

Abstract
Although recent studies indicate that brain-derived neurotrophic factor (BDNF) plays an important role in hippocampal synaptic plasticity, the underlying signaling mechanisms remain largely unknown. Here, we have characterized the signaling events that mediate the BDNF modulation of high-frequency synaptic transmission. Mitogen-associated protein kinase (MAPK), phosphotidylinositol-3 kinase (PI3K), and phospholipase C-γ (PLC-γ) are the three signaling pathways known to mediate neurotrophin signaling in other systems. In neonatal hippocampal slices, application of BDNF rapidly activated MAPK and PI3K but not PLC-γ. BDNF greatly attenuated synaptic fatigue at CA1 synapses induced by a train of high-frequency, tetanic stimulation (HFS). Inhibition of the MAPK and PI3K, but not PLC-γ, prevented the BDNF modulation of high-frequency synaptic transmission. Neurotrophin-3 (NT-3), a close relative of BDNF, did not activate MAPK or PI3K and had no effect on synaptic fatigue in the neonatal hippocampus. Neither forskolin, which activated MAPK but not PI3 kinase, nor ciliary neurotrophic factor (CNTF), which activated PI3K but not MAPK, affected HFS-induced synaptic fatigue. Treatment of the slices with forskolin together with CNTF still had no effect on synaptic fatigue. Thus, although the activation of MAPK and PI3K is required, the two together are not sufficient to mediate the BDNF effect. Inhibition of new protein synthesis by anisomycin or cycloheximide did not prevent the BDNF effect. These data suggest that BDNF modulation of high-frequency transmission is independent of protein synthesis but requires MAPK and PI3K and yet another signaling pathway to act together in the hippocampus.
Studies in the last few years have identified a novel function of neurotrophins: regulation of synaptic function and plasticity (for review, see Lo 1995; Thoenen 1995; Bonhoeffer 1996; Lu and Figurov 1997). Substantial evidence indicates that neurotrophins can influence a wide variety of synapses, not only during development but in the adult nervous system as well. At the neuromuscular junction (NMJ), neurotrophins appear to play two distinctive roles: acute potentiation of transmitter release (Lohof et al. 1993; Stoop and Poo 1995, 1996; Wang and Poo 1997; Xie et al. 1997) and long-term regulation of synapse maturation (Wang et al. 1995; Liou and Fu 1997; Liou et al. 1997; Wang et al. 1998). In the visual cortex, nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and neurotrophin-4/5 (NT-4/5) have been shown to modulate the development of ocular dominance columns in cat and rodents (Maffei et al. 1992; Gu et al. 1994; Cabelli et al. 1995; Riddle et al. 1995; Galuske et al. 1996). Morphological studies indicate that different neurotrophins appear to have differential effects on dendritic growth in the visual cortex (McAllister et al. 1995, 1996, 1997) in a layer-specific fashion.
The function of BDNF in synaptic transmission and plasticity in the hippocampus has been one of the major focuses in recent neurotrophin research. In primary cultures of embryonic hippocampal neurons, acute application of exogenous BDNF rapidly enhances neuronal and synaptic activity and transmitter release (Knipper et al. 1994; Lessmann et al. 1994; Levine et al. 1995; Takei et al. 1997). Although the exact physiological significance of the culture experiments remains unclear, these data suggest that BDNF is capable of modulating synaptic function in the hippocampus. BDNF also seems to play an important role in long-term potentiation (LTP) in the hippocampus. Tetanic stimulation enhances the expression of BDNF mRNA in the hippocampus (Patterson et al. 1992; Castren et al. 1993) and activity-dependent release of BDNF protein has also recently been demonstrated (Goodman et al. 1996; Canossa et al. 1997). Application of exogenous BDNF facilitates tetanus-induced LTP in neonatal hippocampal slices, where the endogenous BDNF levels are low (Figurov et al. 1996). In contrast, inhibition of endogenous BDNF activity, either by the BDNF scavenger TrkB–IgG or by BDNF gene knockout, reduces the magnitude of LTP in adult hippocampus, in which the endogenous BDNF levels are high (Korte et al. 1995; Figurov et al. 1996; Patterson et al. 1996). BDNF may also be involved in the long-term maintenance of LTP, or later phase LTP (Kang et al. 1997; Korte et al. 1998).
In addition to its role in LTP, BDNF has also been shown enhance high-frequency transmission in the hippocampus. Treatment of neonatal slices with BDNF enhances the synaptic responses to high-frequency stimulation (HFS) and attenuates synaptic fatigue in the CA1 synapses (Figurov et al. 1996; Gottschalk et al. 1998). Conversely, inhibition of endogenous BDNF activity by gene knockout or by TrkB–IgG elicits a more pronounced fatigue at these synapses (Figurov et al. 1996; Pozzo-Miller et al. 1999). Whether BDNF can also enhance low frequency, basal synaptic transmission in acute hippocampal slices remains controversial. Several reports have shown that BDNF can rapidly enhance basal synaptic transmission in the CA1 excitatory synapses (Kang and Schuman 1995, 1996; Kang et al. 1996). In contrast, other groups have found that BDNF has minimal or no effect on excitatory transmission but can modulate inhibitory transmission in the CA1 regions (Figurov et al. 1996; Patterson et al. 1996; Tanaka et al. 1997; Frerking et al. 1998; Gottschalk et al. 1998; Huber et al. 1998). This controversy has been related to, at least in part, the observation that BDNF penetration into the slices may be limited (Kang et al. 1996; Patterson et al. 1996; but see Frerking et al. 1998).
The signal transduction mechanisms that mediate the synaptic actions of BDNF in the hippocampus remain unknown. Studies primarily using PC12 cells have identified a number of important signaling pathways that are activated by neurotrophins (Segal and Greenberg 1996). These pathways are initiated by the binding of neurotrophins to specific Trk receptor tyrosine kinases (Barbacid 1994; Kaplan and Stephens 1994). TrkA is activated by NGF, TrkB by BDNF and NT-4/5, and TrkC by NT-3. Once activated, the Trk receptors are autophosphorylated at specific tyrosine residues within the intracellular domains (Kaplan et al. 1991; Lamballe et al. 1991; Soppet et al. 1991). The phosphorylated tyrosines serve as protein interaction sites for SHC, phospholipase C-γ (PLC-γ), and phosphotidylinositol-3 kinase (PI3K), the intracellular molecules that lead to the activation of three major signaling pathways (Vetter et al. 1991; Ohmichi et al. 1992; Obermeier et al. 1993; Stephens et al. 1994). Tyrosine phosphorylation of SHC then triggers SHC/Grb2/Sos interaction, Ras activation, and a series of phosphorylation reactions that include Raf, MEK, and mitogen-associated protein kinase (MAPK) (for review, see Segal and Greenberg 1996). The active PLC-γ cleaves phosphotidylinositol-4,5-biphosphate to generate inositoltrisphosphate (IP3) and diacylglycerol (DAG), which in turn induces the release of Ca2+ from internal stores and activates protein kinase C (PKC), respectively (for review, see Kaplan and Stephens 1994). The putative effector of PI3K is the serine and threonine kinase Akt (Burgering and Coffer 1995; Franke et al. 1995). The cyclic AMP response element-binding protein (CREB), a transcription factor known for its role in synaptic plasticity, appears to be an important downstream mediator for BDNF function, at least in cortical neurons (Finkbeiner et al. 1997). In the present study we examined the signaling mechanisms for the acute effect of BDNF on the CA1 synapses in hippocampal slices. We determined the signaling events activated by acute application of BDNF to the hippocampus. We also tested whether specific inhibitors can block the effect of BDNF on synaptic fatigue for each of the signaling pathways. Our results suggest that both MAPK and PI3K are essential for BDNF modulation of synaptic fatigue in the hippocampus.
Materials and Methods
ELECTROPHYSIOLOGY
The methods used in the present study were essentially the same as described previously (Figurov et al. 1996). Briefly, whole hippocampal slices (400 μm thick) were cut in the transverse plane using a tissue chopper from neonatal Sprague-Dawley rats (Taconics). “Neonatal” is defined as pups that are 12–13 days old (p12–13). In some cases CA1 microslices were prepared by subdissection of this region from the whole hippocampal slices. All slices were maintained in an interface chamber and exposed to an artificial atmosphere of 95% O2 and 5% CO2. Perfusion medium [artificial cerebrospinal fluid (ACSF), 34°C] contained 124 mm NaCl, 3.0 mm KCl, 2.5 mm CaCl2, 1.5 mm MgCl2, 26 mm NaHCO3, 1.25 mm KH2PO4, 10 mm glucose, and 2 mm ascorbate (pH 7.4). The perfusion rate was 15 ml/hr. Electrophysiological or biochemical experiments were performed after slices were placed in the chamber for a recovery period of at least 2 hr. Human recombinant BDNF (kindly provided by Regeneron Pharmaceuticals, Tarrytown, NY) was added directly into the chamber and perfused in a constant volume of ∼3 ml at a final concentration of 2 nm. Other drugs were added to the perfusion medium at the final concentrations as indicated. PD098059 (10 μm), wortmannin (0.5 μm), LY294002 (30 μm), U73122 (5 μm), and forskolin (50 μm) were dissolved first in DMSO and then diluted at least 1000 times into artificial cerebrospinal fluid (ACSF). An extracellular recording electrode was filled with ACSF and placed in the stratum radiatum layer of CA1. Field excitatory postsynaptic potentials (EPSPs) in either whole hippocampal slices or CA1 microslices were monitored by stimulating the Schaffer collaterals alternately at low frequency (1 per min), by two stimulating electrodes positioned on either side of the recording electrode. Repetitive stimulation at 100 Hz was used to examine synaptic fatigue. Responses were digitized (10 kHz) and filtered (1 kHz), analyzed on line, and stored on magnetic media using the acquisition systems DaQSys 1.0 and Process 2.0 (University of Geneva), and pClamp 7.0 (Axon Instruments).
BIOCHEMICAL ANALYSIS OF MAPK, PI3K, AND PLC-γ ACTIVATION
Hippocampal whole slices or CA1 microslices from p12–13 rats were harvested at the end of BDNF and/or drug treatments and quickly frozen in liquid nitrogen. The frozen tissues were sonicated in lysis buffer, which contained 10 mm NaPO4 (pH 7.2), 150 mm NaCl, 1 mm EGTA, 50 mm NaF, 1% NP-40, 1% deoxycholate, 0.1% SDS, 1 mm [4-(2-aminoethyl)-benzenesulfluoride hydrochloride], 10 μg/ml of leupeptin, and 10 μg/ml of aprotinin (English and Sweatt 1996). The protein concentration of lysates was determined by the Bio-Rad protein assay. MAPK activation was measured by Western blot using an antiactive MAPK antibody (1:2000; Promega). Briefly, equal amounts of protein (5 μg per lane) were loaded onto a 4%–20% Tris-glycine gel, separated by electrophoresis, and transferred to an Immobilon P membrane (Millipore). The membranes were first incubated with blocking buffer (0.2 mm Tris, 137 mm NaCl, 0.2% I-Block, and 0.1% Tween 20) for 1 hr and then probed at 4°C overnight with antiactive MAPK antibody (1:2000; Promega). The membranes were rinsed with washing buffer (0.1% Tween 20, 0.2 mm Tris, and 137 mm NaCl) and incubated with HRP-conjugated secondary antibody (1:5000) for 1 hr at room temperature, followed by chemiluminescent detection [(ECL); Pierce]. Two methods were used for the detection of PI3K activation. The first method was identical to that for MAPK activation described above, except an antibody against phosphorylated Akt (PhosphoPlus Akt [Ser-473], 1:2000; New England Biolabs) was used and a blocking buffer containing 5% (wt/vol) nonfat milk instead of I-Block was used. The second method was immunoprecipitation. Briefly, an equal amount of lysate was immunoprecipitated with anti-phosphotyrosine antibody 4G10 (Upstate Biotechnology, Inc). The precipitants were separated on a 10% Tris-glycine gel (10 μg per lane), electrotransferred, and then incubated with an antibody against PI3K p85 subunit (Santa Cruz Biotechnology) overnight at 4°C. The blots were rinsed with washing buffer, incubated with anti-rabbit HRP-conjugated secondary antibody (1:5000) in blocking buffer for 1 hr at room temperature, and detected by an ECL method. For PLC-γ, a similar immunoprecipitation method as that for PI3K was used except that an anti-PLC-γ antibody (Santa Cruz Biotechnology, Inc) was used to detect tyrosine-phosphorylated PLC-γ on the blot. The gels were scanned, and the intensities of the bands were quantified using NIH Image software. Quantitation was based on 3–4 independent experiments (samples), with each repeated at least two times.
Results
BDNF-INDUCED SIGNALING EVENTS IN THE HIPPOCAMPUS
Our previous work demonstrated that BDNF promotes LTP and enhances high-frequency synaptic transmission in the neonatal hippocampus (Figurov et al. 1996; Gottschalk et al. 1998). At least three signal transduction pathways, the MAPK pathway, the PI3K pathway, and the PLC-γ pathway, have been postulated to mediate neurotrophin signaling based on studies using PC12 cells (Segal and Greenberg 1996). To determine which pathway is activated by BDNF in the developing hippocampus, we systematically measured the effects of BDNF on the activities of the key enzymes in these pathways in hippocampal slices. All experiments were performed using slices derived from p12–13 animals, because the effects of exogenous BDNF on LTP and high-frequency transmission have been observed in the hippocampus at this age. Activation of the MAPK pathway was measured by Western blot using antibodies specific for the phosphorylated form of MAPK (Erk1-P and Erk2-P; collectively, pMAPK), which represents the active form of the enzyme. The tyrosine phosphorylation of PLC-γ, and therefore the activation of the enzyme, was detected by immunoprecipitation with an anti-phosphotyrosine antibody, followed by Western blot using an anti-PLC-γ antibody. Two methods were used to measure PI3K activation. The first was to measure the tyrosine-phosphorylated form (the active form) of PI3K by immunoprecipitation of samples with an anti-phosphotyrosine antibody, followed by Western blot using an antibody against PI3K p85 subunit. The second method was to detect the phosphorylation of Akt, the target of PI3K, by Western blot using an antibody that specifically recognizes the phosphorylated Akt. Using these methods, we found that BDNF (2 nm) rapidly activated the MAPK and PI3K pathways in the whole hippocampal slices (Fig. 1). The phosphorylation of MAPK and Akt were both elevated within 30 min after BDNF application (Fig. 1A). However, PLC-γ was not activated after BDNF application in the neonatal hippocampal slices. The tyrosine phosphorylation of PLC-γ did not exhibit any change, whereas that of PI3K showed a marked increase in the same samples under these conditions (Fig. 1B). To confirm this, we also did immunoprecipitation in the reverse order. Control and BDNF-treated slices were first immunoprecipitated with the anti-PLC-γ antibody and, then probed with an anti-tyrosine antibody on the Western blot. Again, we found no increase in tyrosine phosphorylation of PLC-γ (data not shown). Because large amounts of slice tissue were needed to perform immunoprecipitation experiments, we did not examine the time course of PLC-γ activation. Thus, BDNF activates MAPK and PI3K without affecting PLC-γ in these neonatal hippocampal slices.
Figure 1.




Signaling events induced by the acute application of BDNF to neonatal hippocampal slices. Hippocampal whole slices or CA1 microslices from p12 rats were freshly cut, placed in an interface chamber for recovery (2 hr), and then treated with BDNF (2 nm) for 30 min. The slices were harvested and quickly frozen in liquid nitrogen. (A) Activation of pI3K pathway (top) and MAPKK pathway (bottom) by BDNF in the whole hippocampal slices. MAPK and PI3K activities were measured by Western blot using anti-phospho-MAPK antibody (Erk1-P and Erk2-P, or collectively, pMAPK) and anti-phospho-Akt (pAkt) antibody, respectively; n = 5. (B) Effects of BDNF on the PLC-γ pathway (top) and PI3K pathway (bottom). Whole hippocampal slices were treated with or without BDNF for 30 min. The samples were immunoprecipitated with an anti-phosphotyrosine antibody, followed by Western blot using either an anti-PLC-γ antibody (top) or an antibody against PI3K p85 subunit (bottom). Note that BDNF treatment resulted in an increase in tyrosine phosphorylation of PI3K but not that of PLC-γ; n = 3. (C) BDNF (hatched bars) induced MAPK activation in whole hippocampal slices as well as CA1 microslices. Control (open bars). MAPK activity was detected as in A (left), and the intensities of the Erk2-P bands in the Western blots were quantified; n = 4. (D) Time courses of MAPK activation in CA1 microslices; n = 4. BDNF was present during the entire time course. In this and all other figures, the data represent mean ± s.e.m., and the statistics were done using either Student’s t-test for two group comparison or ANOVA test followed by post hoc comparison for multiple groups. Significance: (*) P < 0.01.
Because most, if not all, of the studies on BDNF modulation of hippocampal synapses have focused on CA1 synapses, it is important to determine the signaling events that underlie this modulation in this area. The sensitivity of the MAPK and Akt assays allows detection of MAPK and Akt phosphorylation using only 10 μg of hippocampal tissue. We have developed a method to measure MAPK or Akt activity in CA1 microslices. The CA1 area was dissected from the whole hippocampal slice and was placed in the chamber for recovery before being harvested for the MAPK assay. BDNF elicited almost the same magnitude of MAPK phosphorylation in the microslices as it did in whole hippocampal slices (Fig. 1C). We then compared the time course of BDNF-induced MAPK activation between the whole slices and the microslices. The MAPK phosphorylation exhibited very similar time courses upon BDNF application to the whole slices or the microslices. The phosphorylation reached a peak within 30–40 min and then declined to baseline within 2 hr (Fig. 1D). A similar time course of PI3K activation was observed using the Akt assay in both whole slices as well as microslices (data not shown).
BOTH MAPK AND PI3K PATHWAYS ARE REQUIRED FOR BDNF MODULATION OF SYNAPTIC FATIGUE
An important step in characterizing BDNF signaling is to determine whether the activation of a pathway is specific. We tested the ability of a number of inhibitors to block specific pathways in hippocampal slices. In these experiments, the inhibitors were added to the slice chamber before BDNF application and were present together with BDNF throughout the experiment. BDNF-induced activation of MAPK was prevented by PD098059 (Fig. 2A, 10 μm), a specific inhibitor for MAPK kinase, the enzyme that phosphorylates and activates MAPK (Alessi et al. 1995; Dudley et al. 1995). Treatment with PD098059 alone had no obvious effect on the basal levels of MAPK activity in the slices, suggesting that the endogenous activation of the enzyme is low in the neonatal slices. We then examined the effect of LY294002 (30 μm) and wortmannin (0.5 μm), two compounds that have been shown to inhibit the PI3K activity in a number of systems (Kimura et al. 1994; Sanchez-Margalet et al. 1994). Application of BDNF in the presence of wortmannin (Fig. 2B) and LY294002 (not shown) no longer activated PI3K. In addition, the two compounds alone reduced the basal levels of PI3K in these slices (data not shown). BDNF had no effect on PLC-γ activity in the neonatal hippocampal slices (Fig. 1B). Furthermore, the combination of the PLC-γ inhibitor U73122 and BDNF did not affect the PLC-γ activity either (data not shown).
Figure 2.
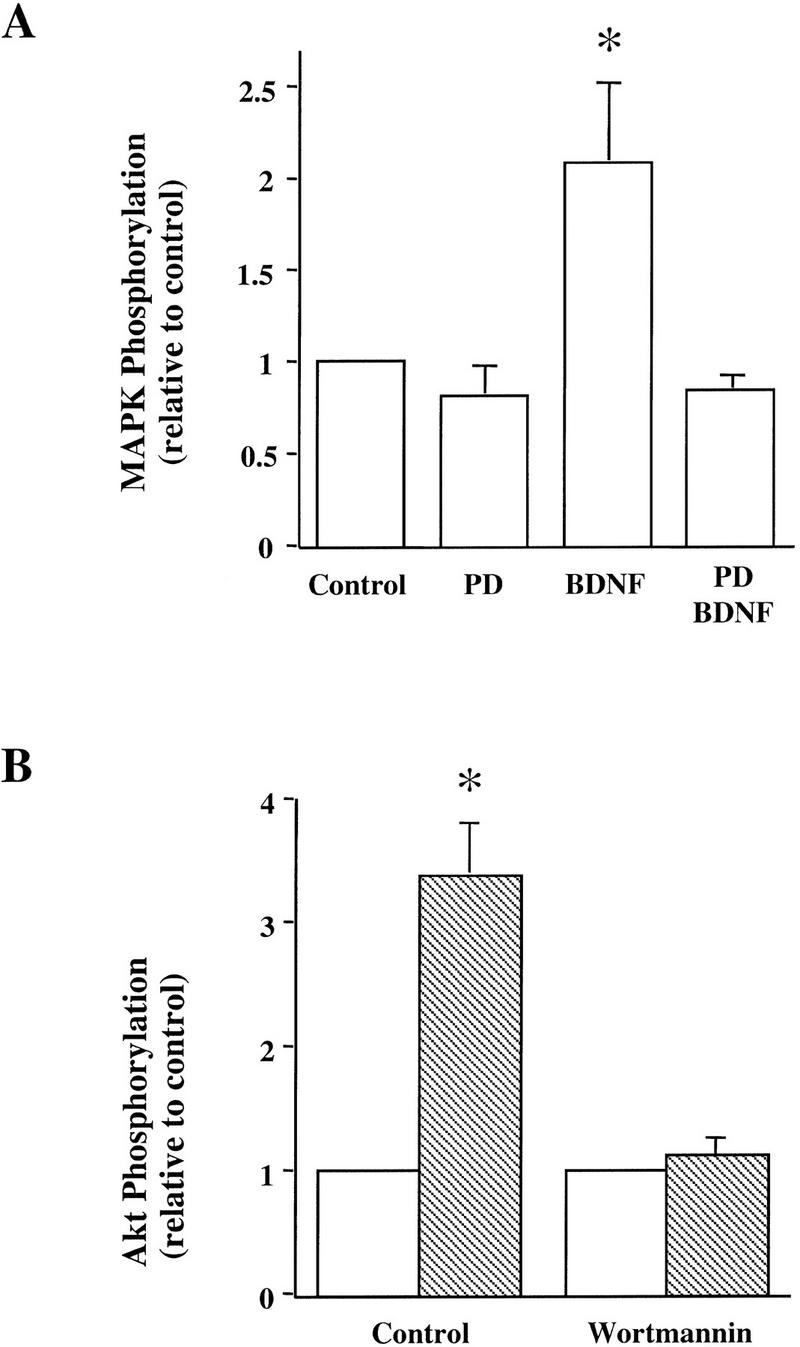
Blockade of BDNF signaling by specific inhibitors. Whole slices from p12–13 hippocampus were pretreated with specific inhibitors for 60 min. The slices were then treated with BDNF for 30 min in the presence of the inhibitors and processed for measurement of specific signaling pathways. (A) Effect of the MAPK kinase inhibitor PD098059 (PD, 10 μm). (B) Effect of the PI3 kinase inhibitor, wortmannin (0.5 μm). (Open bars) Control; (hatched bars) BDNF. These experiments have been repeated four times, and similar results were obtained every time.
BDNF modulates the synaptic fatigue seen during a train of HFS in the neonatal rat hippocampus (Gottschalk et al. 1998). To determine which of the specific signaling events are involved in this modulation, we blocked each of the pathways by the inhibitors identified above and examined whether the BDNF modulation of synaptic fatigue was prevented. As described previously (Gottschalk et al. 1998), we used the percentage of the fortieth EPSP slope over the first EPSP slope as a measure of the synaptic fatigue (from here on termed “response to HFS”) seen during the HFS train. The HFS train elicited a gradual decline of the slope and amplitude of the EPSPs (Fig. 3), and BDNF attenuated this response (Figs. 3 and 4A; control: 30% ± 0.95, n = 8; BDNF: 72% ± 4.2, n = 10). Although BDNF rapidly activated MAPK and PI3K (Fig. 1), it took at least 2.5–3 hr to observe the physiological effects of BDNF (Patterson et al. 1992; Figurov et al. 1996). Application of BDNF in the presence of the MAPK inhibitor PD098059 compound (applied 60 min earlier) prevented the potentiating effect by BDNF, suggesting that MAPK activation is required for BDNF modulation of high-frequency transmission (Figs. 3 and 4A; PD: 30% ± 1.1, n = 8; PD/BDNF: 29% ± 2.4, n = 11). Similarly, pretreatment of the slices with the PI3K inhibitors LY294002 (30 μm) or wortmannin (0.5 μm) blocked the BDNF attenuation of the responses to HFS, whereas treatment with the inhibitors alone had no effect on the synaptic fatigue (Fig. 4B). In contrast, BDNF still potentiated the responses to HFS in slices pretreated with the PLC-γ inhibitor U73122 (5 μm) (Fig. 4B). Taken together, these results suggest that MAPK and PI3K, but not PLC-γ, mediate the BDNF modulation of synaptic fatigue.
Figure 3.

Involvement of MAPK in the BDNF modulation of synaptic fatigue. Examples of EPSPs elicited by a train of HFS (100 Hz) in p12–13 hippocampal slices are shown. Treatment with BDNF attenuated synaptic fatigue, whereas pretreatment with PD098059 (PD) prevented the BDNF effect.
Figure 4.

Specific signaling pathways involved in the BDNF modulation of synaptic fatigue. The inhibitors were applied to p12–13 slices either alone or 60 min before BDNF application. In this and all other figures on synaptic responses to HFS, the slope of the fortieth EPSP in the train is presented as the percentage of the first EPSP slope, and the numbers associated with each column represent the number of recordings. In addition, controls were done for each set of slices. (A) The effect of the MAPK kinase inhibitor PD098059 (PD, 10 μm). (B) The effect of the PI3K inhibitors wortmannin (Wort., 0.5 μm) or LY294002 (LY, 30 μm) and the PLC-γ inhibitor U73122 (5 μm).
We then examined the effects of NT-3, a close relative to BDNF in the neurotrophin family. NT-3 has been shown to activate MAPK in cultured neurons derived from embryonic hippocampus (Marsh and Palfrey 1996). There are several isoforms of TrkC, the receptors for NT-3, only one of which can activate PI3K (Lamballe et al. 1993). Under our experimental conditions, however, neither MAPK nor PI3K was activated by application of 4 nm NT-3 to p12 hippocampal slices (Fig. 5A). Moreover, NT-3 had no modulatory effect on the synaptic responses to HFS (Fig. 5B; control: 43% ± 1.2, n = 5; NT-3: 41% ± 2.5, n = 14). Consistent with this finding, NT-3 has no effect on LTP in the neonatal hippocampus (Figurov et al. 1996). The inability of NT-3 to activate MAPK and PI3K may explain why NT-3 cannot modulate synaptic plasticity in the developing CA1 synapses.
Figure 5.
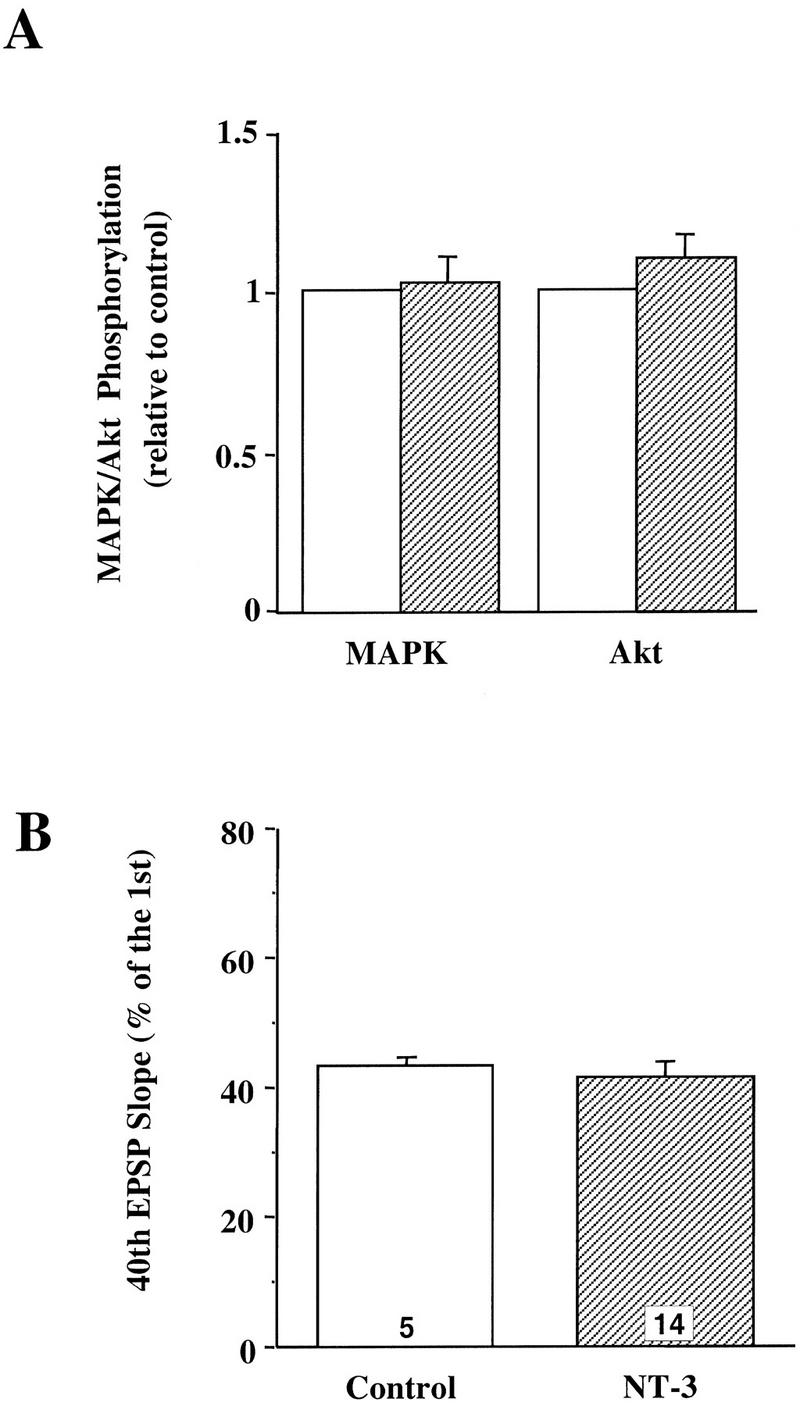
Effects of NT-3 on whole slices from neonatal hippocampus. (A) Effect on the MAPK and PI3K pathways. The whole slices from p12–13 hippocampus were treated with or without NT-3 (4 nm) for 30 min and then harvested for MAPK and Akt assays as described. (B) Effect on synaptic responses to HFS. The p12–13 slices were stimulated with a train of HFS (100 Hz, 1 sec) before and 3 hr after application of NT-3 (4 nm). (Open bars) Control; (hatched bars) NT-3. The synaptic responses to HFS were measured the same way as in Fig. 4.
ACTIVATION OF MAPK AND PI3K TOGETHER IS NOT SUFFICIENT TO POTENTIATE SYNAPTIC REPONSES TO HFS
To distinguish the individual contributions of MAPK and PI3K to the BDNF effect, we needed to identify factors that activated MAPK without affecting PI3K or vice versa. Similar to previous reports (Martin et al. 1997), we found that forskolin (50 μm) elicited a robust increase in MAPK activity (Fig. 6A). The magnitude and the time course of MAPK activation induced by forskolin were similar to those induced by BDNF (Fig. 6B). Despite its robust effect on MAPK, forskolin did not activate PI3K (Fig. 7A). In contrast, ciliary neurotrophic factor [(CNTF) 100 ng/ml] was the only factor we tested that was capable of activating PI3K but had no effect on MAPK activity in whole hippocampal slices (Figs. 6A,C and 7A). The time course and the magnitude of Akt phosphorylation elicited by CNTF were similar to those by BDNF (99% ± 13, peaked around 15–30 min). In contrast, MAPK phosphorylation remained unchanged after CNTF application (Fig. 6A). Thus, forskolin specifically enhances MAPK without affecting PI3K activity, whereas CNTF activates PI3K without affecting MAPK activity.
Figure 6.



Effects of forskolin and CNTF on MAPK activation. (A) Example of MAPK assay gels. p12–13 whole slices were treated with either forskolin (50 μm) or CNTF (4 nm) and then harvested for MAPK assay as described in Fig. 1. (B) Comparison of the time courses of MAPK activation by BDNF(○) and forskolin (●). (C) Quantitation of the MAPK activity 30 min after forskolin or CNTF application. (Open bars) Control; (hatched bars) treatment. Note that the application of forskolin, but not CNTF, activated MAPK.
Figure 7.


Effects of forskolin and CNTF on PI3K activation and HFS in p12–13 hippocampus. (A) The activation of PI3K was detected by measuring the phosphorylation of the PI3K substrate Akt using an anti-phospho-Akt antibody. Note that CNTF, but not forskolin, increased the Akt phosphorylation. (B) Effects of forskolin and/or CNTF on high-frequency synaptic transmission. Forskolin and CNTF were either applied alone or together to p12–13 hippocampal slices, and synaptic responses to HFS were measured the same way as in Fig. 4.
Having found compounds specific for the activation of MAPK activity and PI3K activity individually, we determined whether activation of MAPK or PI3K alone is sufficient to enhance synaptic responses to HFS. Treatment with forskolin alone did not change the synaptic responses to HFS, suggesting that activation of MAPK alone is not sufficient to prevent the synaptic fatigue elicited by HFS (Fig. 7B; forskolin: 39% ± 3.4, n = 32). The application of CNTF alone did not seem to affect the responses to HFS either (Fig. 7B; CNTF: 40% ± 1.4, n = 26). When CNTF and forskolin were applied together, the synaptic responses to HFS were still not significantly elevated (Fig. 7B; CNTF/forskolin: 41% ± 1.9, n = 12). These results suggest that the independent yet simultaneous activation of both MAPK and PI3K is not sufficient to mimic the BDNF modulation of synaptic fatigue.
BDNF MODULATION OF SYNAPTIC FATIGUE IS INDEPENDENT OF PROTEIN SYNTHESIS
Our previous work demonstrated that BDNF modulation of synaptic responses to HFS is mediated through presynaptic mechanisms (Gottschalk et al. 1998). Application of BDNF activated MAPK and PI3K in both whole hippocampal slices and CA1 microslices from p12–13 rats (Fig. 1). Electrophysiology experiments indicated that these microslices exhibit the same degree of synaptic fatigue and that BDNF also attenuated the synaptic fatigue in the microslices (Fig. 8; control: 46% ± 3.5, n = 6; BDNF: 64% ± 5.1, n = 8). These results suggest that BDNF acts on the presynaptic terminals of CA1 synapses and its effect is independent of the cell bodies in the CA3 area. We then determined whether the BDNF modulation of the responses to HFS requires new protein synthesis. Slices were pretreated with the protein synthesis inhibitor anisomycin (40 μm) for 30 min before BDNF was applied to the recording chamber. Anisomycin has been shown to effectively block protein synthesis and later phase LTP in the hippocampal (Stanton and Sarvey 1984; Frey et al. 1988). Incubation of the slices with anisomycin alone did not alter the synaptic fatigue during HFS (Fig. 8). The response to HFS after the application of BDNF to the anisomycin-treated slices was comparable to that observed in slices treated with BDNF alone (Fig. 8). Similar results were seen in slices treated with another inhibitor, cycloheximide (data not shown). Thus, BDNF modulation of high-frequency synaptic transmission is independent of protein synthesis.
Figure 8.

Independence of BDNF effect on protein synthesis. The synaptic responses to HFS were recorded and measured the same way as in Fig. 4. (Left) Potentiation of synaptic responses to HFS by BDNF in CA1 microslices. CA1 microslices were dissected from p12–13 hippocampal whole slices and treated with BDNF for 3 hr. (Right) Effect of protein synthesis inhibitors on BDNF modulation of synaptic fatigue. Anisomycin (40 μm) was applied either alone or 30 min before BDNF application. EPSPs evoked by HFS were compared before and 3 hr. after BDNF and anisomycin application. Note that BDNF is still capable of potentiating the synaptic responses to HFS in the presence of the protein synthesis inhibitor.
Discussion
Signal transduction mechanisms mediating synaptic plasticity are a subject of intense investigation in the field of synaptic physiology. BDNF has now emerged as an important modulator for synaptic transmission and plasticity in the hippocampus and perhaps in other areas of the brain as well. At the developing CA1 synapse, BDNF has been shown to enhance synaptic responses to HFS and to promote LTP (Figurov et al. 1996; Gottschalk et al. 1998; Pozzo-Miller et al. 1999). We have used two approaches to study the signaling mechanisms underlying the synaptic actions of BDNF in the hippocampus. One was to examine which signaling pathways are activated by BDNF in the neonatal hippocampus and the other was to determine whether inhibition of a specific signaling pathway blocks the BDNF modulation of high-frequency synaptic transmission. Our results indicate that both MAPK and PI3K pathways are required but that independently activating the two pathways is not sufficient to elicit the BDNF effect. We also demonstrate that the BDNF modulation of high-frequency transmission is independent of protein synthesis. These results may provide insights into the general signaling mechanisms by which BDNF modulates synaptic plasticity.
Extensive studies using PC12 cells have identified three major signaling pathways activated by neurotrophin: the MAP kinase pathway, the PI3 kinase pathway, and PLC-γ pathway (Kaplan and Stephens 1994; Segal and Greenberg 1996). The signaling pathways involved in a specific function of a trophic factor in a specific neuronal population or under specific conditions, however, need to be investigated individually. For example, NT-3, a close relative of BDNF, has been shown to activate MAPK in embryonic hippocampal neurons in culture (Marsh and Palfrey 1996). In the neonatal hippocampal slices, however, neither MAPK nor PI3K was activated by NT-3. In cultured neurons derived from embryonic hippocampus, BDNF is capable of activating all three signaling pathways (Marsh et al. 1993; Marsh and Palfrey 1996; Qiu et al. 1998). However, BDNF treatment of the neonatal hippocampal slices elicited no increase in tyrosine phosphorylation of the PLC-γ, a common indicator for PLC-γ activation, when measured by immunoprecipitation with an anti-phosphotyrosine antibody followed by Western blot using an anti-PLC-γ antibody. Western blot using an anti-PI3K antibody detected tyrosine phosphorylation of PI3K in the same immunoprecipitants used for PLC-γ assay. Moreover, inhibition of PLC-γ did not affect the ability of BDNF to potentiate synaptic responses to HFS in these slices. It was unclear why PLC-γ was not activated by BDNF in the neonatal slices, but our results reiterate the importance of using the exact same preparation for biochemical and physiological experiments.
BDNF enhances high-frequency synaptic transmission and potentiates LTP in the neonatal hippocampus (Figurov et al. 1996). We found that inhibition of either MAPK or PI3K blocks the BDNF effect, suggesting that both signaling pathways are necessary. The time courses for BDNF-induced MAPK and PI3K activation are shorter than the time needed to see the modulatory effects of BDNF on high-frequency transmission. MAPK and PI3K are the initial signaling events elicited by BDNF, and there may be a number of downstream events that ultimately mediate the BDNF effect. In PC12 cells, for example, application of NGF elicits a rapid increase and decrease in PI3K activity (Kimura et al. 1994; Greene and Kaplan 1995). PI3K is required for NGF-induced neurite outgrowth, which occurs many hours after NGF application. NT-3 does not modulate either synaptic fatigue or LTP in the hippocampal slices, probably owing to its inability to activate these two pathways. However, the simultaneous activation of MAPK by forskolin and PI3K by CNTF does not mimic the BDNF effect either. Thus, these two pathways alone are not sufficient to attenuate synaptic fatigue. It is possible that in addition to MAPK and PI3K, BDNF modulation requires the activation of yet another unidentified signaling pathway. In this context, it is interesting to note that several new substrates of Trk receptor tyrosine kinases have recently been identified (Qian et al. 1998). We cannot exclude the possibility that the targets of BDNF or the magnitude and/or kinetics of MAPK and PI3K activated by BDNF are different from those activated by forskolin and CNTF together.
In addition to attenuating synaptic fatigue, BDNF also promotes tetanus-induced LTP during hippocampal development. In p12–13 slices, tetanic stimulation induced only short-term potentiation (STP) in control but a stable LTP after BDNF treatment (Figurov et al. 1996). An interesting question is whether BDNF elicits certain signaling events that contribute to hippocampal LTP. Extensive studies have identified complex biochemical pathways that lead to LTP in the hippocampus (Roberson et al. 1996). Two important signaling molecules involved in LTP are Ca2+/ calmodulin-dependent protein kinase II (CaMKII) and protein kinase C (PKC), both of which require Ca2+ for their activation. MAPK is also activated by tetanic stimulation and is required for LTP in the adult hippocampus (English and Sweatt 1996). PLC-γ has been shown to be involved in the priming of hippocampal LTP (Okada et al. 1989; Cohen et al. 1998). An activation of MAPK by BDNF may help CA1 synapses to induce LTP. BDNF-induced potentiation of synaptic responses to tetanus may result in more sustained depolarization and more Ca2+ influx, which in turn would lead to more sustained activation of CaMKII and/or PKC. Although our experiments showed that BDNF does not activate PLC-γ in neonatal hippocampus, it is possible that application of BDNF together with tetanic stimulation will activate the pathway. Thus, it is reasonable to imagine that BDNF may facilitate LTP in the neonatal hippocampus by enhancing some of the signaling events necessary for LTP induction.
Because the synaptic fatigue seen during HFS is a presynaptic phenomenon (Highstein and Bennnet 1975; Heuser and Reese 1979; Zucker 1989; Dobrunz and Stevens 1997; Schikorski and Stevens 1997; Wang and Kaczmarek 1998), BDNF modulation of synaptic fatigue must be presynaptic. BDNF could act either directly on the presynaptic terminals or indirectly by stimulating the postsynaptic production of some other factor or messenger that in turn may feedback onto the presynaptic site. We have demonstrated recently that hippocampal CA1 synapses in BDNF knockout mice exhibit a severe synaptic fatigue due to a reduction in the number of docked vesicles at the active zones (Pozzo-Miller et al. 1999). Interestingly, among all the synaptic proteins tested, the levels of two proteins, synaptophysin and synaptobrevin, are markedly reduced only in the synaptosomes derived from the mutant mice. No change has been found in any of the synaptic proteins in the whole hippocampal homogenates, suggesting that the decrease in the two proteins occurs only at the nerve terminals and not in the cell bodies. Moreover, treatment of the mutant slices with BDNF restores the impairments in high-frequency transmission and in the terminal levels of synaptobrevin and synaptophysin within 3 hr. These results suggest that the BDNF modulation may not require the synthesis of new proteins within the cell bodies. In the present study we show that BDNF still enhances high-frequency transmission in CA1 microslices, which are free from the presynaptic CA3 cell bodies. Furthermore, we found that BDNF still enhances high-frequency transmission in the presence of protein synthesis inhibitors. Thus, regardless of whether the postsynaptic cells are involved or not, BDNF modulation of high-frequency transmission is independent of protein synthesis. Our present result is in marked contrast with a previous report, which suggested that BDNF acutely potentiated basal synaptic transmission by new synthesis of proteins in isolated CA1 synapses (Kang and Schuman 1996).
In summary, our results suggest that the BDNF modulation of high-frequency transmission requires the activation of MAPK and PI3K but is independent of protein synthesis. However, activation of both MAPK and PI3K together by other factors is not sufficient to mimic the BDNF effect on synaptic fatigue. This result implies that another signaling pathway may be necessary to act together with MAPK and PI3K. Alternatively, it is possible that the simultaneous activation of MAPK and PI3K by other individual factors may not have the same magnitude and/or kinetics or act on the same populations of cells as that of BDNF. Further work is needed to distinguish these possibilities.
Acknowledgments
We express our gratitude to Serena Dudek and Daniel Feldman and to members of the Lu laboratory for helpful discussions and critical comments on the manuscript. We would also like to thank Regeneron Pharmaceuticals for providing recombinant BDNF. This work is supported in part by a grant from Shanghai Science and Technology Commission and National Natural Science Foundation of China.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
References
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Barbacid M. The Trk family of neurotrophin receptors. J Neurobiol. 1994;25:1386–1403. doi: 10.1002/neu.480251107. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer T. Neurotrophins and activity-dependent development of the neocortex. Curr Opin Neurobiol. 1996;6:119–126. doi: 10.1016/s0959-4388(96)80017-1. [DOI] [PubMed] [Google Scholar]
- Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction [see comments] Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- Cabelli RJ, Horn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- Canossa M, Griesbeck O, Berninger B, Campana G, Kolbeck R, Thoenen H. Neurotrophin release by neurotrophins: Implications for activity-dependent neuronal plasticity. Proc Natl Acad Sci. 1997;94:13279–13286. doi: 10.1073/pnas.94.24.13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castren E, Pitkanen M, Sirvio J, Parsadanian A, Lindholm D, Thoenen H, Riekkinen PJ. The induction of LTP increases BDNF and NGF mRNA but decreases NT-3 mRNA in the dentate gyrus. NeuroReport. 1993;4:895–898. doi: 10.1097/00001756-199307000-00014. [DOI] [PubMed] [Google Scholar]
- Cohen AS, Raymond CR, Abraham WC. Priming of long-term potentiation induced by activation of metabotropic glutamate receptors coupled to phospholipase C. Hippocampus. 1998;8:160–170. doi: 10.1002/(SICI)1098-1063(1998)8:2<160::AID-HIPO8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long-term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- ————— A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller L, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: A major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1047. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Frerking M, Malenka RC, Nicoll RA. Brain-derived neurotrophic factor (BDNF) modulates inhibitory, but not excitatory, transmission in the CA1 region of the hippocampus. J Neurophysiol. 1998;80:3383–3386. doi: 10.1152/jn.1998.80.6.3383. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988;452:57–65. doi: 10.1016/0006-8993(88)90008-x. [DOI] [PubMed] [Google Scholar]
- Galuske RA, Kim DS, Castren E, Thoenen H, Singer W. Brain-derived neurotrophic factor reversed experience-dependent synaptic modifications in kitten visual cortex. Eur J Neurosci. 1996;8:1554–1559. doi: 10.1111/j.1460-9568.1996.tb01618.x. [DOI] [PubMed] [Google Scholar]
- Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Heffti F. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci. 1996;7:223–228. doi: 10.1006/mcne.1996.0017. [DOI] [PubMed] [Google Scholar]
- Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene LA, Kaplan DR. Early events in neurotrophin signalling via Trk and p75 receptors. Curr Opin Neurobiol. 1995;5:579–587. doi: 10.1016/0959-4388(95)80062-x. [DOI] [PubMed] [Google Scholar]
- Gu Q, Liu Y, Cynader MS. Nerve growth factor-induced ocular dominance plasticity in adult cat visual cortex. Proc Natl Acad Sci. 1994;91:8408–8412. doi: 10.1073/pnas.91.18.8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE, Reese TS. Synaptic-vesicle exocytosis captured by quick freezing. In: Schmitt FO, Worden FG, editors. The neurosciences: Fourth study program. Cambridge, MA: MIT Press; 1979. pp. 573–600. [Google Scholar]
- Highstein SM, Bennnet MVL. Fatigue and recovery of transmission at the Mauthner fiber-giant fiber synapse of the hatchet-fish. Brain Res. 1975;98:229–242. doi: 10.1016/0006-8993(75)90003-7. [DOI] [PubMed] [Google Scholar]
- Huber KM, Sawtell NB, Bear MF. Brain-derived neurotrophic factor alters the synaptic modification threshold in visual cortex [In Process Citation] Neuropharmacology. 1998;37:571–579. doi: 10.1016/s0028-3908(98)00050-1. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- ————— A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- Kang H, Jia L, Suh K, Tang L, Schuman EM. Determinants of BDNF-induced hippocampal synaptic plasticity: Role of the TrkB receptor and the kinetics of neurotrophin delivery. Learn & Mem. 1996;3:188–196. doi: 10.1101/lm.3.2-3.188. [DOI] [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: Different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Stephens RM. Neurotrophin signal transduction by the Trk receptor. J Neurobiol. 1994;25:1404–1417. doi: 10.1002/neu.480251108. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Hempstead BL, Martin-Zanca D, Chao MV, Parada LF. The trk proto-oncogene product: A signal transducing receptor for nerve growth factor [see comments] Science. 1991;252:554–558. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- Kimura K, Hattori S, Kabuyama Y, Shizawa Y, Takayanagi J, Nakamura S, Toki S, Matsuda Y, Onodera K, Fukui Y. Neurite outgrowth of PC12 cells is suppressed by wortmannin, a specific inhibitor of phosphatidylinositol 3-kinase. J Biol Chem. 1994;269:18961–18967. [PubMed] [Google Scholar]
- Knipper M, da Penha Berzaghi M, Blochl A, Breer H, Thoenen H, Lindholm D. Positive feedback between acetylcholine and the neurotrophins nerve growth factor and brain-derived neurotrophic factor in the rat hippocampus. Eur J Neurosci. 1994;6:668–671. doi: 10.1111/j.1460-9568.1994.tb00312.x. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Lindholm D. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Kang H, Bonhoeffer T, Schuman E. A role for BDNF in the late-phase of hippocampal long-term potentiation [In Process Citation] Neuropharmacology. 1998;37:553–559. doi: 10.1016/s0028-3908(98)00035-5. [DOI] [PubMed] [Google Scholar]
- Lamballe F, Klein R, Barbacid M. trkC, a new member of the trk family of tyrosine protein kinases, is a receptor for neurotrophin-3. Cell. 1991;66:967–979. doi: 10.1016/0092-8674(91)90442-2. [DOI] [PubMed] [Google Scholar]
- Lamballe F, Tapley P, Barbacid M. trkC encodes multiple neurotrophin-3 receptors with distinct biological properties and substrate specificities. EMBO J. 1993;12:3083–3094. doi: 10.1002/j.1460-2075.1993.tb05977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurons. NeuroReport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou JC, Fu WM. Regulation of quantal secretion from developing motoneurons by postsynaptic activity-dependent release of NT-3. J Neurosci. 1997;17:2459–2468. doi: 10.1523/JNEUROSCI.17-07-02459.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou JC, Yang RS, Fu WM. Regulation of quantal secretion by neurotrophic factors at developing motoneurons in Xenopus cell cultures. J Physiol (Lond) 1997;503:129–139. doi: 10.1111/j.1469-7793.1997.129bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo DC. Neurotrophic factors and synaptic plasticity. Neuron. 1995;15:979–981. doi: 10.1016/0896-6273(95)90085-3. [DOI] [PubMed] [Google Scholar]
- Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- Lu B, Figurov A. Role of neurotrophins in synapse development and plasticity. Rev Neurosci. 1997;8:1–12. doi: 10.1515/revneuro.1997.8.1.1. [DOI] [PubMed] [Google Scholar]
- Maffei L, Berardi N, Domenici L, Parisi V, Pizzorusso T. Nerve growth factor (NGF) prevents the shift in ocular dominance distribution of visual cortical neurons in monocularly deprived rats. J Neurosci. 1992;12:4651–4662. doi: 10.1523/JNEUROSCI.12-12-04651.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh HN, Palfrey HC. Neurotrophin-3 and brain-derived neurotrophic factor activate multiple signal transduction events but are not survival factors for hippocampal pyramidal neurons. J Neurochem. 1996;67:952–963. doi: 10.1046/j.1471-4159.1996.67030952.x. [DOI] [PubMed] [Google Scholar]
- Marsh HN, Scholz WK, Lamballe F, Klein R, Nanduri V, Bartacid M, Palfrey HC. Signal transduction events mediated by the BDNF receptor gp 145trkB in primary hippocampal pyramidal cell culture. J Neurosci. 1993;13:4281–4292. doi: 10.1523/JNEUROSCI.13-10-04281.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER. MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron. 1997;18:899–912. doi: 10.1016/s0896-6273(00)80330-x. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron. 1996;17:1057–1064. doi: 10.1016/s0896-6273(00)80239-1. [DOI] [PubMed] [Google Scholar]
- ————— Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron. 1997;18:767–778. doi: 10.1016/s0896-6273(00)80316-5. [DOI] [PubMed] [Google Scholar]
- Obermeier A, Lammers R, Wiesmuller KH, Jung G, Schlessinger J, Ullrich A. Identification of Trk binding sites for SHC and phosphatidylinositol 3′- kinase and formation of a multimeric signaling complex. J Biol Chem. 1993;268:22963–22966. [PubMed] [Google Scholar]
- Ohmichi M, Decker SJ, Saltiel AR. Activation of phosphatidylinositol-3 kinase by nerve growth factor involves indirect coupling of the trk proto-oncogene with src homology 2 domains. Neuron. 1992;9:769–777. doi: 10.1016/0896-6273(92)90039-g. [DOI] [PubMed] [Google Scholar]
- Okada D, Yamagishi S, Sugiyama H. Differential effects of phospholipase inhibitors in long-term potentiation in the rat hippocampal mossy fiber synapses and Schaffer/commissural synapses. Neurosci Lett. 1989;100:141–146. doi: 10.1016/0304-3940(89)90674-5. [DOI] [PubMed] [Google Scholar]
- Patterson S, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: A stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller, L., W.A. Gottschalk, L. Zhang, K. McDermott, J. Du, R. Gopalakrishnan, C. Oho, Z. Shen, and B. Lu. 1999. Impairments in high frequency transmission, synaptic vesicle docking and synaptic protein distribution in the hippocampus of BDNF knockout mice. J. Neurosci. (in press). [DOI] [PMC free article] [PubMed]
- Qian X, Riccio A, Zhang Y, Ginty DD. Identification and characterization of novel substrates of Trk receptors in developing neurons. Neuron. 1998;21:1017–1029. doi: 10.1016/s0896-6273(00)80620-0. [DOI] [PubMed] [Google Scholar]
- Qiu YH, Zhao X, Hayes RL, Perez-Polo JR, Pike BR, Huang L, Clifton GL, Yang K. Activation of phosphatidylinositol 3-kinase by brain-derived neurotrophic factor gene transfection in septo-hippocampal cultures. J Neurosci Res. 1998;52:192–200. doi: 10.1002/(SICI)1097-4547(19980415)52:2<192::AID-JNR7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Riddle DR, Lo DC, Katz LC. NT-4-mediated rescue of lateral geniculate neurons from effects of monocular deprivation. Nature. 1995;378:189–191. doi: 10.1038/378189a0. [DOI] [PubMed] [Google Scholar]
- Roberson ED, English JD, Sweatt JD. A biochemist’s view of long-term potentiation. Learn & Mem. 1996;3:1–24. doi: 10.1101/lm.3.1.1. [DOI] [PubMed] [Google Scholar]
- Sanchez-Margalet V, Goldfine ID, Vlahos CJ, Sung CK. Role of phosphatidylinositol-3-kinase in insulin receptor signaling: Studies with inhibitor, LY294002. Biochem Biophys Res Commun. 1994;204:446–452. doi: 10.1006/bbrc.1994.2480. [DOI] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci. 1997;17:5858–5867. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- Soppet D, Escandon E, Maragos J, Middlemas DS, Reid SW, Blair J, Burton LE, Stanton BR, Kaplan DR, Hunter T, et al. The neurotrophic factors, brain-derived neurotrophic factor, and neurotrophin-3 are ligands for the trkB tyrosine kinase receptor. Cell. 1991;65:895–903. doi: 10.1016/0092-8674(91)90396-g. [DOI] [PubMed] [Google Scholar]
- Stanton PK, Sarvey JM. Blockade of long-term potentiation in rat hippocampal CA1 region by inhibitors of protein synthesis. J Neurosci. 1984;4:3080–3088. doi: 10.1523/JNEUROSCI.04-12-03080.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens RM, Loeb DM, Copeland TD, Pawson T, Greene LA, Kaplan DR. Trk receptors use redundant signal transduction pathways involving SHC and PLC-gamma 1 to mediate NGF responses. Neuron. 1994;12:691–705. doi: 10.1016/0896-6273(94)90223-2. [DOI] [PubMed] [Google Scholar]
- Stoop R, Poo MM. Potentiation of transmitter release by ciliary neurotrophic factor requires somatic signaling. Science. 1995;267:695–699. doi: 10.1126/science.7839148. [DOI] [PubMed] [Google Scholar]
- ————— Synaptic modulation by neurotrophic factors: Differential and synergistic effects of brain-derived neurotrophic factor and ciliary neurotrophic factor. J Neurosci. 1996;16:3256–3264. doi: 10.1523/JNEUROSCI.16-10-03256.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei N, Sasaoka K, Inoue K, Takahashi M, Endo Y, Hatanaka H. Brain-derived neurotrophic factor increases the stimulation-evoked release of glutamate and the levels of exocytosis-associated proteins in cultured cortical neurons from embryonic rats. J Neurochem. 1997;68:370–375. doi: 10.1046/j.1471-4159.1997.68010370.x. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Saito H, Matsuki N. Inhibition of GABAa synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–596. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- Vetter ML, Martin-Zanca D, Parada LF, Bishop JM, Kaplan DR. Nerve growth factor rapidly stimulates tyrosine phosphorylation of phospholipase C-gamma 1 by a kinase activity associated with the product of the trk protooncogene. Proc Natl Acad Sci. 1991;88:5650–5654. doi: 10.1073/pnas.88.13.5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- Wang T, Xie KW, Lu B. Neurotrophins promote maturation of developing neuromuscular synapses. J Neurosci. 1995;15:4796–4805. doi: 10.1523/JNEUROSCI.15-07-04796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XH, Poo MM. Potentiation of developing synapses by postsynaptic release of neurotrophin-4. Neuron. 1997;19:825–835. doi: 10.1016/s0896-6273(00)80964-2. [DOI] [PubMed] [Google Scholar]
- Wang X, Berninger B, Poo M. Localized synaptic actions of neurotrophin-4. J Neurosci. 1998;18:4985–4992. doi: 10.1523/JNEUROSCI.18-13-04985.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie K, Wang T, Olafsson P, Mizuno K, Lu B. Activity-dependent expression of NT-3 in muscle cells in culture: Implication in the development of neuromuscular junctions. J Neurosci. 1997;17:2947–2958. doi: 10.1523/JNEUROSCI.17-09-02947.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]