

Abstract
Neurotrophic factors, including BDNF and NT-3, have been implicated in the regulation of synaptic transmission and plasticity. Previous attempts to analyze synaptic transmission and plasticity in mice lacking the NT-3 gene have been hampered by the early death of the NT-3 homozygous knockout animals. We have bypassed this problem by examining synaptic transmission in mice in which the NT-3 gene is deleted in neurons later in development, by crossing animals expressing the CRE recombinase driven by the synapsin I promoter to animals in which the NT-3 gene is floxed. We conducted blind field potential recordings at the Schaffer collateral–CA1 synapse in hippocampal slices from homozygous knockout and wild-type mice. We examined the following indices of synaptic transmission: (1) input-output relationship; (2) paired-pulse facilitation; (3) post-tetanic potentiation; and (4) long-term potentiation: induced by two different protocols: (a) two trains of 100-Hz stimulation and (b) theta burst stimulation. We found no difference between the knockout and wild-type mice in any of the above measurements. These results suggest that neuronal NT-3 does not play an essential role in normal synaptic transmission and some forms of plasticity in the mouse hippocampus.
Neurotrophic factors, including BDNF and NT-3, have been implicated in the regulation of synaptic transmission and plasticity (for a recent review, see Schuman 1999). For example, BDNF and NT-3 have been found to enhance synaptic transmission at the neuromuscular junction (Lohof et al. 1993) and in area CA1 of the hippocampus (Kang and Schuman 1995). In addition, the induction of long-term potentiation (LTP) leads to increases in both BDNF and NT-3 mRNA levels in area CA1 of the hippocampus (Patterson et al. 1992). Mice with a targeted disruption of the BDNF gene show diminished LTP in the hippocampus (Korte et al. 1995, 1996; Patterson 1996 ). In addition, application of TrkB-IgG fusion proteins can interfere with both early (Figurov et al. 1996; Kang et al. 1997) and late (Kang et al. 1997) LTP in the hippocampus and visual cortex (Akaneya et al. 1997). Unfortunately, similar studies using genetic or pharmacological disruptions of NT-3 function have not been reported owing to mice mortality and lack of reagent availability, respectively.
Although most of the homozygous NT-3 knockout mice die within a few days of birth (Ernfors et al. 1994), the heterozygous animals live throughout adulthood, permitting an analysis of synaptic transmission. At the lateral perforant path–granule cell synapse in the dentate gyrus, slices from heterozygous (+/−) knockout animals exhibited normal LTP, but diminished paired-pulse facilitation and post-tetanic potentiation (Kokaia et al. 1998). As of yet, synaptic transmission and plasticity in area CA1 of the hippocampus has not been examined in these mice.
NT-3 is expressed in both neurons and glia (Tessarollo et al. 1993). Studies using pharmacological manipulations to examine the contribution of a particular molecule to plasticity are often compromised by an inability to address the source or site of action of the molecule of interest. A similar concern exists for genetic deletion experiments in which a gene is deleted in all cell types. By engineering a transgenic mouse in which the NT-3 gene is selectively deleted in neurons, the present study allowed us to address the contribution of neuronal NT-3 to both synaptic transmission and plasticity in the hippocampus.
Materials and Methods
PCR FOR NT-3-LOXP DELETION
PCR primer P1 5′-TTACCTGCTCATGAAGAAGCCTTGTTGAGC-3′ and primer P3 5′-GCATGGTTTCTGGCAGTCATAGATGCTTCC-3′ were used to detect the deletion of NT-3–loxp allele. Before deletion, these two primers were 5 kb apart and would not generate a visible band with the following cycling conditions. After deletion, these two primers would be only 200 bp apart and would give a 200-bp PCR band in 1.5% agarose gel. PCR cycling conditions were 94°C, 30 sec; 65°C, 30 sec; 72°C, 30 sec for 32 cycles.
GENERATION OF NT-3 CONDITIONAL KNOCKOUT MICE
Targeting vector construction, embryonic stem (ES) cell experiments, and chimeric mice will be described in detail elsewhere (L. Ma, J. Marth, and L.F. Parada, in prep.). After germ-line transmission of the NT-3–loxp allele was obtained, these mice were mated with different Cre-expressing mouse lines. In the present case, a transgenic mouse line expressing Cre recombinase under the control of the Synapsin I promoter (SynI–Cre) provided by Dr. J. Marth (University of California, San Diego), was used to generate a conditional knockout of NT-3 specifically in the central nervous system. lacZ reporter mice (Tsien et al. 1996) were mated with SynI–Cre transgenic mice to generate lacZ-expressing mice recapitulating Cre expression. We have estimated the activation time of the synapsin I promoter in our mice by examining its ability to drive expression of lacZ. At embryonic day 11.5 (E11.5) a very weak expression of lacZ can be observed, but robust expression of lacZ starts at E12.5. Analysis of the expression pattern of lacZ indicates strongest expression is in differentiated neurons.
SOUTHERN BLOT HYBRIDIZATION
Mouse brain genomic DNA (10–30 μg) from 5-week-old animals was digested with EcoRI and resolved on 0.8% agarose gel, blotted onto nylon membranes (Amersham), and hybridization was performed using either 2.2-kb 5′ or 1.4-kb 3′ external probes as detailed in Figure 1a. The filters were then washed once with 1× SSC, 0.1% SDS at 65°C for 1 hr, then once with 0.5× SSC, 0.1% SDS at 65°C for 30 min (Sambrook et al. 1989).
Figure 1.

Strategy for generating NT-3 conditional knockout mice. (a) Genomic structure of mouse NT-3 gene. (b) Homologous recombinant allele. Targeting vector was transfected into CJ7 ES cells and the details of selection were as described previously (Tessarollo et al. 1994). Homologous recombination was screened with Southern blot using both 5′ and 3′ probes. (c) Generation of cko+ allele. Recombinant ES clones were transfected with a CMV–Cre-expressing plasmid to delete the TK-neo selection cassette (Kuhn et al. 1995; Ramirez-Solis et al. 1995). Southern blot was carried out to screen for NT-3 allele (NT-3–loxp allele) flanked by two loxp sites. ES clones with NT-3–loxp were injected into blastocysts for germ-line transfer. (d) Generation of cko−allele. Mice with NT-3–loxp allele were then crossed with Synapsin I–Cre (SynI–Cre) (Schoch et al. 1996) transgenic mice to produce NT-3 conditional knockout mice with cko allele. From a–d, restriction sites are (B) BamHI, (E) EcoRI.
NORTHERN BLOT HYBRIDIZATION
Mouse brain total RNAs from 5-week-old animals were prepared using Trizol (GIBCO-BRL) and separated on 2.2 m formaldehyde and 1% agarose gels. Northern blot was performed on Nylon membranes. An NT-3 cDNA fragment (0.8 kb) was used as probe for hybridization. Washing conditions were as described for Southern blot hybridization (above). After exposure for NT-3 mRNA signals, the membrane was stripped and rehybridized with GAPDH probe (Sambrook et al. 1989).
PHOSPHORIMAGER QUANTIFICATION
Either Southern or Northern membranes were exposed to PhosphorImager screens and the signal intensity of each band was measured as described by the manufacturer (Bio-Rad). Percentage of NT-3–loxp (cko−) deletion or NT-3 mRNA reduction was calculated based on the relative signal intensities.
X-GAL STAINING OF BRAIN SECTIONS
PO mouse pups were perfused with 4% paraformaldehyde. Whole brains were embedded in chicken albumin and sectioned as 100-mm coronal sections with a virbratome. The brain sections were stained in 1× PBS, 2 mm MgCl2, 50 mm potassium ferri/ferrocyanide, 1 mg/ml X-gal at 25°C for 48 hr.
ELECTROPHYSIOLOGICAL RECORDING AND ANALYSIS
Hippocampal slices (400 μm) were prepared from young adult (31- to 45-day-old) male mice using a vibratome. Immediately after dissection, individual slices were placed in a well of a 24-well plate containing oxygenated artificial cerebral spinal fluid (ACSF). The plate was stored in an enclosed chamber saturated with 95% O2 and 5% CO2. The ACSF was then replaced by 250 μl of oxygenated slice medium consisting of ACSF (119 mm NaCl, 2.5 mm KCl, 1.3 mm MgSO4, 2.5 mm CaCl2, 2.0 mm NaH2PO4, 26.2 mm NaHCO3, 20.0 mm glucose) supplemented with B-27 (GIBCO). Slices were incubated for at least 2 hr before recording. For electrophysiological recordings, slices were submerged in a stream of ACSF maintained at 29 ± 1°C and gassed with 95% O2 and 5% CO2. Field excitatory postsynaptic potentials/currents (EPSPs) measured in stratum radiatum were evoked by stimulation of the Schaffer collateral–commissural afferents at a rate of 0.033 Hz using bipolar stimulating electrodes. To be included for further analysis baseline field EPSPs of slope −0.2 to −0.3 mV/msec had to be elicited with stimulation currents ≤50μA and have fiber volley amplitudes ≤0.15 mV. Extracellular recording electrodes were filled with 3m NaCl. The initial slope of the EPSP was measured for all experiments. Input–output (I/O) relations were monitored by measuring the EPSP slope in response to increasing stimulation currents. Paired-pulse facilitation was measured at four different intervals: 10, 25, 50, and 100 msec. Tetanic stimulation was delivered at the test intensity in 1-sec trains at 100 Hz, with two trains delivered 15 sec apart. Theta-burst stimulation (TBS) consisted of one episode of the following: 10 bursts of stimuli, each of four pulses at 100 Hz; interburst interval, 200 msec. Paired or unpaired t-tests were used to calculate the statistical significance of within group or between group comparisons, respectively. A one-way analysis of variance was used to calculate the statistical significance of potential differences between the I/O curves. P values greater than 0.05 were considered not significant (N.S.). All of the electrophysiology and analysis were conducted with the experimenter blind to the genotype of the animal.
Results
GENERATION OF NT-3 CONDITIONAL KNOCKOUT MICE
The NT-3 conditional knockout mice were generated as illustrated in Figure 1 and will be described in greater detail elsewhere (L. Ma, J. Marth, and L.F. Parada, in prep.) The design of the recombination cassette provided for the single NT-3 coding exon, as well as the TK-neo selection cassette, to be flanked by loxP sites (Fig. 1b). Removal of the TK-neo cassette was achieved by transient transfection of the homologous recombinant ES cells with a Cre-expressing vector pMC-Cre followed by a Southern blot screen of cell clones for selective deletion of the TK-neo cassette (Fig. 1c; data not shown; see below). cko+ cell clones were blastocyst injected and subsequently introduced into the mouse germ line (data not shown). Mice homozygous for the cko+ allele or compound heterozygotes for the cko+ and the null allele (Tessarollo et al. 1994) have been maintained for more than four (>1.5 years) generations with no apparent physical or behavioral abnormalities (L. Ma, J. Marth, and L.F. Parada, in prep.).
NT-3 and its receptor trkC are widely expressed in the peripheral and central nervous systems in both neural and glial cells (Tessarollo et al. 1993). To examine the role of NT-3 expression exclusively in post-mitotic neurons, we crossed our NT-3 cko+ mice to a transgenic line bearing a Synapsin I promoter-driven Cre transgene (Schoch et al. 1996). The SynI–cre mouse strain was crossed with cko+ mice to generate cko− progeny (Fig. 1d). Figure 2a indicates that dramatic reduction of NT-3 cko+ to cko− was observed in brain of mice of dual SynI–Cre, cko+/+ genotype with minor cko− appearance in other tissues (i.e., heart and kidney). This low level of cko− may reflect leakiness of the Syn–Cre transgene or, alternatively, minor contamination of the tissue preparations with sympathetic or parasympathetic neurons (Fig. 2a). Figure 2a further demonstrates that no other mouse genotypes exhibited even traces of the cko− allele. Because the Synapsin I promoter has been characterized previously as neuron specific (Li et al. 1993; Schoch et al. 1996) and neurons consist of ∼20% of the cell population in the central nervous system, mice with a SynI–Cre/cko− genotype should have NT-3 ablated specifically in these cells. To examine this possibility, Southern blot hybridization was carried out to estimate the extent of cko+ deletion in adult mouse brains (5 weeks). As shown in Figure 2b,c, we estimate that as much as 60% of the cko+ allele was transformed to cko− in the double transgenic mice. This result indicates that either the SynI–Cre transgenes expressed ectopically in non-neuronal cells or at low levels in progenitor cells, which may be common to neural and non-neural lineages. We further measured the expression of NT-3 mRNA in SynI–Cre/cko− mice by Northern blot hybridization of whole brain total RNA (Fig. 2d,e). In agreement with the genomic analysis, SynI–Cre/cko− mice had low levels of NT-3 mRNA compared to cko+ mice (Fig 2e). Finally, a reporter mouse strain designed to activate the β-galactosidase gene when Cre is expressed (Tsien et al. 1996) was crossed with SynI–Cre mice to analyze the expression pattern of Cre in the brain. As shown in Figure 2f and g, lacZ reporter gene expression was observed in cortex, hippocampus, and diencephalon. Although neurons appear to be stained uniformly, it is also likely that some non-neural cells have also ablated the NT-3 gene in these mice.
Figure 2.
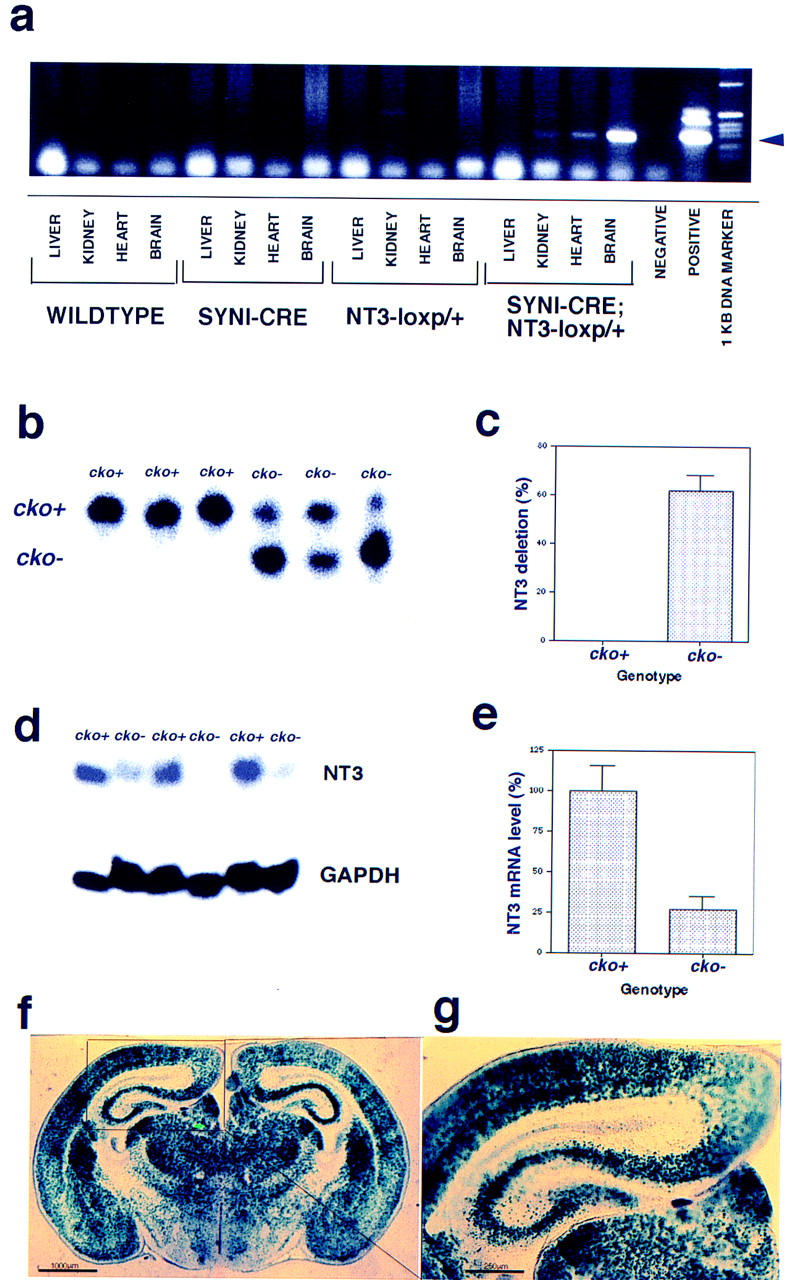
Characterization of SynI–Cre, NT-3 conditional knockout mice. (a) PCR assay from tissue genomic DNAs indicating the appearance in brain of Cre-specific NT-3–loxp (cko−) allele (see arrowhead) indicating tissue specific cre-mediated looping out of the NT-3 coding exon. (b) Southern blot of whole brain genomic DNA to show the extent of the NT-3 deletion by SynI–Cre to transform the cko+ allele into cko−. (c) Approximately 63% of the whole brain genomic cko+ allele was transformed into cko− NT-3 gene by SynI–Cre. (d) Northern blot analysis of whole brain RNA with NT-3 probe to show the decrease of NT-3 mRNA after exposure to SynI–Cre. GAPDH mRNA was used as an internal loading control. (e) Quantification of NT-3 mRNA levels with PhosphorImager indicates a 75% reduction brain NT-3 mRNA. (f,g) SynI–Cre mice were crossed with a lacZ reporter mouse strain (Tsien et al. 1996) to show the regions of Cre expression, which was recapitulated by lacZ expression. (g) Higher magnification of hippocampal region of f. (f) Bar, 1000 μm; (g) bar, 250 μm.
SYNAPTIC TRANSMISSION AND PLASTICITY ARE NORMAL IN THE NT-3 CONDITIONAL KNOCKOUT MICE
To test the role of neuronal NT-3 in synaptic transmission and plasticity, we examined several indices of synaptic transmission in the conditional knockout (SynI–Cre/cko−) and control mice (cko+/cko+) at the Schaffer collateral–CA1 synapse in the hippocampus. Basal synaptic transmission in slices from the conditional knockout mice was indistinguishable from that in the control mice (Fig. 3). This was monitored by measuring the slope of the fEPSP (field EPSP) in response to electrical stimulation of varying current amplitudes. As shown in Figure 3, the mean responses obtained from conditional knockout slices were not significantly different from those obtained from control slices (N.S.).
Figure 3.

Input–output relations are normal in hippocampal slices from conditional neuronal NT-3 mutant mice. Shown are the mean fEPSP responses to stimuli of increasing strengths for both the knockout (SynI–cre/cko−) and control (cko+/cko+) slices. There was no significant difference between the two groups. (n) Number of slices. The number of animals from which the slices were taken was eight and eight, for the knockout and controls, respectively.
In addition, two measures of short-term synaptic plasticity, paired-pulse facilitation (PPF) and post-tetanic potentiation (PTP), were also examined. PPF, observed at many different synapses, involves an augmentation of the synaptic response to the second of two closely spaced stimuli. It is likely mediated by residual intraterminal Ca2+ at the time of the second stimulus (e.g., Katz and Miledi 1968). The PPF observed in slices from the NT-3 knockout animals did not differ significantly from the controls at any of the interstimulus intervals (ISI) examined (Fig. 4) [PPF, mean percent of first response at 10 msec ISI; knockout, 142.5 ± 9.4% (n = 5); control, 142.2 ± 10.1% (n =5); (P = N.S.); 25 msec ISI: knockout, 144.9 ± 4.7% (n = 8; control, 149.9 ± 9.1% (n = 8); (P = N.S.); 50 msec ISI knockout, 151.7 ± 4.2% (n = 20); control, 142.6 ± 2.5% (n = 21; P = N.S.); 100 msec ISI knockout, 141.7 ± 2.4% (n = 20); control, 137.2 ± 1.7% (n = 21); (P = N.S.)]. In addition, analysis of the synaptic response immediately after 100 Hz or TBS (PTP) revealed no significant difference between slices from the knockout and control animals [mean percent of baseline after 100 Hz: knockout 255.2 ± 15.3% (n = 11); control 275.8 ± 15.6% (n = 10) (P = N.S.); following theta burst: knockout, 211.1 ± 18.1% (n = 9); control, 241.9 ± 24.9% (n = 11) (P = N.S.)]. We then addressed whether long-lasting plasticity was intact in slices from the neuronal NT-3 knockout mice. We found that two trains of 100 Hz stimulation (1 sec each) produced LTP in the knockout slices that was indistinguishable from that seen in the control slices (Fig. 5) mean percent of baseline 50–60 min after tetanus: knockout, 159.4 ± 8.2% (n = 11); control, 169.1 ± 7.3% (n = 10) (P = N.S.)]. A recent study of the BDNF/TrkB dependence of LTP demonstrated that the stimulation parameters used to induce LTP are an important determinant of whether TrkB is involved (Kang et al. 1997). LTP induced by 100 Hz stimulation was not blocked by function-blocking TrkB antibodies, whereas LTP induced by TBS was inhibited (Kang et al. 1997). As such, we also tested whether LTP induced by TBS (see Methods) was sensitive to neuronal NT-3 deletion. We found, however, that TBS-induced LTP was also normal in the NT-3 knockout slices [mean percent of baseline 50–60 min after TBS: knockout, 147.0 ± 10.2% (n = 9); control, 142.1 ± 9.8% (n = 11) (P = N.S.)]. Taken together, these results indicate that neuronal NT-3 is not required for synaptic transmission or short- or long-term plasticity at the Schaffer collateral–CA1 synapse in the mouse hippocampus.
Figure 4.

Paired-pulse facilitation is normal in hippocampal slices from conditional neuronal NT-3 mutant mice. Shown is the mean for three different interstimulus intervals. There was no significant difference between the knockout (solid bars) and control (open bars) slices at any of the intervals tested. The number of slices used for each interval is as follows (knockout/control): 10 msec (5,5); 25 msec (8,8); 50 msec (20,21); 100 msec (20,21). The number of animals from which the slices were taken was 8 and 8 for the 50 and 100 msec ISI, 3 and 3 for the 25 msec ISI, and 2 and 2 for 10 msec ISI for the knockout and controls, respectively.
Figure 5.
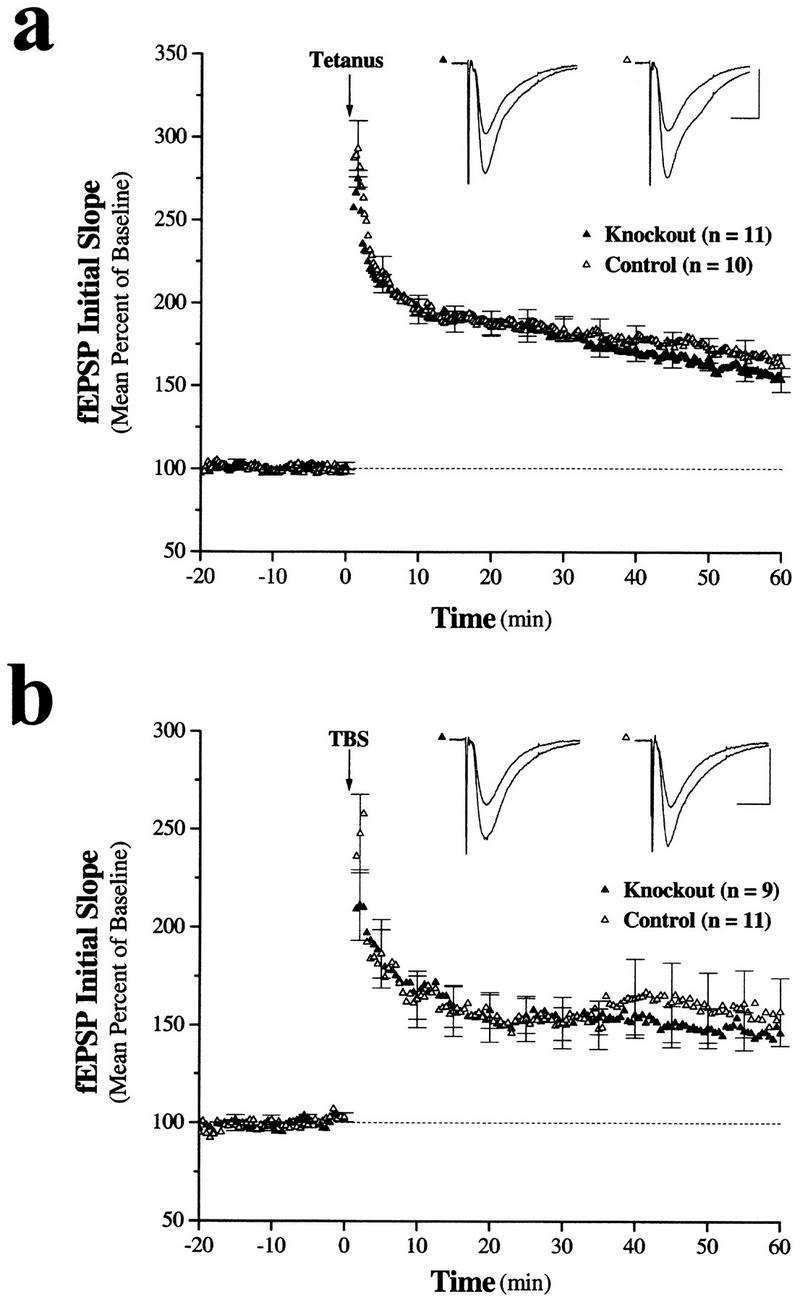
LTP is normal in hippocampal slices form conditional neuronal NT-3 mutant mice. (a) Ensemble averages for slices exposed to two epochs of 100 Hz stimulation (tetanus) for 1 sec each. Both the control and the mutant mice exhibited LTP, which was of a similar magnitude. In the mutant mice the mean fEPSP slope was −0.25 ± 0.02 mV/msec and −0.38 ± 0.02 mV/msec, before and after LTP induction by 100 Hz. These slices exhibited significant potentiation (P < 0.001). In the control mice the mean fEPSP slope was −0.24 ± 0.01 mV/msec and −0.41 ± 0.02 mV/msec, before and after LTP induction by 100 Hz. These slices also exhibited significant potentiation (P < 0.001). (n) Number of slices. The number of animals from which the slices were obtained was four and four for knockout and control mice, respectively. (b) Ensemble averages for slices exposed to TBS. Both the control and the mutant mice exhibited LTP, which was of a similar magnitude. In the mutant mice the mean fEPSP slope was −0.25 ± 0.02 mV/msec and −0.36 ± 0.03 mV/msec, before and after LTP induction by TBS. These slices exhibited significant potentiation (P < 0.001). In the control mice the mean fEPSP slope was −0.23 ± 0.02 mV/msec and −0.32 ± 0.03 mV/msec, before and after LTP induction by TBS. These slices also exhibited significant potentiation (P < 0.001). Bar, 0.5 mV/100 msec. (n) Number of slices. The number of animals from which the slices were obtained was four and four for knockout and control mice, respectively.
Discussion
Some of our SynI–Cre/cko− colony have surpassed a year of age with no apparent deleterious phenotypes (behavioral or pathological) resulting from ablation in most neurons and in a proportion of additional neural cells. We have also maintained NT-3 compound heterozygotes for the conditional and null alleles in the context of the Syn–Cre transgene. These mice have a complete somatic ablation of one NT-3 allele in addition to the ablation in all neurons and some glial cells of the second cko− allele. We have yet to observe any overt pathological or behavioral phenotype in these mice (L. Ma and L. Parada, pers. comm.). Recently, Bates et al. (1999) reported the generation of a similar conditional NT-3 mutant mouse. Through use of a nestin promoter to drive Cre expression, NT-3 conditional deletion in the central nervous system and additional sites was reported. Unlike the Syn–Cre/cko− described here, Bates and colleagues observed gait and behavioral abnormalities that they associated with cerebellar malfunction. We have not assessed directly the cerebellar morphology of our Syn–Cre/cko− mice to determine the extent of neural and non-neural ablation in this tissue.
The lack of effect of neuronal NT-3 deletion on synaptic transmission or plasticity differs from recent observations in NT-3+/− mice, in which a decrease in paired-pulse facilitation was observed in the dentate gyrus (Kokaia et al. 1998). These NT-3+/− mice show ∼30% reduction in NT-3 mRNA levels in the dentate granule neurons (Elmer et al. 1997), but NT-3 levels in area CA1 have not been reported. Taken together these results suggest, however, that neuronal NT-3 plays different roles at Schaffer collateral–CA1 synapses versus dentate granule synapses. This is further substantiated by the observation that LTP induction results in up-regulation of NT-3 in CA1 neurons (Patterson et al. 1992), but results in NT-3 down-regulation in dentate granule cells (Castren et al. 1993).
Our earlier studies demonstrated that application of either BDNF or NT-3 to hippocampal slices is sufficient to enhance synaptic transmission (Kang and Schuman 1995, 1996). To what extent does the neurotrophin-induced synaptic enhancement involve cellular mechanisms that overlap with LTP? Early experiments (Kang and Schuman 1995) examined the extent to which previous potentiation by either BDNF or NT-3 influenced the subsequent ability to obtain LTP at the same synapses. These experiments showed that LTP (induced by several trains of 100-Hz stimulation) could still be elicited at synapses previously potentiated by neurotrophin. This implied that neurotrophin-induced potentiation and LTP used at least partially independent mechanisms. A later study (Kang et al. 1997) revealed, however, that the degree to which BDNF/TrkB-dependent potentiation overlaps with LTP depends on the particular stimulation protocols used to induce LTP. That is, LTP induced by 100-Hz stimulation was found to be independent of TrkB function, whereas LTP induced by more “subtle” stimulation protocols such as TBS or pairing required TrkB function (Kang et al. 1997). It was with this subtlety in mind that we tested both 100-Hz and TBS-induced LTP in the NT-3 neuronal knockout. We found, however, that LTP was intact in both cases, indicating that neuronal NT-3 is not required for these forms of LTP.
Taken together with our earlier work (Kang and Schuman 1995), these data imply that NT-3 is sufficient, but not necessary, for synaptic potentiation in area CA1 of the hippocampus. One caveat concerns the species differences in these two sets of studies: NT-3-induced potentiation has been observed in rat hippocampus, but has not yet been reported in mouse hippocampus. An additional potentially noteworthy feature of our previous work with neurotrophins in hippocampal slices is the degree to which the potentiation elicited by either BDNF or NT-3 appeared to use similar cellular mechanisms. For example, both neurotrophins decreased PPF (Kang and Schuman 1995), required receptor tyrosine kinase activity (Kang and Schuman 1995a) and protein synthesis (Kang and Schuman 1996) for potentiation, and did not require NMDA receptor function for potentiation (H. Kang and E.M. Schuman, unpubl.). Although the synaptic enhancement elicited by BDNF is prevented completely by TrkB receptor blockade (Kang et al. 1997), preliminary experiments indicated that NT-3-induced potentiation is prevented only partially by blockade of TrkC function (H. Kang and E.M. Schuman, unpubl.). This raises the possibility that some of the potentiation produced by the application of exogenous NT-3 may be mediated by interactions with TrkB. If so, this could explain why the absence of neuronal NT-3 did not affect LTP in the present study, as other TrkB ligands, such as BDNF, could compensate.
The hippocampus is a prominent site of NT-3 expression (Ernfors et al. 1990). Our Northern blot analysis indicates that the conditional knockout resulted in ∼75% reduction of the NT-3 mRNA in brain. Moreover, the lacZ expression pattern in the reporter mouse (Fig. 2f,g) indicates that the Cre recombinase was expressed robustly in the pyramidal cell layers of areas CA3 and CA1. These data, however, do not exclude the possibility that a non-neuronal source could provide the NT-3 required to sustain both synaptic transmission and plasticity at the Schaffer collateral–CA1 synapse. An analysis of mice in which NT-3 is deleted in glia will likely provide the answer to this question. In addition, given the abundance of neuronal NT-3 in the hippocampus, a more thorough analysis of the present conditional knockout mice may elucidate a previously unappreciated function for NT-3 in synaptic transmission.
Acknowledgments
We are grateful to Jamey Marsh for providing the syn-cre mouse, which will be described in detail elsewhere. L.F.P. and L.M. were supported by National Institutes of Health R01 NS33199. E.M.S. is a Pew Scholar and HHMI Assistant Investigator.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
References
- Akaneya Y, Tsumoto T, Kinoshita S, Hatanaka H. Brain-derived neurotrophic factor enhances long-term potentiation in rat visual cortex. J Neurosci. 1997;17:6707–6716. doi: 10.1523/JNEUROSCI.17-17-06707.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates B, Rios M, Trumpp A, Chen C, Fan G, Bishop JM, Jaenisch R. Neurotrophin-3 is required for proper cerebellar development. Nat Neurosci. 1999;2:115–117. doi: 10.1038/5669. [DOI] [PubMed] [Google Scholar]
- Castren E, Pitkanen M, Sirvio J, Parsadanian A, Lindholm D, Thoenen H, Riekkinen P. The induction of LTP increases BDNF and NGF mRNA but decreases NT-3 mRNA in the dentate gyrus. Neuroreport. 1993;4:895–898. doi: 10.1097/00001756-199307000-00014. [DOI] [PubMed] [Google Scholar]
- Elmer E, Kokaia M, Ernfors P, Ferencz I, Kokaia Z, Lindvall O. Suppressed kindling epileptogenesis and perturbed BDNF and TrkB gene regulation in NT-3 mutant mice Exp. Neurol. 1997;145:93–103. doi: 10.1006/exnr.1997.6478. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Wetmore C, Persson H. Identification of cells in rat brain and peripheral tissues expressing mRNA for members of the nerve growth factor family. Neuron. 1990;5:511–526. doi: 10.1016/0896-6273(90)90090-3. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Lee K-F, Kucera J, Jaenisch R. Lack of neurotrophin-3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell. 1994;77:503–512. doi: 10.1016/0092-8674(94)90213-5. [DOI] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- ————— A requirement for local protein synthesis in neurotrophin-induced synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- Kang H, Shelton D, Welcher A, Schuman EM. Neurotrophins and time: Different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol. 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokaia M, Asztely F, Olofsdotter K, Sindreu CB, Kullman DM, Lindvall O. Endogenous neurotrophin-3 regulates short-term plasticity at lateral perforant path-granule cell synapses. J Neurosci. 1998;18:8730–8739. doi: 10.1523/JNEUROSCI.18-21-08730.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem E, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T. Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci. 1996;93:12547–12552. doi: 10.1073/pnas.93.22.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1129. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- Li L, Suzuki T, Mori N, Greengard P. Identification of a functional silencer element involved in neuron-specific expression of the synapsin I gene. Proc Natl Acad Sci. 1993;90:1460–1464. doi: 10.1073/pnas.90.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohof AM, Ip N, Poo M-M. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: A stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TAS, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Ramirez-Solis R, Liu P, Bradley A. Chromosome engineering in mice. Nature. 1995;378:720–724. doi: 10.1038/378720a0. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 1989. [Google Scholar]
- Schoch S, Cibelli G, Thiel G. Neuron-specific gene expression of synapsin I. Major role of a negative regulatory mechanism. J Biol Chem. 1996;271:3317–3323. doi: 10.1074/jbc.271.6.3317. [DOI] [PubMed] [Google Scholar]
- Schuman EM. Neurotrophin regulation of synaptic transmission. Curr Opin Neurobiol. 1999;9:105–109. doi: 10.1016/s0959-4388(99)80013-0. [DOI] [PubMed] [Google Scholar]
- Tessarollo L, Tsoulfas P, Martin-Zanca D, Gilbert DJ, Jenkins NA, Copeland NG, Parada LF. trkC, a receptor for neurotrophin-3, is widely expressed in the developing nervous system and in non-neuronal tissues. Development. 1993;118:463–475. doi: 10.1242/dev.118.2.463. [DOI] [PubMed] [Google Scholar]
- Tessarollo L, Vogel KS, Palko ME, Reid SW, Parada LF. Targeted mutation in the neurotrophin-3 gene results in loss of muscle sensory neurons. Proc Natl Acad Sci. 1994;91:11844–11848. doi: 10.1073/pnas.91.25.11844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]