

Abstract
Neocortical pyramidal neurons respond to prolonged activity blockade by modulating their balance of inward and outward currents to become more sensitive to synaptic input, possibly as a means of homeostatically regulating firing rates during periods of intense change in synapse number or strength. Here we show that this activity-dependent regulation of intrinsic excitability depends on the neurotrophin brain-derived neurotrophic factor (BDNF). In experiments on rat visual cortical cultures, we found that exogenous BDNF prevented, and a TrkB–IgG fusion protein reproduced, the change in pyramidal neuron excitability produced by activity blockade. Most of these effects were also observed in bipolar interneurons, indicating a very general role for BDNF in regulating neuronal excitability. Moreover, earlier work has demonstrated that BDNF mediates a different kind of homeostatic plasticity present in these same cultures: scaling of the quantal amplitude of AMPA-mediated synaptic inputs up or down as a function of activity. Taken together, these results suggest that BDNF may be the signal controlling a coordinated regulation of synaptic and intrinsic properties aimed at allowing cortical networks to adapt to long-lasting changes in activity.
Many developing neurons experience dramatic changes in the number or strength of the synaptic connections they receive (Shatz 1990; Constantine-Paton and Cline 1998). At the same time they must prevent firing rates from falling too low or saturating, both to remain responsive to their inputs and to allow activity to selectively strengthen or weaken individual synaptic connections (Bear and Malenka 1994). This problem can be solved if the excitability of each neuron in a network can be independently adjusted to keep firing rates within functional boundaries (Turrigiano 1999). Recent work has identified two distinct homeostatic mechanisms that may stabilize neuronal activity in the face of large changes in synaptic drive. First, ongoing activity can adjust the intrinsic excitability of cortical neurons by modifying the balance of voltage-dependent conductances (Desai et al. 1999a,b). Second, neurons can scale the strength of excitatory synaptic inputs up or down in response to changes in activity (Lissin et al. 1998; O’Brien et al. 1998; Turrigiano et al. 1998). By stabilizing neuronal firing rates, these two forms of homeostatic plasticity should help neurons remain responsive to their inputs during periods of change in intrinsic neuronal properties or in the number and strength of synaptic inputs (Turrigiano 1999).
Activity-dependent synaptic scaling and activity-dependent changes in intrinsic ionic conductances both increase the excitability of pyramidal neurons in response to activity blockade (Turrigiano et al. 1998; Desai et al. 1999a,b). The former does this by globally increasing the amplitude of excitatory postsynaptic currents in a multiplicative manner (Turrigiano et al. 1998). The latter does it by selectively regulating the density of individual ionic currents to increase the firing rate and lower the spike threshold of activity-deprived neurons; sodium currents increase in size, persistent potassium currents decrease, and calcium and transient potassium currents remain the same (Desai et al. 1999a,b). Both forms of plasticity will tend to keep firing rates homeostatically within bounds, suggesting that they act synergistically. This raises the important question of whether they are regulated by the same or by different signaling pathways.
Synaptic scaling is mediated in part by the activity-dependent release of the neurotrophin brain-derived neurotrophic factor (BDNF) (Rutherford et al. 1998). BDNF is produced by cortical pyramidal neurons, and the high affinity BDNF receptor TrkB is present on both pyramidal neurons and interneurons (Kokaia et al. 1993; Miranda et al. 1993; Cabelli et al. 1996; Cellarino et al. 1996). BDNF expression, and probably release, is tightly regulated by activity (Isackson et al. 1991; Zafra et al. 1991; Castrén et al. 1992; Ghosh et al. 1994; Wetmore et al. 1994; Blochl and Thoenen 1995). Long-term manipulations of BDNF in visual cortex disrupt ocular dominance column segregation (Cabelli et al. 1995, 1997) and experience-dependent modifications of ocular dominance among cortical neurons (Galuske et al. 1996). In addition, long-term manipulation of BDNF in vitro influences cortical dendritic growth (McAllister et al. 1995, 1996, 1997) and the amount of inhibition received by cortical pyramidal neurons (Rutherford et al. 1997), suggesting that BDNF plays an important role in the development of cortical connectivity. We have shown previously that incubating in exogenous BDNF prevents, whereas scavenging endogenous TrkB ligands mimics, the increase in pyramidal neuron excitatory synaptic strengths produced by activity blockade (Rutherford et al. 1998). These data demonstrate that activity blockade scales excitatory synaptic strengths up by reducing the release of endogenous TrkB ligands.
In addition to a role in the long-term regulation of synaptic strengths, BDNF has been shown to influence the expression of voltage-dependent conductances in both cell lines and neurons (Gonzalez and Collins 1997; Lesser et al. 1997; Oyelese et al. 1997; Sherwood et al. 1997). Because activity blockade increases the intrinsic excitability of cortical neurons by selectively regulating the balance of voltage-dependent conductances (Desai et al. 1999a), this suggested that BDNF might also be the signal linking changes in activity to changes in intrinsic neuronal excitability.
Here we explore the possibility that activity regulates the intrinsic excitability of cortical neurons through changes in the release of BDNF. We find that the leftward and upward shift in pyramidal neuron FI curves produced by activity blockade can be prevented by coapplication of BDNF and that a TrkB–IgG fusion protein mimics the effects of activity blockade. These data suggest that the same activity-dependent signal, BDNF, produces a coordinated change in both the amplitude of excitatory synaptic inputs and the function relating that amplitude to the firing rate of the postsynaptic neuron.
Materials and Methods
CORTICAL CULTURES
Rat visual cortical cultures were prepared from rat pups at postnatal days 4–6 as described (Rutherford et al. 1997). In some experiments cultures were grown in astrocyte-conditioned serum-free minimum essential medium (GIBCO Life Technologies) supplemented with 2% B27 supplement (GIBCO Life Technologies), 20 mm dextrose, 2 mm l-glutamine, and 50 U/ml penicillin/streptomycin. No differences were noted for the different plating conditions. Experiments were performed after 7–9 days in vitro. All data were obtained in parallel on treated and age-matched sister control cultures. Activity was blocked with 0.5 μm tetrodotoxin (TTX), which was refreshed after 24 hr. Human recombinant BDNF was obtained from Promega and was used at a concentration of 20 ng/ml. TrkB–IgG and TrkA–IgG were obtained from Genentech and were used at a concentration of 10 μg/ml. BDNF and the Trk–IgGs were aliquoted, stored at −80°C, and thawed just prior to use.
PHYSIOLOGY
Whole-cell patch recordings from pyramidal neurons were obtained in artificial cerebrospinal fluid (ACSF) at room temperature as described (Rutherford et al. 1998; Turrigiano et al. 1998; Desai et al. 1999a). Immediately prior to experimentation, pharmacological agents in which test cultures had been incubated (TTX, BDNF, TrkB–IgG, or TrkA–IgG) were washed out completely. Recordings with resting potentials greater than −55 mV or series resistances larger than 20 mΩ were excluded. All average data are reported as mean ± s.e.m. for the number of neurons indicated. Statistical tests were two-tailed Student’s t-tests, and P < 0.05 was considered significant. Data for each experimental condition were obtained from 2–4 separate platings, with each plating contributing measurements from 3–8 cells. In all experiments, synaptic transmission was blocked with 20 μm 6-cyano-7-nitoquinoxaline-2,3-dione (CNQX), 20 μm bicuculline, and 50 μm D(−)-amino-7-phosphonovaleric acid (AP5). Pyramidal and interneurons were distinguished as previously described (Rutherford et al. 1997, 1998; Turrigiano et al. 1998).
To measure firing rates and threshold, neurons were held in a current clamp using an Axoclamp 2B amplifier. If the resting potential of a neuron differed from −60 mV, a DC current was injected to bring the membrane potential to −60 mV. Constant current pulses of 500 msec duration were then injected. The instantaneous firing frequency for the first and subsequent interspike intervals was calculated for each neuron. Input resistance was measured from the steady-state voltage deflection produced by current steps of −10 pA.
SOLUTIONS
ACSF contained 126 mm NaCl, 3 mm KCl, 2 mm MgSO4, 1 mm NaH2PO4, 25 mm NaHCO3, 2 mm CaCl2, and 14 mm dextrose. The pH was buffered to 7.4 by bubbling continuously with 5% CO2/95% O2 The final osmolality was 305–315 mOsm. The internal pipette solution contained 130 mm KMeSO4, 10 mm KCl, 10 mm HEPES/K-HEPES, 2 mm MgSO4, 0.5 mm EGTA, and 3 mm ATP. The pH was adjusted to 7.3 with KOH.
Results
As reported previously (Desai et al. 1999a), blocking activity for 2 days with TTX produced a significant increase in the intrinsic excitability of pyramidal neurons. The FI curves for activity-blocked neurons, measured immediately after the TTX was washed out, were shifted upward and to the left relative to control neurons, and the increase in firing rate was significant for all current injections over 20 pA (Fig. 1a,b). This increase in firing rate to a given current injection was prevented by coapplication of 20 ng/ml BDNF during the period of activity blockade (Fig. 1a,b, BDNF/TTX). For all current injections, BDNF/TTX was not significantly different from control and was significantly different from TTX alone for current injections over 50 pA. This concentration of BDNF is likely to be saturating (Rutherford et al. 1998), but we have not yet determined the threshold concentration for the effect. The slope of the FI curve (measured from the initial, linear part of the curve) was significantly increased by 100 ± 14% following TTX treatment, and this increase was blocked by BDNF (Fig. 1c, initial slope). In addition, TTX reduced the threshold current to 55 ± 12% of control values, and this reduction was also prevented by BDNF (Fig. 1c, threshold current). As reported previously (Rutherford et al. 1997, 1998; Turrigiano et al. 1998; Desai et al. 1999a), TTX treatment for 2 days with or without BDNF, or treatment with the TrkB–IgG, had no significant effect on the passive properties of pyramidal neurons (Fig. 2). We have also shown that these manipulations have no significant effect on neuronal survival (Rutherford et al. 1997).
Figure 1.
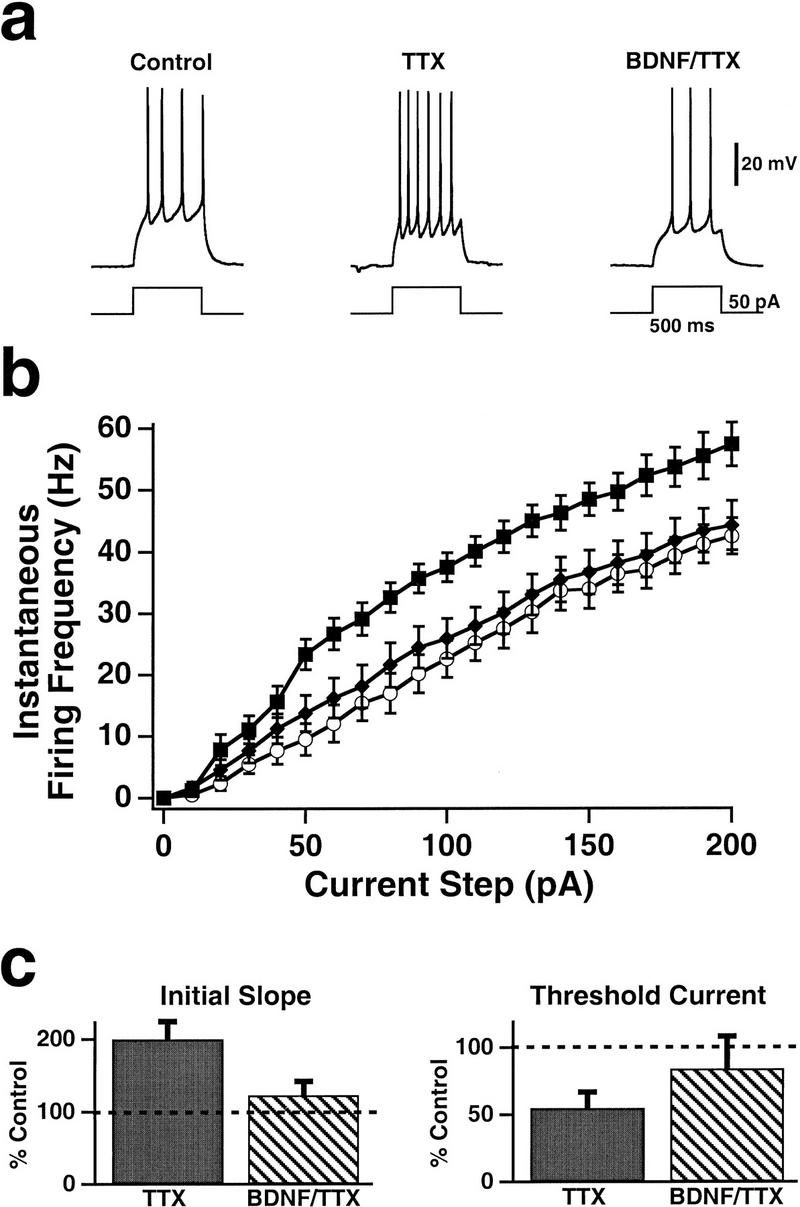
The increase in pyramidal neuron excitability produced by prolonged activity blockade was prevented by BDNF. (a) Sample spike trains evoked by somatic current injections in pyramidal neurons that had been maintained in control cultures (left), in cultures that had been treated with TTX for 2 days before experimentation (middle), and in cultures that had been treated with both TTX and BDNF over the same period (right). (b) Average FI curves measured in control (●; n = 18), TTX-treated (█; n = 18), and BDNF/TTX-treated (♦; n = 10) pyramidal neurons. (c) The slope of the initial, linear part of the FI curve (left) and the threshold current (right) for treated neurons, plotted as a percentage of those for control neurons (0.28 ± 0.01 Hz/pA and 39 ± 3 pA, respectively).
Figure 2.

Passive properties were unaltered by two-day incubation in TTX, BDNF/TTX, TrkB–IgG, or TrkA–IgG. (a) The resting input resistance was the same across conditions for both pyramidal neurons and interneurons. (b) The resting potential was also the same across conditions. None of the differences in any of the figures is significant. The numbers of cells in each condition are given in Figs. 1, 3, and 4.
To determine the role of the endogenous release of TrkB ligands on the intrinsic excitability of cortical neurons, we incubated cultures with a TrkB–IgG fusion protein. This chimeric protein binds endogenous TrkB ligands, which include BDNF and NT-4/5 with high affinity, and prevents them from activating endogenous TrkB receptors (Shelton 1995; Cabelli 1997; Rutherford et al. 1998). Treatment for two days with the TrkB–IgG fusion protein increased the intrinsic excitability of pyramidal neurons, so that once the TrkB–IgG was washed out, they fired faster than control neurons for a given current injection (Fig. 3a). The initial slope of the FI curve was increased by the TrkB–IgG, and the threshold current was reduced (Fig. 3c). In contrast, incubation with a TrkA–IgG, which binds NGF with high affinity, had no effect on intrinsic excitability (Fig. 3b,c), indicating that the increased excitability produced by the TrkB–IgG is not a nonspecific effect of the fusion protein.
Figure 3.
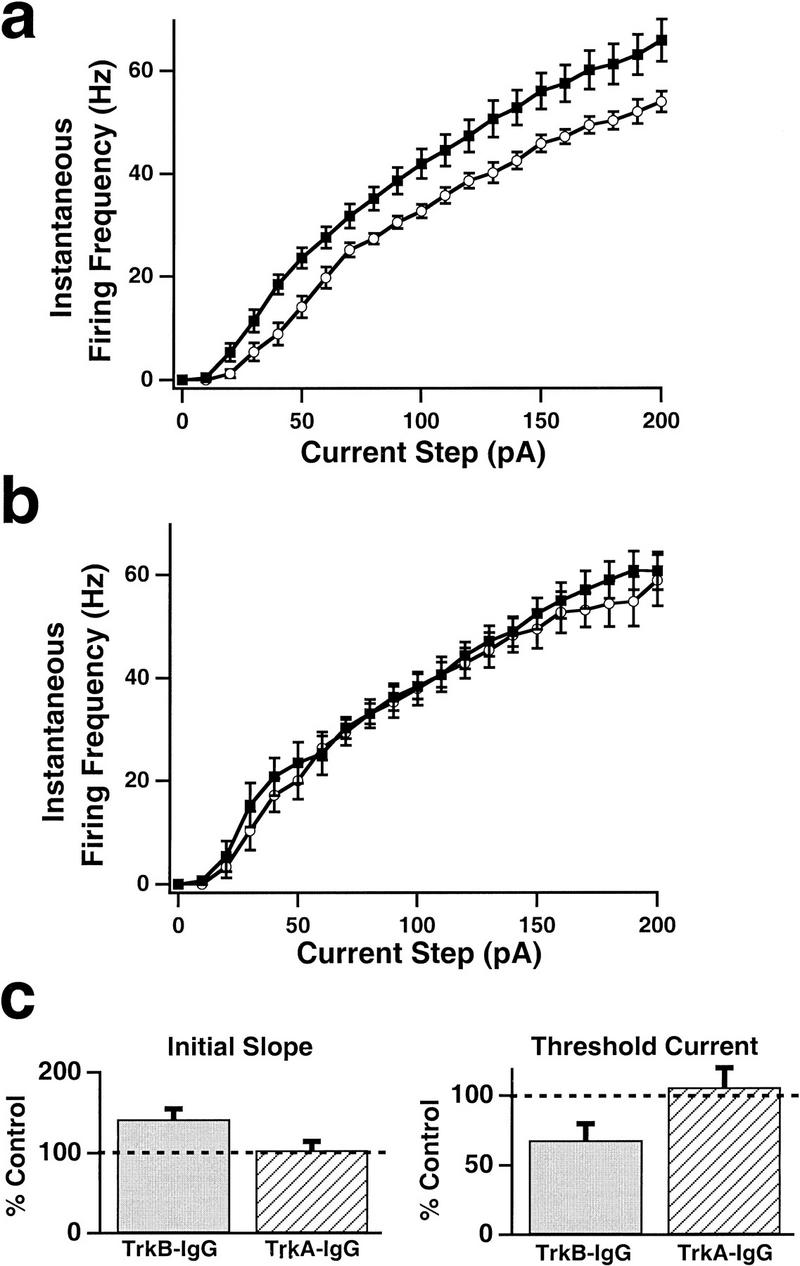
Reducing the endogenous activation of TrkB receptors for 2 days increased pyramidal neuron excitability, but reducing that of TrkA receptors had no effect. (a) Average FI curves measured in control (○; n = 13) and TrkB–IgG-treated (█; n = 14) pyramidal neurons. (b) Average FI curves measured in control (○; n = 7) and TrkA–IgG-treated (█; n = 8) pyramidal neurons. (c) The initial FI curve slope (left) and threshold current (right) of neurons treated with TrkB–IgG and TrkA–IgG fusion proteins, plotted as a percentage of control values.
An interesting aspect of activity-dependent scaling of synaptic strengths is that the direction of change of excitatory synapses depends on the identity of the postsynaptic neuron. Whereas activity blockade increases the strength of excitatory synapses onto pyramidal neurons, it has no effect on the strength of the excitatory synapses onto interneurons (Turrigiano et al. 1998). In addition, BDNF scales pyramidal excitatory synaptic strengths down, but scales interneuronal excitatory synaptic strengths up (Rutherford et al. 1998). This raises the question of whether the changes in intrinsic excitability produced by activity blockade also depend on neuronal identity.
To address this, we incubated cultures for 2 days with TTX and then recorded from a group of readily identifiable interneurons, with a bipolar morphology characterized by oval somata with one or two processes extending from either end (Rutherford et al. 1997, 1998). This population of cells likely includes more than one morphological subtype among inhibitory interneurons, with bipolar and bitufted interneurons predominating (Fairen et al. 1984; Gonchar and Burkhalter 1997; Sekirnjak et al. 1997). They also exhibited features stereotypically associated with inhibitory interneurons in cortex (McCormick et al. 1985; Connors and Gutnick 1990; Benardo and Wong 1995; Thomson et al. 1996): They were observed to be GABA-positive when immunostained against GABA (Rutherford et al. 1997), and dual recordings in culture revealed that this group of cells formed inhibitory connections onto pyramidal neurons. As was the case for pyramidal neurons, activity blockade increased the intrinsic excitability of these bipolar interneurons (Fig. 4). The FI curve was shifted left and up in activity-deprived neurons relative to control neurons, with a significant difference in firing rate for all current injections between 60 and 150 pA (Fig. 4a,b), and the initial slope of the FI curve was significantly increased (Fig. 4c). As for pyramidal neurons, these effects were prevented by coapplication of BDNF during the period of activity blockade (Fig. 4). Although the change in the FI curve was in the same direction as for pyramidal neurons, the magnitude of the effect was smaller. The increase in the slope of the FI curve was only 38 ± 8% for interneurons (Fig. 4c), compared to 100 ± 14% for pyramidal neurons (Fig. 1c), and for interneurons the reduction in the threshold current was not significant (Fig. 4c).
Figure 4.
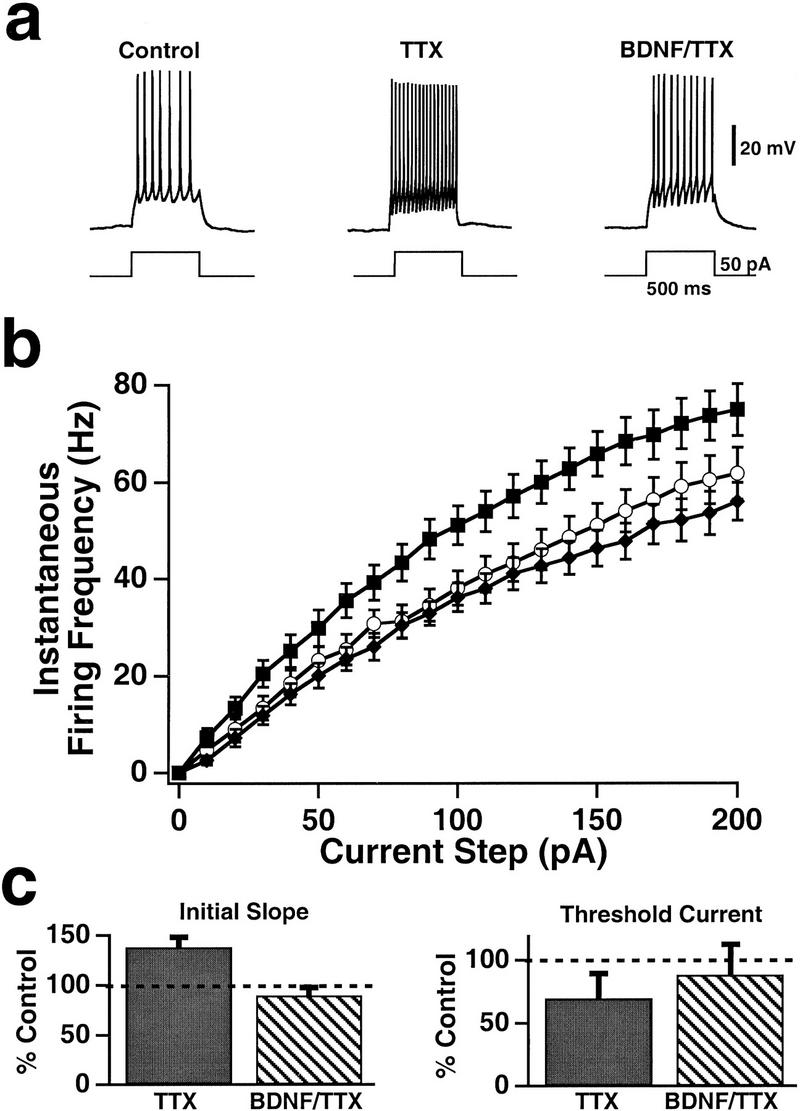
Activity blockade increased the excitability of interneurons, and this increase was prevented by BDNF. (a) Sample spike trains evoked by somatic current injections in bipolar interneurons that had been maintained in control cultures (left), in cultures that had been treated with TTX for 2 days before experimentation (middle), and in cultures that had been treated with both TTX and BDNF over the same period (right). (b) Average FI curves measured in control (○; n = 16), TTX-treated (█; n = 16), and BDNF/TTX-treated (♦; n = 12) interneurons. (c) The initial FI curve slope (left) and threshold current (right) for treated interneurons, plotted as a percentage of those for control interneurons (0.42 ± 0.02 Hz/pA and 25 ± 4 pA, respectively).
Discussion
We have shown that activity modifies the intrinsic excitability of cultured cortical neurons through a BDNF-dependent mechanism. In both pyramidal and interneurons, activity blockade produces a leftward shift in the FI curve, increases the slope of the curve, and reduces the threshold current. These effects are prevented by coapplication of BDNF during the period of activity blockade. In addition, scavenging BDNF and other endogenous TrkB ligands mimics the effects of activity blockade. Taken together these data strongly suggest that reduced activity increases the intrinsic excitability of cortical neurons by reducing the release of endogenous ligands of TrkB.
Cortical networks, both in vivo and in vitro, are composed of extensive recurrent excitatory and inhibitory networks. BDNF appears to have a crucial role in the homeostatic regulation of cortical excitability, by selectively modifying excitation and inhibition within these networks (Rutherford et al. 1997, 1998). When activity rises and BDNF production increases, the properties of in vitro cortical networks are modified to favor inhibition, whereas when activity falls and BDNF production is reduced, the properties of these networks are altered to favor excitation (Rutherford et al. 1998). This shift in the balance of excitation and inhibition occurs through modifications in many properties of the network, including changes in the amount of inhibitory current received by pyramidal neurons (Rutherford et al. 1997) and the differential regulation of excitatory synaptic strengths onto pyramidal and interneurons (Rutherford et al. 1998; Turrigiano et al. 1998). Here we show that BDNF also modifies the intrinsic excitability of cortical neurons. One important remaining question is whether the effects of activity on both synaptic scaling and intrinsic excitability are mediated directly by BDNF or by another TrkB ligand, NT-4/5. Experiments with the TrkB–IgG fusion protein are consistent with endogenous release of either neurotrophin being the determining factor, though BDNF seems the more likely candidate because of its well-established role in neuronal plasticity and because of its demonstrated ability to block both activity-dependent processes. What is clear from these data is that the relevant TrkB ligand exerts its effects on network excitability through a complex and coordinated regulation of many distinct properties of cortical networks.
Interestingly, the intrinsic excitabilities of interneurons and pyramidal neurons are regulated in the same direction by BDNF. This is in contrast to the effects of BDNF on excitatory synaptic strengths, which change in opposite directions, depending on whether the target is a pyramidal or an interneuron (Rutherford et al. 1998). This demonstrates that in the case of interneurons, excitatory synaptic strengths and the function relating current to firing rate can be differentially regulated. This may be important, because changes in synaptic strength and changes in intrinsic excitability are unlikely to be functionally equivalent. Whereas synaptic scaling can modify the gain of excitation and inhibition independently, a change in the slope of the FI curve will alter the effectiveness of both excitatory and inhibitory inputs, thus modifying the sensitivity of the neuron to all synaptic drive. This raises the interesting possibility that activity-dependent regulation of neuronal FI curves is a general mechanism for adjusting the sensitivity of a neuron to all of its inputs, whereas synaptic scaling may be a more selective process that can differentially adjust the gain of different classes of synaptic input to modify the balance of excitation and inhibition within a network.
Theoretical and experimental work has suggested that the ability of neurons to selectively regulate different classes of voltage-dependent conductances as a function of activity may have a number of important consequences. Such selective regulation can change neuronal firing patterns from tonic firing to bursting (LeMasson et al. 1993; Turrigiano et al. 1994, 1995), could help to equalize the effectiveness of inputs coming into different parts of a dendritic arbor (Siegel et al. 1994), and could allow neurons to adapt the range of firing rates they use to match the range of their inputs (Stemmler and Koch 1999). Although we have not examined here which ionic conductances are affected by BDNF or TrkB–IgG treatment, we can be certain that the neurotrophin-dependent changes in excitability are mediated by changes in voltage-gated currents, as passive properties were unaltered by these treatments. Moreover, the most likely explanation of the present results is that BDNF acts on the sodium and persistent potassium currents because these were the currents that were selectively regulated by TTX treatment (Desai et al. 1999a,b). These studies suggest that by differentially regulating the magnitudes of voltage-dependent conductances, neurotrophins could have an important role in modifying the integrative properties of cortical neurons.
Acknowledgments
This work was supported by grants K02 NS01893 and RO1 NS36853. N.S.D. was supported by an individual National Research Service Award. G.G.T. is an Alfred P. Sloan Fellow.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
References
- Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Benardo LS, Wong RKS. Inhibition in the cortical network. In: Gutnick MJ, Mody I, editors. The cortical neuron. New York, NY: Oxford University Press; 1995. pp. 141–155. [Google Scholar]
- Blochl A, Thoenen H. Characterization of nerve growth factor (NGF) release from hippocampal neurons: Evidence for a constitutive and an unconventional sodium-dependent regulated pathway. Eur J Neurosci. 1995;7:1220–1228. doi: 10.1111/j.1460-9568.1995.tb01112.x. [DOI] [PubMed] [Google Scholar]
- Cabelli RJ, Hohn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- Cabelli RJ, Allendoerfer KL, Radeke MJ, Welcher AA, Feinstein SC, Shatz CJ. Changing patterns of expression and subcellular localization of TrkB in the developing visual system. J Neurosci. 1996;14:7965–7980. doi: 10.1523/JNEUROSCI.16-24-07965.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabelli RJ, Shelton DL, Segal RA, Shatz CJ. Blockade of endogenous ligands of TrkB inhibits formation of ocular dominance columns. Neuron. 1997;19:63–76. doi: 10.1016/s0896-6273(00)80348-7. [DOI] [PubMed] [Google Scholar]
- Castrén E, Zafra F, Thoenen H, Lindholm D. Light regulates expression of brain-derived neurotrophic factor mRNA in rat visual cortex. Proc Natl Acad Sci. 1992;89:9444–9448. doi: 10.1073/pnas.89.20.9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellarino A, Maffei L, Domenici L. The distribution of brain-derived neurotrophic factor and its receptor TrkB in parvalbumin-containing neurons of the rat visual cortex. Eur J Neurosci. 1996;8:1190–1197. doi: 10.1111/j.1460-9568.1996.tb01287.x. [DOI] [PubMed] [Google Scholar]
- Connors BW, Gutnick MJ. Intrinsic firing patterns of diverse neocortical neurons. Trends Neurosci. 1990;13:99–104. doi: 10.1016/0166-2236(90)90185-d. [DOI] [PubMed] [Google Scholar]
- Constantine-Paton M, Cline HT. LTP and activity-dependent synaptogenesis: The more alike they are, the more different they become. Curr Opin Neurobiol. 1998;8:139–148. doi: 10.1016/s0959-4388(98)80017-2. [DOI] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG. Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat Neurosci. 1999a;2:515–520. doi: 10.1038/9165. [DOI] [PubMed] [Google Scholar]
- Desai, N.S., X.-J. Wang, and G.G. Turrigiano. 1999b. Activity-dependent changes in the balance of inward and outward currents can account for changes in pyramidal neuron excitability. Nat. Neurosci. (web supplement, http://neurosci.nature.com).
- Fairen A, DeFelipe J, Regidor J. Nonpyramidal neurons. In: Peters A, Jones EG, editors. Cerebral cortex. Vol. 1. New York, NY: Plenum Press; 1984. pp. 201–253. [Google Scholar]
- Galuske RA, Kim DS, Castrén E, Thoenen H, Singer W. Brain-derived neurotrophic factor reverses experience-dependent synaptic modifications in kitten visual cortex. Eur J Neurosci. 1996;8:1554–1559. doi: 10.1111/j.1460-9568.1996.tb01618.x. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- Gonchar Y, Burkhalter A. Three distinct families of GABAergic neurons in rat visual cortex. Cereb Cortex. 1997;7:347–358. doi: 10.1093/cercor/7.4.347. [DOI] [PubMed] [Google Scholar]
- Gonzalez M, Collins WF., III Modulation of motoneuron excitability by brain-derived neurotrophic factor. J Neurophysiol. 1997;77:502–506. doi: 10.1152/jn.1997.77.1.502. [DOI] [PubMed] [Google Scholar]
- Isackson PJ, Huntsman MM, Murray KD, Gall CM. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: Temporal patterns of induction distinct from NGF. Neuron. 1991;6:937–948. doi: 10.1016/0896-6273(91)90234-q. [DOI] [PubMed] [Google Scholar]
- Kokaia Z, Bengzon J, Metsis M, Kokaia M, Persson H, Lindvall O. Coexpression of neurotrophins and their receptors in neurons of the central nervous system. Proc Natl Acad Sci. 1993;90:6711–6715. doi: 10.1073/pnas.90.14.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeMasson G, Marder E, Abbott LF. Activity-dependent regulation of conductances in model neurons. Science. 1993;259:1915–1917. doi: 10.1126/science.8456317. [DOI] [PubMed] [Google Scholar]
- Lesser SS, Sherwood NT, Lo DC. Neurotrophins differentially regulate voltage-gated ion channels. Mol Cell Neurosci. 1997;10:173–183. doi: 10.1006/mcne.1997.0656. [DOI] [PubMed] [Google Scholar]
- Lissin DV, Gomperts SN, Carroll RC, Christine CW, Kalman D, Kitamura M, Hardy S, Nicoll RA, Malenka RC, von Zastrow M. Activity differentially regulates the surface expression of AMPA and NMDA glutamate receptors. Proc Natl Acad Sci. 1998;95:7097–7102. doi: 10.1073/pnas.95.12.7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- ————— Neurotrophin regulation of cortical dendritic growth requires activity. Neuron. 1996;17:1057–1064. doi: 10.1016/s0896-6273(00)80239-1. [DOI] [PubMed] [Google Scholar]
- ————— Opposing roles for BDNF and NT-3 in regulating cortical dendritic growth. Neuron. 1997;18:767–778. doi: 10.1016/s0896-6273(00)80316-5. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Connors BW, Lighthall JW, Prince DA. Comparative electrophysiology of pyramidal and sparsely spiny stellate neurons of the neocortex. J Neurophysiol. 1985;54:782–806. doi: 10.1152/jn.1985.54.4.782. [DOI] [PubMed] [Google Scholar]
- Miranda RC, Sohrabji F, Toran-Allerand CD. Neuronal colocalization of mRNAs for neurotrophins and their receptors in the developing central nervous system suggests a potential for autocrine interactions. Proc Natl Acad Sci. 1993;90:6439–6443. doi: 10.1073/pnas.90.14.6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- Oyelese AA, Rizzo MA, Waxman SG, Kocsis JD. Differential effects of NGF and BDNF on axotomy-induced changes in GABAA-receptor-mediated conductance and sodium currents in cutaneous afferent neurons. J Neurophysiol. 1997;78:31–42. doi: 10.1152/jn.1997.78.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford LC, DeWan A, Lauer HM, Turrigiano GG. BDNF mediates the activity-dependent regulation of inhibition in neocortical cultures. J Neurosci. 1997;17:4527–4535. doi: 10.1523/JNEUROSCI.17-12-04527.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- Sekirnjak C, Martone ME, Weiser M, Deernick T, Bueno E, Rudy B, Ellisman M. Subcellular localization of the K+ channel subunit Kv3.1b in selected rat CNS neurons. Brain Res. 1997;766:173–187. doi: 10.1016/s0006-8993(97)00527-1. [DOI] [PubMed] [Google Scholar]
- Shatz CJ. Impulse activity and the patterning of connections during CNS development. Neuron. 1990;5:745–746. doi: 10.1016/0896-6273(90)90333-b. [DOI] [PubMed] [Google Scholar]
- Shelton DL, Sutherland J, Gripp J, Camerato T, Armanini MP, Phillips HS, Carroll L, Spencer SD, Levinson AD. Human trks: Molecular cloning, tissue distribution, and expression of extracellular domain immunoadhesins. J Neurosci. 1995;15:477–491. doi: 10.1523/JNEUROSCI.15-01-00477.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood NT, Lesser SS, Lo DC. Neurotrophin regulation of ionic currents and cell size depends on cell context. Proc Natl Acad Sci. 1997;94:5917–5922. doi: 10.1073/pnas.94.11.5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel M, Marder E, Abbott LF. Activity-dependent current distributions in model neurons. Proc Natl Acad Sci. 1994;91:11308–11312. doi: 10.1073/pnas.91.24.11308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmler M, Koch C. How voltage-dependent conductances can adapt to maximize the information encoded by neuronal firing rate. Nat Neurosci. 1999;2:521–527. doi: 10.1038/9173. [DOI] [PubMed] [Google Scholar]
- Thomson AM, West DC, Hahn J, Deuchars J. Single axon IPSPs elicited in pyramidal cells by three classes of interneurones in slices of rat neocortex. J Physiol. 1996;496:81–102. doi: 10.1113/jphysiol.1996.sp021667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. Homeostatic plasticity in neuronal networks: The more things change, the more they remain the same. Trends Neurosci. 1999;22:221–227. doi: 10.1016/s0166-2236(98)01341-1. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Abbott LF, Marder E. Activity-dependent changes in the intrinsic properties of cultured neurons. Science. 1994;264:974–977. doi: 10.1126/science.8178157. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, LeMasson G, Marder E. Selective regulation of current densities underlies spontaneous changes in the activity of cultured neurons. J Neurosci. 1995;15:3640–3652. doi: 10.1523/JNEUROSCI.15-05-03640.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Wetmore C, Olson L, Bean AJ. Regulation of brain-derived neurotrophic factor (BDNF) expression and release from hippocampal neurons is mediated by non-NMDA type glutamate receptors. J Neurosci. 1994;14:1688–1700. doi: 10.1523/JNEUROSCI.14-03-01688.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra F, Castrén E, Thoenen H, Lindholm D. Interplay between glutamate and gamma-aminobutyric acid transmitter systems in the physiological regulation of brain-derived neurotrophic factor and nerve growth factor synthesis in hippocampal neurons. Proc Natl Acad Sci. 1991;88:10037–10041. doi: 10.1073/pnas.88.22.10037. [DOI] [PMC free article] [PubMed] [Google Scholar]