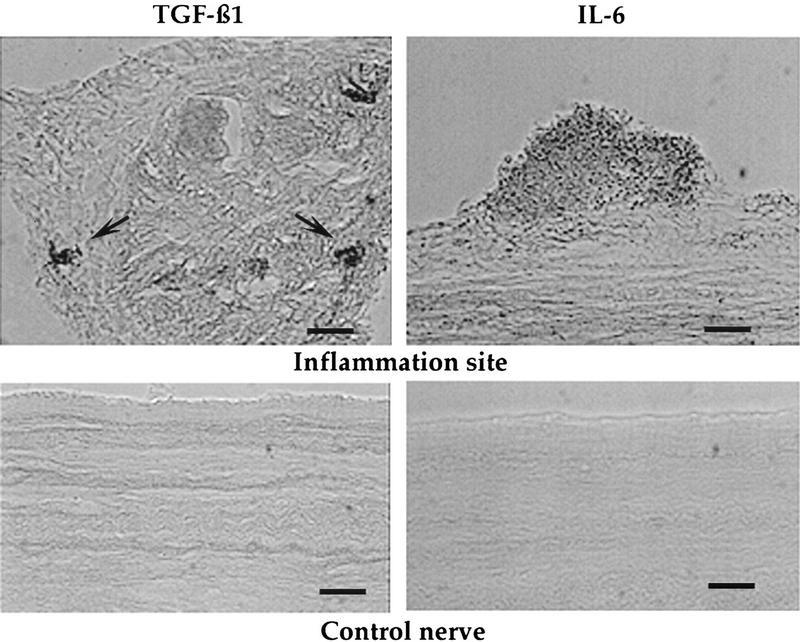
Immunocytochemical localization of cells containing TGFβ1 and IL-6. An inflammation was elicited by tying a loose ligation around nerve p9. Four days later the nerve segment immediately below the ligation was fixed, frozen, and sectioned in the longitudinal plane. A segment of a control nerve from the same animal was treated similarly. Sections were exposed to the primary antibody followed by an HRP-coupled secondary antibody. Images were captured using an Argus 20 system. Groups of TGFβ1 immunopositive cells were detected under the connective tissue sheath and adjacent to the axon bundle (arrow). This section was taken near the end of the nerve segment. In contrast, large accumulations of IL-6-positive cells were found attached to the outer border of the sheath. There was no staining of control nerves. Bars, 30 μm.
THE ANTIBODY TO TGFβ1 RECOGNIZES A POLYPEPTIDE IN HEMOCYTES AND AT THE SITE OF INFLAMMATION
To begin to identify the antigen recognized by the anti-TGFβ1 antibody, Western blots of hemocyte extracts were probed with the antibody. A single polypeptide, at 28-kD, was recognized consistently (Fig. 4). This constituent is soluble because when the hemocytes were fractionated by differential centrifugation, the 28-kD polypeptide was in the supernatant (not shown). We also looked for the factor at a site of inflammation. Gauze that elicited a foreign body reaction after 4 days in an animal was extracted with detergent. Western blots of the extract showed that the 28-kD polypeptide was present (Fig. 4B). The antibody detected higher molecular weight polypeptides on some blots.
Figure 4.

Recognition of a TGFβ1-like polypeptide on Western blots. (A) A blot, probed with the anti-TGFβ-1 antibody, showing the prominent 28-kD polypeptide (arrow) in an extract of hemocytes, and authentic recombinant vertebrate TGFβ1 (arrowhead). (B) Blot of hemocytes and a detergent extract of gauze that had been implanted in an animal for 4 days. The anti-TGFβ1 antibody detected a prominent 28-kD polypeptide in the gauze extract (arrow). Much of the antigen in the hemocyte sample was found in high molecular weight polypeptides.
TGFβ1 INDUCES HYPEREXCITABILITY IN THE SENSORY NEURONS
The presence of a putative 28-kD TGFβ1-homolog in hemocytes and at sites of inflammation prompted us to investigate a possible role for TGFβ1 in response to inflammation. To avoid the effects of nerve injury, which induces LTH in the sensory neurons (Walters et al. 1991), we took advantage of the fact that the neurons with axons in pedal nerve p9 to the skin comprise a discrete group within the sensory cluster (Billy and Walters 1989). p9 is the longest nerve in the animal (Fig. 1), and severing the nerve at the body wall while bathing the nervous system in a high divalent cation medium precludes the possibility that fast and slow injury signals will reach the cell bodies located 5–6 cm away (Gunstream et al. 1995).
Using this preparation, one-half of the CNS was exposed to 100 ng/ml active recombinant vertebrate TGFβ1 as described previously (Zhang et al. 1997). The contralateral side served as a control. Twenty-four hours later, we washed both ganglia with ASW and recorded from 10 p9 neurons in each cluster. In all four experiments the sensory neurons exposed to the TGFβ1 fired significantly more spikes than the control cells (experimental = 8.1 ± 0.6, control = 4.5 ± 0.4, P < 0.05; Table 1). The TGFβ1 also caused a significant reduction in the threshold (P < 0.01; Table 1).
TGFβ1 ACTIVATES PKC INAXOPLASM
TGFβ1 initiates kinase cascades in cells (Lin et al. 1992) and we wanted to determine whether it would activate PKC, which has been implicated in the induction of electrical changes, including hyperexcitability, in the sensory neurons (Sugita et al. 1992, 1997; Manseau et al. 1998). Aplysia neurons contain only one calcium-dependent and one calcium-independent form of PKC (Sossin et al. 1993). We also examined effects on IAK-1(Injury-activatedkinase-1), an axoplasmic kinase that is phosphorylated after injury (Y.J. Sung, M. Povelones, X.P. Zhang, D.F. Zhu, and R.T. Ambron, unpubl.). The intact nervous system was placed into a chamber containing three compartments so that the p9 nerves were separated from each other and from the ganglia (see Materials and Methods). p9 in one compartment was exposed to 100 ng/ml TGFβ1, and 30 min later equal-size segments of control and experimental nerves were assayed for PKC activity (Table 2). In all three experiments PKC was much more active in the nerves exposed to the factor than in controls. By comparing the activity elicited by TGFβ1 to that in identical samples treated with exogenous phorbol ester and PS, to maximally activate both isoforms, we found that exposure to TGFβ1 had elicited maximal activity. In contrast, only a small percentage of total kinase in the control nerves was constitutively active (Table 2). All of the activity was blocked with the PKC-specific inhibitor chelerythrin. To determine whether PKC was present in axons, we extruded axoplasm from the pedal nerves and found that both PKC isoforms were present. In significant contrast to the robust stimulation of PKC, TGFβ1 had no effect on IAK-1.
Table 2.
Activation of PKC by TGFβ-1
| 1
|
2
|
3
|
||||
|---|---|---|---|---|---|---|
| Experiment
|
Con
|
TGFβ1
|
Con
|
TGFβ1
|
Con
|
TGFβ1
|
| Substrate | 0.20 | 3.1 | 0.21 | 3.10 | 0.38 | 1.26 |
| + Activator | 2.57 | 2.5 | 2.67 | 2.60 | 0.97 | 1.15 |
Nerve p9, exposed to recombinant vertebrate TGFβ1 for 30 min, and the contralateral control p9 (Con), were homogenized separately and extracts were assayed for PKC activity by measuring the incorporation of 32P from γ-labeled ATP into a peptide substrate containing a PKC phosphorylation site (see Materials and Methods). Incorporation was assessed by scintillation and ranged from 8,000 to 100,000 cpm depending on the specific activity of the ATP. PKC activity is expressed as pmoles of 32P incorporated into the peptide/min per μg protein. Phorbol ester and PS (activator) were added to a duplicate tube in each experiment to maximally activate the PKC in the sample.
HEMOCYTES INTERACT WITH NEURONS IN CULTURE
The cells that contain TGFβ1 are found near axons at the site of an inflammation where they could influence the neurons by direct contact with their axons (Fig. 3). To begin to explore this possibility, we grew Aplysia neurons in vitro and then added hemocytes while the neurons were actively extending neurites. The hemocytes settled on the substratum and became motile. From studies of >200 neurons in 30 dishes we observed that the migrating hemocytes contacted and adhered to the neurites, often forming clusters at the neurite tip (Fig. 5). Hemocytes also came into contact with the neuronal cell soma but did not remain attached. These interactions were all observed when the hemocytes and neurons were removed from the same and from different animals.
Figure 5.
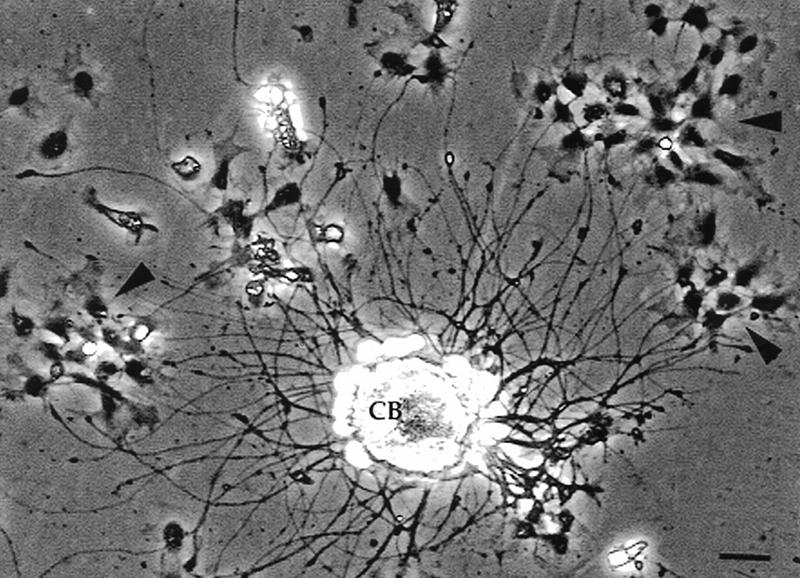
Interaction between hemocytes and neurons in vitro. The motile, phase-dense hemocytes (arrowheads) preferentially attach to the neurites and not the cell body (CB). Here the hemocytes formed clusters near the growing tips of the neurites. Bar, 15 μm.
Discussion
Tissue damage, nerve injury, and nerve inflammation all cause hyperalgesia in humans. Although pain is a subjective human experience, lower animals are thought to exhibit pain-like states through specific behaviors, most notably withdrawal reflexes. Walters (1994) has argued persuasively that the site-specific sensitization seen after injury to invertebrates, such as Aplysia, is a primitive form of hyperalgesia that has evolved as a means of protecting the injured area from additional damage. It is relevant, therefore, that damage to the body wall of Aplysia (Lewin and Walters 1999), injury to its nerves (Walters et al. 1991), or inflammation in these nerves (Clatworthy et al. 1994) all elicit similar long-lasting defensive behaviors. Moreover, in both mammalian and Aplysia nociceptive systems the behavioral response reflects the induction of LTH in the nociceptive neurons. This raises the important questions as to which intracellular pathways are elicited by each stimulus and where do they converge to induce the LTH? We have attempted to answer these questions by comparing and contrasting the responses to inflammation with those reported previously for nerve injury (Ambron and Walters 1996; Liao et al 1999; Y.J. Sung, M. Povelones, X.-P. Zhang, D.-F. Zhu, and R.T. Ambron, unpubl.).
A concomitant of the induction of LTH by inflammation in Aplysia is the accumulation of hemocytes at the site of the lesion (Clatworthy et al. 1994). Activated immunocytes release many types of factors, and it was reported that when Aplysia hemocytes are exposed to an inflammatory agent they release a factor that causes LTH in the sensory neurons (A.L. Clatworthy and E.F. Grose, unpubl). It was of interest, therefore, that Zhang et al. (1997) found that recombinant vertebrate TGFβ1 was able to induce synaptic facilitation in these same neurons. The factor did not have an effect on excitability, however. The appearance of LTH in the sensory neurons is regulated by events elicited by nerve injury (Walters et al. 1991; Gunstream et al. 1995; Ambron and Walters 1996) and it was unclear whether Zhang et al. (1997) controlled for this in their studies. We therefore exposed the sensory neurons to vertebrate TGFβ1 under conditions in which injury reactions were minimized and found that the factor caused the neurons to become hyperexcitable (Table 1). Chin et al. (1999) have obtained similar results. The LTH induced by TGFβ1 was manifest both as an increase in firing in response to a stimulus and a decrease in threshold (Table 1). The latter was not observed after inflammation in vivo (Table 1), and it is likely that multiple factors are released during inflammation. 5-HT, for example, would have a substantial effect on the excitability of the sensory neurons (Dale et al. 1987: Lewin and Walters 1999; Liao et al. 1999) and we have immunocytochemical evidence that 5-HT is present in some hemocytes (Fig. 2).
The fact that sensory neurons respond to vertebrate TGFβ1 in ways that mimic responses to natural stimuli and that Aplysia neurons contain a receptor that recognizes vertebrate TGFβ1 (Zhang et al. 1997) strongly suggest that Aplysia contains a TGFβ1-like factor that resembles its vertebrate counterpart. One source of this factor appears to be hemocytes, as vesicles in these cells contain an antigen that is recognized by an antibody to vertebrate TGFβ1 (Fig. 2). Furthermore, hemocytes containing the antigen were present within the inflamed nerve where they could release the factor directly into the area traversed by axons of sensory neurons. These findings mesh nicely with the idea that the Aplysia TGFβ1 homolog is released from hemocytes at the site of an inflammation to contribute to the induction of the LTH. This hypothesis would be strengthened if the molecular identity of the homolog were known. Franchini et al. (1996) used immunocytochemical methods to demonstrate the presence of a TGFβ1-like antigen in hemocytes of other invertebrates, but the antigen was not characterized further. We found that the antibody to TGFβ1 consistently recognized a polypeptide of 28-kD on Western blots prepared from extracts of hemocytes (Fig. 4). This constituent was soluble and was also present in gauze that had caused a foreign body reaction (Fig. 4). The presence of higher molecular weight polypeptides on some of the blots suggested that the 28-kD protein might be processed from precursors. Vertebrate TGFβ1 is initially secreted as part of a large complex that is subsequently cleaved in stages by metalloproteinases to yield a 25-kD homodimer (Miyazono et al. 1988). Tolloid, a metalloproteinase present in the Aplysia nervous system (Liu et al. 1997), would presumably be available to process the Aplysia TGFβ1. The processing must be different from that in vertebrates, however, as the Aplysia factor is a 28-kD monomer. Resolution of these issues awaits further chracterization of the 28-kD constituent.
The finding that recombinant TGFβ1 activated PKC in the nerves is important for several reasons. First, it extends previous studies, mentioned above, indicating that a TGFβ1-like factor modulates the activity of the sensory neurons (Zhang et al. 1997; Chin et al. 1999). Second, studies of vertebrate nociceptive neurons have shown that PKC is activated in axons in response to agents that cause pain (Cesare and McNaughton 1997). The activated kinase is postulated to phosphorylate channels locally to increase sensitivity. By analogy, the activation of PKC in Aplysia axons could increase axon excitability, thereby contributing to a state of general arousal (Sugita et al. 1992, 1997). We found that TGFβ1 activated all of the PKC in the nerve, meaning that the factor affected both the calcium-dependent and -independent forms of the enzyme (Table 2). How this occurs and which substrates are phosphorylated in the axon is now under investigation. Third, Manseau et al. (1998) have reported a link between hyperexcitability and the activation of PKC in Aplysia, so that our work serves as a bridge between the PKC cascade and an extrinsic factor that might be released during various kinds of stress. Interestingly, TGFβ1 did not affect IAK-1, which is a kinase in the axon that is activated by injury (Y.-J. Sung, M. Povelones, X.-P. Zhang, D.-F. Zhu, and R.T. Ambron, unpubl.). Thus, in spite of the similarity in the effects on the neurons, injury and inflammation differ in the kinase cascades that are initiated.
Vertebrate TGFβ1 has a broad spectrum of activities. For example, it induces macrophages to migrate to the site of a lesion (Wahl et al. 1987), enhances phagocytosis of macrophages, and causes the release of IL-1 and tumor necrosis factor-α (TNF-α; Bayne 1983). Consequently, the Aplysia factor could have multiple roles, especially in cells that were found near axons in the nerve. We have started to examine the interactions between hemocytes and neurons in an in vitro preparation and found that the hemocytes adhere selectively to the neurites (Fig. 5). A more detailed description of this process is in preparation (J. Farr, D. Valchich, and R.T. Ambron). The specificity of hemocyte/neuron interactions gives credence to the idea that hemocytes, like vertebrate macrophages, have multiple roles when the nervous system is physically injured or assaulted by a foreign body.
Acknowledgments
We thank Dr. Wayne Sossin for his advice on the PKC assays and for supplying the peptide substrate. We also thank Dr. Matt Lewin and Mr. Michael Povelones for help in assessing the effects of TGFβ1 on sensory neuron excitability. This work was supported by Javits Investigator Award NS-22150.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
References
- Ambron RT, Walters ET. Priming events and retrograde injury signals: A new perspective on the cellular and molecular biology of nerve regeneration. Mol Neurobiol. 1996;13:61–79. doi: 10.1007/BF02740752. [DOI] [PubMed] [Google Scholar]
- Ambron RT, Zhang XP, Gunstream JD, Povelones M, Walters ET. Intrinsic injury signals enhance growth, survival and excitability of Aplysia neurons. J Neurosci. 1996;16:7469–7477. doi: 10.1523/JNEUROSCI.16-23-07469.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayne CJ. Molluscan immunobiology. In: Saleuddin ASM, Wilbur KM, editors. The mollusca, Vol. 5, Physiology 2. New York, NY: Academic Press; 1983. pp. 407–486. [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in mammals. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Billy AJ, Walters ET. Long-term expansion and sensitization of mechanosensory receptive fields in Aplysia support an activity-dependent model of whole-cell sensory plasticity. J Neurosci. 1989;9:1254–1262. doi: 10.1523/JNEUROSCI.09-04-01254.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare P, McNaughton P. Peripheral pain mechanisms. Curr Opin Neurobiol. 1997;7:493–499. doi: 10.1016/s0959-4388(97)80028-1. [DOI] [PubMed] [Google Scholar]
- Chin, J., A. Angers, L.J. Cleary, A. Eskin, and J.H. Byrne. 1999. TGF-β1 in Aplysia: Role in long-term changes in the excitability of sensory neurons and distribution of TβR-II-like immunoreactivity. Learn. & Memory (this issue). [PMC free article] [PubMed]
- Clatworthy AL, Castro GA, Budelmann BU, Walters ET. Induction of a cellular defense reaction is accompanied by an increase in sensory neuron excitability in Aplysia. J Neurosci. 1994;14:3263–3270. doi: 10.1523/JNEUROSCI.14-05-03263.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N, Kandel ER, Schacher S. Serotonin produces long-term changes in the excitability of Aplysia sensory neurons in culture that depend on new protein synthesis. J Neurosci. 1987;7:2232–2238. doi: 10.1523/JNEUROSCI.07-07-02232.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchini A, Kletsas D, Ottaviani E. Immunocytochemical evidence of PDGF- and TGF-β-like molecules in invertebrate and vertebrate immunocytes: An evolutionary approach. Histochem J. 1996;28:599–605. doi: 10.1007/BF02331380. [DOI] [PubMed] [Google Scholar]
- Gunstream J, Castro GA, Walters ET. Retrograde transport of plasticity signals in Aplysia sensory neurons following axonal injury. J Neurosci. 1995;15:439–448. doi: 10.1523/JNEUROSCI.15-01-00439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illich PA, Walters ET. Mechanosensory neurons innervating Aplysia siphon encode noxious stimuli and display nociceptive sensitization. J Neurosci. 1997;17:459–469. doi: 10.1523/JNEUROSCI.17-01-00459.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger KE, Sossin WS, Sacktor TC, Bergold PJ, Beushausen S, Schwartz JH. Cloning and characterization of Ca-dependent and Ca -independent forms of PKCs expressed in Aplysia sensory cells. J Neurosci. 1991;11:2303–2313. doi: 10.1523/JNEUROSCI.11-08-02303.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamotte RH, Zhang J-M, Petersen M. Alterations in the functional properties of dorsal root ganglion cells with unmyelinated axons after a chronic nerve constriction in the rat. Prog Brain Res. 1996;10:105–111. doi: 10.1016/s0079-6123(08)62568-6. [DOI] [PubMed] [Google Scholar]
- Lewin MR, Walters ET. Cyclic GMP pathway is critical for inducing long-term sensitization of nociceptive sensory neurons. Nat Neurosci. 1999;2:18–23. doi: 10.1038/4520. [DOI] [PubMed] [Google Scholar]
- Liao Y, Gunstream JD, Lewin M, Ambron RT, Walters ET. Activation of protein kinase A contributes to the expression but not the induction of long-term hyperexcitability caused by axotomy of sensory neurons. J Neurosci. 1999;19:1247–1256. doi: 10.1523/JNEUROSCI.19-04-01247.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Wang X-F, Ng-Eaton E, Weinberg RA, Lodish HF. Expression cloning of the TGF-β type II receptor, a functional transmembrane serine/threonine kinase. Cell. 1992;68:775–785. doi: 10.1016/0092-8674(92)90152-3. [DOI] [PubMed] [Google Scholar]
- Liu Q-R, Hattar H, Endo S, MacPhee K, Zhang H, Cleary LJ, Byrne JH, Eskin A. A developmental gene (Tolloid, BMB-1) is regulated in Aplysia neurons by treatments that induce long-term sensitization. J Neurosci. 1997;12:755–764. doi: 10.1523/JNEUROSCI.17-02-00755.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manseau F, Sossin WS, Castellucci VF. Long-term changes in excitability by protein kinase C activation in Aplysia sensory neurons. J Neurophysiol. 1998;79:1210–1218. doi: 10.1152/jn.1998.79.3.1210. [DOI] [PubMed] [Google Scholar]
- Miyazono K, Hellman U, Wernstedt C, Heldin C-H. Latent high molecular weight complex of transforming growth factor β. J Biol Chem. 1988;263:6407–6415. [PubMed] [Google Scholar]
- Ottaviani E, Cossarizza A. Immunocytochemical evidence of vertebrate bioactive peptide-like molecules in the immuno cell types of the freshwater snail Planobarius corneus (L.) (Gastropoda, Pulmonata) FEBS Lett. 1990;267:250–252. doi: 10.1016/0014-5793(90)80937-e. [DOI] [PubMed] [Google Scholar]
- Ottaviani E, Franceschi C. The neuroimmunology of stress from invertebrates to man. Prog Neurobiol. 1996;48:421–440. doi: 10.1016/0301-0082(95)00049-6. [DOI] [PubMed] [Google Scholar]
- Ottaviani E, Caselgrandi E, Petraglia F, Francheschi C. Stress response in freshwater snail Planobarius corneus (L.) (Gatropoda, Pulmonata): Interaction between CRF, ACTH, and Biogenic Amines. Gen Comp Endocrinol. 1992;87:354–360. doi: 10.1016/0016-6480(92)90041-h. [DOI] [PubMed] [Google Scholar]
- Sossin WS, Schwartz JH. Selective activation of Ca++ activated PKCs in Aplysia neurons by 5HT. J Neurosci. 1992;12:1160–1168. doi: 10.1523/JNEUROSCI.12-04-01160.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossin WS, Dias-Arrastia R, Schwartz JH. Characterization of two isoforms of PKC in the nervous system of Aplysia californica. J Biol Chem. 1993;268:5763–5768. [PubMed] [Google Scholar]
- Stefano GB. Invertebrate and vertebrate neuroimmune and autoimmune commonalities involving opiod peptides. Cell Mol Neurobiol. 1992;12:357–366. doi: 10.1007/BF00711538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita S, Goldsmith JR, Baxter DA, Byrne JH. Involvement of protein kinase C in serotonin-induced spike broadening and synaptic facilitation in sensorimotor connections of Aplysia. J Neurophysiol. 1992;68:643–651. doi: 10.1152/jn.1992.68.2.643. [DOI] [PubMed] [Google Scholar]
- Sugita S, Baxter DA, Byrne JH. Modulation of a cAMP/protein kinase A cascade by protein kinase C in sensory neurons of Aplysia. J Neurosci. 1997;17:7237–7244. doi: 10.1523/JNEUROSCI.17-19-07237.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl SM, Hunt DA, Wakefield LM, McCartney-Francis N, Wahl LM, Roberts AB, Sporn MB. Transforming growth factor-β (TGF-β) induces monocyte chemotaxis and growth factor production. Proc Natl Acad Sci. 1987;84:5788–5792. doi: 10.1073/pnas.84.16.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters ET. Site-specific sensitization of defensive reflexes in Aplysia: A simple model of long-term hyperalgesia. J Neurosci. 1987a;7:400–407. doi: 10.1523/JNEUROSCI.07-02-00400.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Multiple sensory neuronal correlates of site-specific sensitization in Aplysia. J Neurosci. 1987b;7:408–417. doi: 10.1523/JNEUROSCI.07-02-00408.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Injury-related behavior and neuronal plasticity: An evolutionary perspective on sensitization, hyperalgesia, and analgesia. Int Rev Neurobiol. 1994;36:325–427. doi: 10.1016/s0074-7742(08)60307-4. [DOI] [PubMed] [Google Scholar]
- Walters ET, Byrne JH, Carew TJ, Kandel ER. Mechanoafferent neurons innervating the tail of Aplysia. I. Response properties and synaptic connections. J Neurophysiol. 1983;50:1522–1542. doi: 10.1152/jn.1983.50.6.1522. [DOI] [PubMed] [Google Scholar]
- Walters ET, Alizadeh H, Castro EA. Similar neuronal alterations induced by axonal injury and learning in Aplysia. Science. 1991;253:797–799. doi: 10.1126/science.1652154. [DOI] [PubMed] [Google Scholar]
- Zhang F, Endo S, Cleary LJ, Eskin A, Byrne JH. Role of transforming growth factor-β in long-term synaptic facilitation in Aplysia. Science. 1997;275:1318–1320. doi: 10.1126/science.275.5304.1318. [DOI] [PubMed] [Google Scholar]
- Zimmermann M, Herdegen T. Plasticity of the nervous system at the systemic, cellular and molecular levels: A mechanism of chronic pain and hyperalgesia. Prog Brain Res. 1996;110:233–260. doi: 10.1016/s0079-6123(08)62578-9. [DOI] [PubMed] [Google Scholar]