

Abstract
The induction of long-term potentiation (LTP) and long-term depression (LTD) at excitatory synapses in the hippocampus can be strongly modulated by patterns of synaptic stimulation that otherwise have no direct effect on synaptic strength. Likewise, patterns of synaptic stimulation that induce LTP or LTD not only modify synaptic strength but can also induce lasting changes that regulate how synapses will respond to subsequent trains of stimulation. Collectively known as metaplasticity, these activity-dependent processes that regulate LTP and LTD induction allow the recent history of synaptic activity to influence the induction of activity-dependent changes in synaptic strength and may thus have an important role in information storage during memory formation. To explore the cellular and molecular mechanisms underlying metaplasticity, we investigated the role of metaplasticity in the induction of LTP by υ-frequency (5-Hz) synaptic stimulation in the hippocampal CA1 region. Our results show that brief trains of υ-frequency stimulation not only induce LTP but also activate a process that inhibits the induction of additional LTP at potentiated synapses. Unlike other forms of metaplasticity, the inhibition of LTP induction at potentiated synapses does not appear to arise from activity-dependent changes in NMDA receptor function, does not require nitric oxide signaling, and is strongly modulated by β-adrenergic receptor activation. Together with previous findings, our results indicate that mechanistically distinct forms of metaplasticity regulate LTP induction and suggest that one way modulatory transmitters may act to regulate synaptic plasticity is by modulating metaplasticity.
Studies of the potential cellular mechanisms responsible for information storage during learning have focused on forms of synaptic plasticity that involve activity-dependent changes in synaptic strength, such as those seen in long-term potentiation (LTP) and long-term depression (LTD). Accumulating evidence suggests, however, that even patterns of synaptic activity that have no direct effect on synaptic strength can nonetheless modify synaptic function by inducing long-lasting changes that regulate LTP and/or LTD induction by subsequent patterns of synaptic activity (Abraham and Bear 1996; Abraham and Tate 1997). For instance, some patterns of synaptic stimulation that have no direct effect on synaptic strength at lateral perforant path synapses onto granule cells in the dentate gyrus can enhance the induction of LTP by subsequent υ burst stimulation (Christie et al. 1995). Conversely, in the hippocampus some patterns of synaptic activity that have no lasting, direct effect on synaptic strength at Schaffer collateral fiber synapses onto CA1 pyramidal cells can induce a persistent inhibition of LTP induction by subsequent high-frequency stimulation (Huang et al. 1992; Fujii et al. 1996; Abraham and Huggett 1997). This regulation of LTP and LTD induction by prior pre- and postsynaptic activity is known as metaplasticity (Abraham and Bear 1996; Abraham and Tate 1997).
Compared with our current understanding of the cellular and molecular processes responsible for synaptic plasticity, relatively little is known about the mechanisms underlying metaplasticity. The inhibition of LTP induction by prior synaptic stimulation is blocked when the conditioning stimulation is delivered in the presence of NMDA receptor antagonists (Huang et al. 1992; Fujii et al. 1996; Abraham and Huggett 1997), suggesting that NMDA receptor activation during conditioning stimulation has a crucial role in this form of metaplasticity (see also Coan et al. 1989). Although the signaling pathways downstream of NMDA receptor activation have not yet been well characterized, these pathways may inhibit the induction of LTP by producing a persistent inhibition of NMDA receptor activity that raises the threshold for LTP induction (Selig et al. 1995). Because nitric oxide synthase (NOS) inhibitors can both prevent the inhibition of LTP induction produced by transient NMDA receptor activation (Izumi et al. 1992a) and facilitate NMDA receptor-mediated synaptic currents in CA1 pyramidal cells (Kato and Zorumski 1993), it has been suggested that some forms of metaplasticity may arise from a NO-dependent, negative-feedback modulation of NMDA receptors (Abraham and Tate 1997) or other targets (Zorumski and Izumi 1998). Consistent with this notion, Murphy and Bliss (1999) have recently shown that a brief pulse of photolytically released NO induces both a long-lasting depression of NMDA receptor-mediated synaptic potentials and a persistent inhibition of LTP induction in the CA1 region of hippocampal slices.
Although NMDA receptor modulation may contribute to the metaplastic inhibition of LTP induction by some patterns of synaptic stimulation, it seems likely that other processes are also involved. For instance, the induction of LTP by high-frequency stimulation is not typically associated with a depression of NMDA receptor-mediated synaptic responses in CA1 pyramidal cells (Kauer et al. 1988; Muller et al. 1988; Muller and Lynch 1990; Perkel and Nicoll 1993; but see Bernard and Wheal 1995). Under some experimental conditions, high-frequency stimulation can induce LTP of NMDA receptor-mediated synaptic responses (Bashir et al. 1991; Asztely et al. 1992; Clark and Collingridge 1995; see also O'Conner et al. 1995). Yet, high-frequency synaptic stimulation can induce metaplastic changes that inhibit the induction of additional LTP (Frey et al. 1995) and/or facilitate the subsequent induction of LTD (i.e., depotentiation) (Barrionuevo et al. 1980; Staubli and Lynch 1990; Fujii et al. 1991; Wexler and Stanton 1993; O'Dell and Kandel 1994; Wagner and Alger 1995). Thus, although long-term modifications of NMDA receptor function may be involved in some forms of metaplasticity, it is unclear what role, if any, these modifications might have in the inhibition of additional LTP induction and facilitation of LTD induction at potentiated synapses.
In this study we investigated the mechanisms underlying the inhibition of additional LTP induction at previously potentiated synapses. Our results indicate that neither NO signaling nor an activity-dependent down-regulation of NMDA receptor activity has an important role in this form of metaplasticity. In contrast to a previous report showing that β-adrenergic receptor activation has no effect on the inhibition of LTP induction produced by low levels of NMDA receptor activation (Izumi et al. 1992b), we find that the β-adrenergic receptor agonist isoproterenol (ISO) re-enables the induction of LTP at potentiated synapses. Our results suggest that distinct metaplastic mechanisms regulate LTP induction at excitatory synapses onto hippocampal CA1 pyramidal cells.
Materials and Methods
ELECTROPHYSIOLOGY
Standard techniques were used to obtain 400-μm thick hippocampal slices from halothane anesthetized, 4- to 6-week-old, male C57BL/6 mice. Slices were maintained in an interface-type slice chamber (Fine Science Tools, Inc.) perfused (2–3 ml/min) with a warm (30°C) artificial cerebrospinal fluid (ACSF) containing 124 mm NaCl, 4.4 mm KCl, 25 mm NaHCO3, 1 mm NaH2PO4, 1.2 mm MgSO4, 2.0 mm CaCl2, and 10 mm glucose that was gassed with 95% O2/5% CO2. Slices were allowed to recover for at least 1 hr following slice preparation, and unless indicated otherwise, all experiments were done at 30°C. Techniques described previously were used to record Schaffer collateral/commissural fiber stimulation-evoked excitatory postsynaptic potentials (EPSPs) in the CA1 pyramidal cells (Thomas et al. 1998; Makhinson et al. 1999). In general, field EPSPs (fEPSPs) evoked at 0.02 Hz were recorded using glass microelectrodes (filled with ACSF, resistance = 5–10 MΩ) placed in stratum radiatum. In all experiments the intensity of presynaptic fiber stimulation was adjusted to evoke fEPSPs that were 50% of the maximal fEPSP amplitude determined at the beginning of each experiment. Baseline measurements of fEPSPs were collected for at least 20 min prior to any experimental manipulation.
Whole-cell voltage-clamp techniques were used to record NMDA receptor-mediated excitatory postsynaptic currents (EPSCs) evoked by presynaptic stimulation pulses delivered at 0.05 Hz. In these experiments the CA3 region was removed, slices were placed in a submerged-slice recording chamber, and experiments were performed at room temperature. Low resistance (3–5 MΩ) patch-clamp electrodes were filled with a solution containing 122.5 Cs gluconate, 17.5 mm CsCl, 10 mm TEA-Cl, 0.2 mm EGTA, 2 mm Mg-ATP, 0.3 mm GTP, and 10 mm HEPES (pH 7.2). Cells were voltage-clamped at −60 mV using an Axopatch 1-D amplifier, and the NMDA receptor-mediated component of the EPSC was isolated by bathing slices in a modified ACSF containing 3.5 mm CaCl2, 0.5 mm MgSO4, 10 μm CNQX or NBQX, and 100 μm picrotoxin. In control experiments we confirmed that the EPSCs recorded in these conditions were blocked by the NMDA receptor antagonist 2-amino-5-phosphonovaleric acid (d,l-APV, 100 μm, N = 5; data not shown). In all voltage-clamp experiments, a 30-msec duration voltage step (2 mV) was applied 50–60 msec before each synaptic stimulation pulse to monitor input and access resistance throughout the experiment. Experiments where access resistance (typically 15–25 MΩ) changed by >16% were excluded from the analysis.
DATA ANALYSIS
The delivery of presynaptic fiber stimulation pulses as well as data acquisition and analysis were performed using the Experimenter's Workbench and Common Processing software package (Data Wave Technologies). All values are listed as means ± s.e. Complex spike bursting during 5-Hz stimulation was measured as described previously (Thomas et al. 1998). For statistical comparisons, responses recorded at various time points before and after stimulation were averaged over 5-min periods. Statistical significance of within-group comparisons in experiments in which more than one stimulation train was delivered were determined using one-way repeated measures ANOVAs (multiple pairwise comparisons were performed using Student–Newman–Keuls tests). Paired t-tests were used for within-group comparisons in experiments in which only one stimulation protocol was delivered. Unpaired t-tests or, where appropriate, one-way ANOVAs were used to compare results between different experiments (Bonferroni tests were used for multiple comparisons to control groups).
Results
AN NMDA RECEPTOR-DEPENDENT FORM OF METAPLASTICITY SUPPRESSES THE INDUCTION OF LTP BY MULTIPLE TRAINS OF 5-hz stimulation
We investigated the metaplastic effects of LTP-inducing patterns of synaptic stimulation using brief trains of υ-frequency (5-Hz) stimulation that produce a modest, submaximal level of LTP in the CA1 region of mouse hippocampal slices (Thomas et al. 1996, 1998). As shown in Figure 1A, whereas a high-frequency stimulation protocol consisting of two, 100-pulse trains of 100-Hz stimulation (intertrain interval = 10 sec) induced an approximately twofold potentiation of synaptic transmission, a single 5-Hz stimulation train consisting of 75 pulses produced a robust, but more modest, level of potentiation. To determine whether additional 5-Hz stimulation trains could progressively increase synaptic strength up to some saturating level, we examined the amount of LTP induced by two 75-pulse trains of 5-Hz stimulation delivered with an intertrain interval of 20 min. As shown in Figure 1B, although the first 5-Hz stimulation train induced a significant potentiation, no additional potentiation was induced by the second 5-Hz stimulation train. The amount of potentiation observed at the end of these experiments (fEPSPs were 154.1 ± 6.9% of baseline 35–40 min after the first 5-Hz train) was not significantly different (unpaired t-test) from that seen in slices where only a single 5-Hz stimulation train was delivered (fEPSPs were 143.4 ± 6.6% of baseline 35–40 minutes after the first 5-Hz train). A shorter train of 5-Hz stimulation (25 pulses) that induced a smaller, but significant (P < 0.01, paired t-test), initial potentiation (fEPSPs were potentiated to 118.7 ± 3.8% of baseline in control experiments, N = 6) also inhibited the induction of significant additional LTP by 75 pulses of 5-Hz stimulation. In these experiments a 75-pulse train of 5-Hz stimulation delivered 20 min later induced only a small (≅13%), nonsignificant increase in synaptic strength above that induced by the initial 25-pulse 5-Hz stimulation train (20 min after the second 5-Hz stimulation train fEPSPs were 131.4 ± 6.7% of baseline, N = 5). Thus, 5-Hz stimulation of naive synapses not only induces LTP but also appears to activate a metaplastic process that inhibits the induction of additional LTP by subsequent 5-Hz stimulation. In experiments in which two stimulation electrodes were used to activate independent groups of presynaptic fibers, we found that a 75-pulse train of 5-Hz stimulation to one pathway did not prevent the induction of normal levels of LTP by a 75-pulse train of 5-Hz stimulation delivered 20 min later to the second pathway (N = 7; data not shown). This indicates that the inhibition of LTP induction by prior 5-Hz stimulation is a homosynaptic form of metaplasticity that suppresses the induction of LTP only at previously potentiated synapses.
Figure 1.
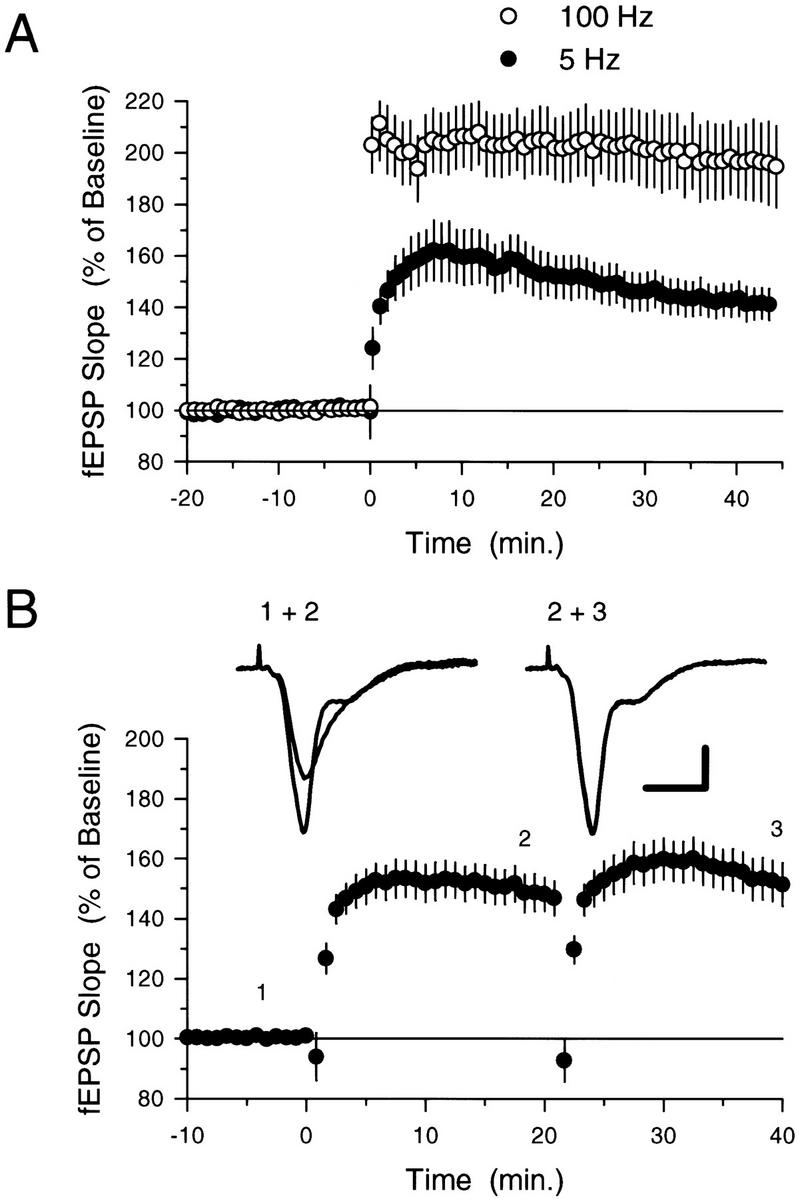
Five-hertz stimulation induces a submaximal, but “saturating,” level of LTP. (A) Following a 20-min period of baseline recording, LTP was induced by either two 100-pulse trains of 100-Hz stimulation (○, N = 9) or 75 stimulation pulses delivered at 5 Hz (●, N = 11). Although 5-Hz stimulation induced a significant potentiation of synaptic transmission (fEPSPs were potentiated to 142.3 ± 6.3% of baseline, P < 0.001 compared with pre-5-Hz stimulation baseline, paired t-test), a larger level of potentiation was generated by 100-Hz stimulation (45 min after 100-Hz stimulation fEPSPs were 195.9 ± 15.5% of baseline). (B) Seventy-five pulses of 5-Hz stimulation delivered to potentiated synapses fails to induce additional LTP. Two trains of 5-Hz stimulation (75 pulses) were delivered with an intertrain interval of 20 min starting at time = 0. Only the last 10 min of the 20-min baseline recording period are shown. Note that although a clear potentiation was induced by the first train of 5-Hz stimulation (fEPSPs were 150.0 = 5.9 % of baseline 20 min after 5-Hz stimulation), the second train had little effect on synaptic strength. The traces show fEPSPs recorded in a typical experiment at the times indicated by the numbers. Calibration bars, 2 mV and 5 mSec.
Previous studies of the inhibition of LTP induction produced by synaptic stimulation protocols that otherwise fail to induce persistent changes in synaptic strength have revealed a number of key characteristics of this form of metaplasticity (for review, see Abraham and Tate 1997). First, although the LTP-inhibiting effects of prior synaptic stimulation are long lasting, the suppression of LTP induction decays with time such that normal levels of LTP can be induced by high-frequency stimulation delivered 60 min or more after prior conditioning stimulation (Huang et al. 1992; Fujii et al. 1996; Abraham and Huggett 1997; see also Frey et al. 1995). Consistent with this, we found that significant levels of additional LTP could be produced at potentiated synapses if the second train of 5-Hz stimulation was delivered 50, rather than 20, min after the first 5-Hz stimulation train (Fig. 2A,B). Second, the mechanisms responsible for the metaplastic inhibition of LTP induction by prior synaptic stimulation do not produce a complete block of LTP induction but instead appear to increase the threshold level of synaptic activity required to induce LTP (Huang et al. 1992). Similarly, we found that significant amounts of additional LTP could be induced by higher frequency synaptic stimulation trains delivered 20 min after LTP was first induced by 5-Hz stimulation (Fig. 2C). In these experiments, fEPSPs potentiated to 151.5 ± 13.5% of baseline 15–20 min after LTP was initially induced by 75 pulses of 5-Hz stimulation and then further increased to 184.9 ± 14.9% of baseline 15–20 min after a 15-sec long train of 20-Hz stimulation (N = 6, P < 0.05, Student–Newman–Keuls comparison to potentiated level induced by 5-Hz stimulation; see Fig. 2C). Likewise, we observed that two 100-pulse trains of 100-Hz stimulation [intertrain interval (ITI) = 10 sec, trains delivered 20 min after LTP induction] also induced significant levels of additional LTP (Fig. 2C). In these experiments fEPSPs potentiated to 158.1 ± 13.7% of baseline 15–20 min after LTP was initially induced by 75 pulses of 5-Hz stimulation and then further increased to 222.9 ± 18.7% of baseline 15–20 min after 100-Hz stimulation (N = 9, P < 0.05, Student–Newman–Keuls comparison to potentiated level induced by 5-Hz stimulation). Finally, previous studies have shown that conditioning stimulation delivered in the presence of NMDA receptor blockers does not inhibit subsequent LTP induction, suggesting that NMDA receptor activation during conditioning stimulation is required for the induction of this form of metaplasticity (Huang et al. 1992; Fujii et al. 1996; Abraham and Huggett 1997). Likewise, we found that the when the first train of 5-Hz stimulation was delivered in the presence of 50 μm d,l-APV, the induction of LTP was completely blocked, and following APV washout, a second train of 5-Hz stimulation delivered 20 min after the first train induced normal levels of LTP (Fig. 3A).
Figure 2.
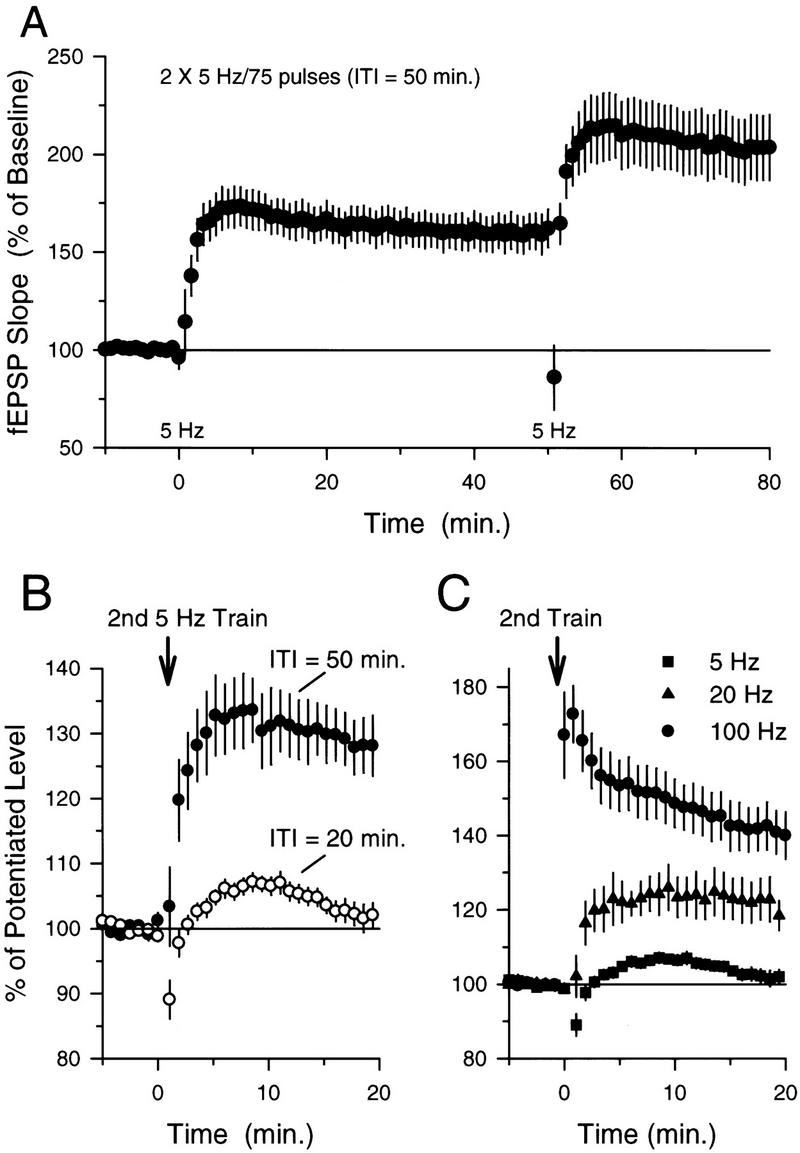
The induction of LTP by 5-Hz stimulation induces a transient inhibition of additional LTP induction that can be overcome with higher frequency synaptic stimulation. (A) LTP was induced by delivering 75 pulses of 5-Hz stimulation at time = 0 (only the last 10 min of the 20-min baseline recording period is shown). A second train of 5-Hz stimulation delivered 50 min later induces a significant (P < 0.05, Student–Newman–Keuls test) additional potentiation of synaptic transmission. (B) Summary of the effects of ITI on LTP induction by multiple trains of 5-Hz stimulation. The effects of a second train of 5-Hz stimulation on synaptic transmission delivered either 20 min after LTP induction (○) or 50 min after LTP induction (●) are shown superimposed for comparison. Although a second train of 5-Hz stimulation delivered 20 min after LTP was induced had no effect on synaptic strength (fEPSPs were 102.2 ± 1.7% of the potentiated baseline), a clear potentiation was induced when the second train of 5-Hz stimulation was delivered 50 min after LTP was initially induced (fEPSPs potentiated to 124 ± 4% above the potentiated baseline). Data are from experiments shown in Figs. 1B and 2A (this figure). fEPSPs have been normalized to the size of the potentiated fEPSPs recorded over the last 5 min prior to the second train of 5-Hz stimulation. (C) Summary of the effects of different patterns of synaptic stimulation on the induction of LTP at potentiated synapses. Responses have been normalized as in B. The second train of synaptic stimulation was delivered at time = 0 (corresponding to 20 min after the initial induction of LTP). Whereas 75 pulses of 5-Hz stimulation induces no further potentiation of synaptic transmission (█; data from B), both 300 pulses of 20-Hz stimulation (▴, N = 6, fEPSPs potentiated to 121.5 ± 5.3% of the potentiated baseline) and two 1-sec-long trains of 100-Hz stimulation (●, N = 9, fEPSPs potentiated to 140.9 ± 6.1% of the potentiated baseline) induce significant levels of additional LTP (P < 0.5, Student–Newman–Keuls test).
Figure 3.
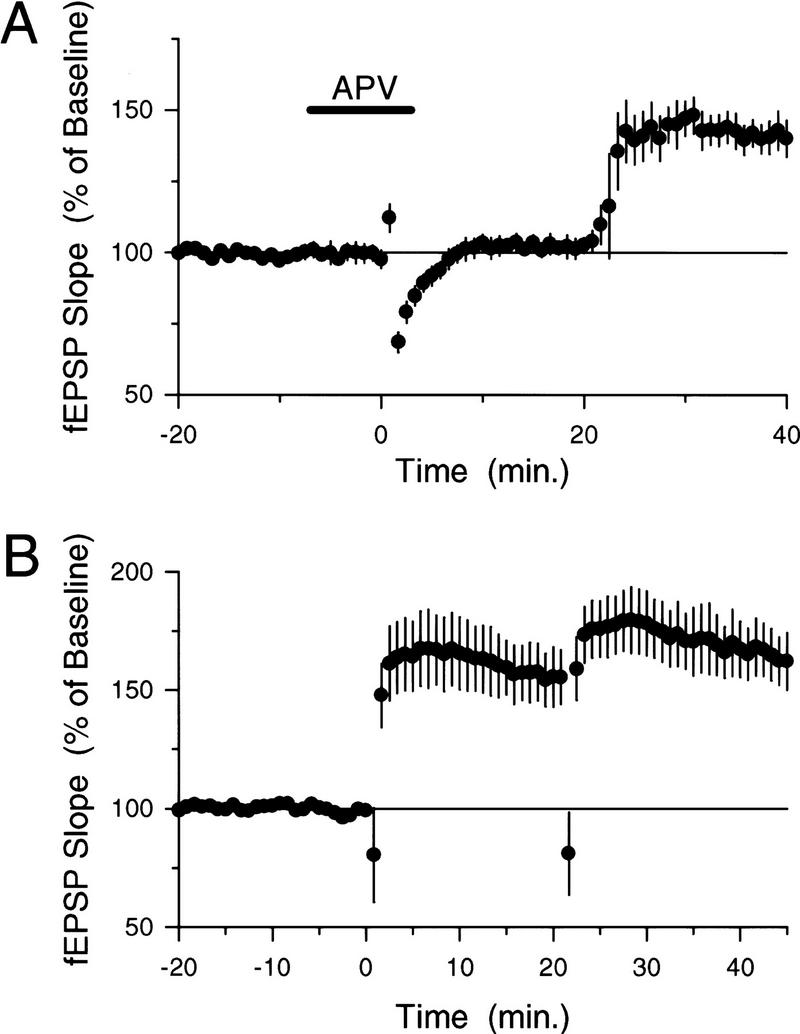
NMDA receptor activation, but not NO signaling, is required for the metaplastic inhibition of LTP induction by 5-Hz stimulation. (A) The NMDA receptor blocker d,l-APV (50 μm) was applied for the duration indicated by the bar to block NMDA receptors during the first of two trains of 5-Hz stimulation (first train delivered at time = 0, ITI = 20 min). APV completely blocked the induction of LTP by the first train of 5-Hz stimulation (fEPSPs were 101.7 ± 3.2% of baseline 20 min after the first 5-Hz stimulation train, N = 6). Following APV washout, the second train of 5-Hz stimulation induced a clear potentiation of synaptic transmission (fEPSPs potentiated to 140.6 ± 5.2% of baseline 20 min after the second 5-Hz stimulation train). (B) The NOS inhibitor L-NAME does not prevent the inhibition of additional LTP induction at potentiated synapses. Slices (N = 6) were continuously bathed in L-NAME (100 μm), and two trains of 5-Hz stimulation (ITI = 20 min) were delivered starting at time = 0. Whereas the first train of 5-Hz stimulation induced a significant potentiation in the presence of L-NAME (fEPSPs were potentiated to 156.3 ± 12% of baseline 15–20 min after the first train, P < 0.05 compared with pre-5-Hz stimulation baseline, Student–Newman–Keuls test), the second 5-Hz stimulation train failed to significantly enhance synaptic transmission above the potentiated level (Student–Newman–Keuls comparison with the level of potentiation induced by the first train, fEPSPs were 167.1 ± 11.8 % of baseline 15–20 min after the second train).
A NO-DEPENDENT INHIBITION OF NMDA RECEPTOR ACTIVITY IS NOT RESPONSIBLE FOR THE SUPPRESSION OF ADDITIONAL LTP INDUCTION AT POTENTIATED SYNAPSES
Because previous studies have suggested that a persistent depression of NMDA receptor-mediated synaptic responses (Selig et al. 1995), perhaps mediated by NO (Murphy and Bliss 1999), may be involved in at least some forms of metaplasticity (Abraham and Tate 1997), we examined whether this mechanism might contribute to the inability of 5-Hz stimulation to induce additional LTP at potentiated synapses. To explore the potential role of NO signaling, we examined the effects of the NOS inhibitor l-nitroarginine methyl ester (L-NAME) on the amount of potentiation induced by two trains of 5-Hz stimulation delivered with a 20-min ITI. L-NAME (100 μm) had no effect on the amount of potentiation induced by the first train of 5-Hz stimulation, and, as in control experiments (Fig. 1B), the second train of 5-Hz stimulation failed to induce a further increase in synaptic strength (Fig. 3B). Because the high concentration of L-NAME used in our experiments inhibits the induction of LTP by some protocols (Böhme et al. 1991; Haley et al. 1992; Ko and Kelly 1999), the results shown in Figure 3B suggest that the inhibition of additional LTP induction at potentiated synapses does not arise from mechanisms that require NO production. Furthermore, the lack of an effect of L-NAME on the amount of LTP induced by the first train of 5-Hz stimulation in these experiments indicates that 5-Hz stimulation induces an NO-independent form of LTP in the CA1 region of the hippocampus (at least during the first 20 min after 5-Hz stimulation), a finding consistent with previous observations that NO is not required for the induction of LTP by some patterns of synaptic stimulation (Gribkoff and Lum-Ragan 1992; Chetkovich et al. 1993; Haley et al. 1993; O'Dell et al. 1994; Malen and Chapman 1997).
To determine whether an activity-dependent inhibition of NMDA receptor-mediated EPSCs might contribute to the suppression of additional LTP at potentiated synapses, we examined the effects of 5-Hz stimulation on NMDA receptor-mediated EPSCs. After recording a stable baseline period of EPSCs, we paired a 75-pulse train of presynaptic fiber stimulation delivered at 5 Hz with postsynaptic depolarization to −40 mV (postsynaptic depolarization was used to partially mimic the depolarization produced by postsynaptic complex spike bursting induced by 5 Hz stimulation under normal conditions; see Thomas et al. 1998). Seventy-five stimulation pulses delivered at 5-Hz failed to induce a long-lasting depression of NMDA receptor-mediated EPSCs (Fig. 4A), suggesting that the inhibition of additional LTP induction at potentiated synapses is not due to a persistent modification of synaptic NMDA receptors produced by the first train of 5-Hz stimulation. The conditions in these voltage-clamp experiments were different from those used in our extracellular recording studies of 5-Hz stimulation-induced LTP, however, and it is possible that some of these differences may have influenced the effects of 5-Hz stimulation on NMDA receptor activity. We thus also examined the effects of 5-Hz stimulation on NMDA receptor-mediated fEPSPs using the approach outlined by Selig et al. (1995) (see also Muller et al. 1988). In these experiments we used two stimulating electrodes to activate independent groups of presynaptic fibers and induced LTP by a single train of 5-Hz stimulation in only one pathway. Twenty minutes after 5-Hz stimulation, a modified ACSF containing 10 μm NBQX, 100 μm picrotoxin, and 0.1 mm MgSO4 was applied onto the slices to pharmacologically isolate the NMDA receptor-mediated component of the fEPSPs. As shown in Figure 4B, the magnitude of the NMDA receptor component of the fEPSP (relative to the AMPA-receptor component recorded during baseline) was similar in both the unstimulated pathway and in the pathway that received 5-Hz stimulation. In control experiments (no 5-Hz stimulation) we found that the relative magnitude of the NMDA receptor-mediated EPSPs in two independent pathways was identical (not shown) as reported previously by Selig et al. (1995). Thus, even under the same conditions used in our studies of LTP induction we find no evidence supporting a role for LTD of NMDA receptor-mediated responses in the metaplastic effects of 5-Hz stimulation on LTP induction.
Figure 4.
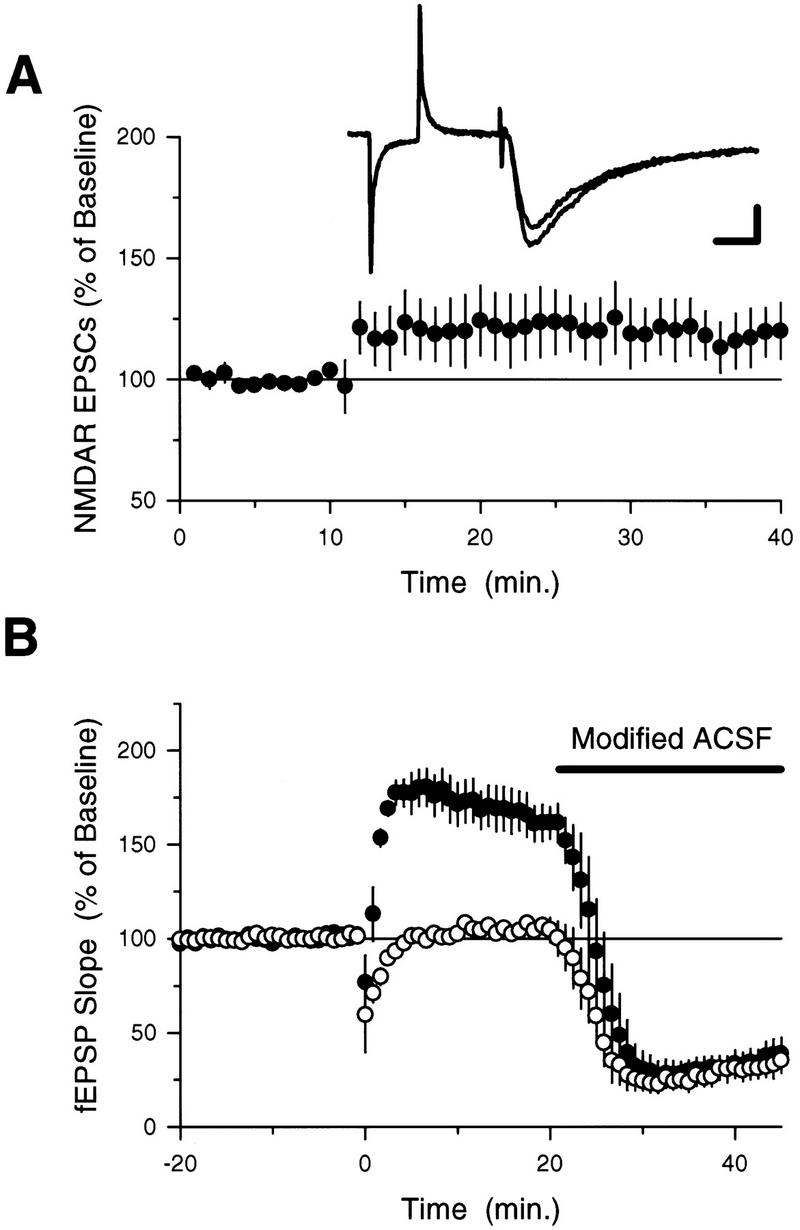
Brief trains of 5-Hz stimulation do not induce a lasting depression of NMDA receptor-mediated synaptic responses. (A) NMDA receptor-mediated EPSCs were recorded in CA1 pyramidal cells voltage-clamped at −60 mV. After a 10-min baseline period, 75 stimulation pulses delivered at 5 Hz were paired with postsynaptic depolarization to −40 mV (responses during 5-Hz stimulation not shown). On average, EPSCs were 114.8 ± 11.2% of baseline 40 min after pairing (N = 9) and not significantly different (paired t-test) from baseline. The traces show the average of three EPSCs recorded just prior to, and 30 min after (larger response), 5-Hz stimulation. Calibration bars, 25 pA and 25 msec. (B) Two pathway experiment investigating potential changes in NMDA receptor-mediated fEPSPs following 5-Hz stimulation. Two stimulating electrodes were used to activate independent groups of presynaptic fibers. At time = 0, LTP was induced in one pathway using 75 pulses of 5-Hz stimulation (●). Twenty minutes later a modified ACSF containing 10 μm NBQX, 100 μm picrotoxin, and 0.1 mm MgSO4 was applied to isolate the NMDA receptor component of the fEPSPs (fEPSPs recorded under these conditions were blocked by the NMDA receptor antagonist d,l-APV, 100 μm, N = 4; data not shown). Responses have been normalized to the AMPA receptor-mediated fEPSPs recorded during baseline. The NMDA component of the fEPSPs (normalized to the AMPA component recorded during baseline) in the potentiated pathway and the unstimulated pathway (no 5-Hz stimulation) are not significantly different (t-test).
β-adrenergic receptor activation overcomes the metaplastic inhibition of ltp induction induced by 5-hz stimulation
Because β-adrenergic receptor (βAR) activation has potent, activity-dependent effects on LTP induction in the CA1 region of the hippocampus (Thomas et al. 1996; Katsuki et al. 1997; Moody et al. 1998; Makhinson et al. 1999), we examined whether βAR activation might modulate 5-Hz stimulation-induced metaplasticity. Consistent with our previous observations (Thomas et al. 1996), we found that a 10-min application of the βAR agonist ISO (1.0 μm) had no effect on the amount of LTP induced by 75 pulses of 5-Hz stimulation delivered to naive synapses (Fig. 5A). Despite the fact that βAR activation has no effect on the induction of LTP by this protocol, ISO enabled the induction of additional LTP by a second 5-Hz stimulation train delivered to synapses potentiated 20 min earlier by the first 5-Hz train (Figs. 5B and 6A). ISO also enabled the induction of additional LTP by 5-Hz stimulation at synapses where a small amount of LTP was induced 20 min earlier by a 25-pulse train of 5-Hz stimulation (Fig. 6B). In control experiments, a 10-min application of 1.0 μm ISO delivered 10 min after LTP induction had no effect on the amount of potentiation induced by single trains of either 75 pulses or 25 pulses of 5-Hz stimulation (Fig. 6A,B). Thus, although activation of βAR has no effect on the amount of LTP induced by 75 pulses of 5-Hz stimulation at naive synapses and has no persistent effect on synaptic strength at potentiated synapses when applied after LTP induction, it opposes the metaplastic processes that suppress 5-Hz stimulation-induced LTP at potentiated synapses.
Figure 5.
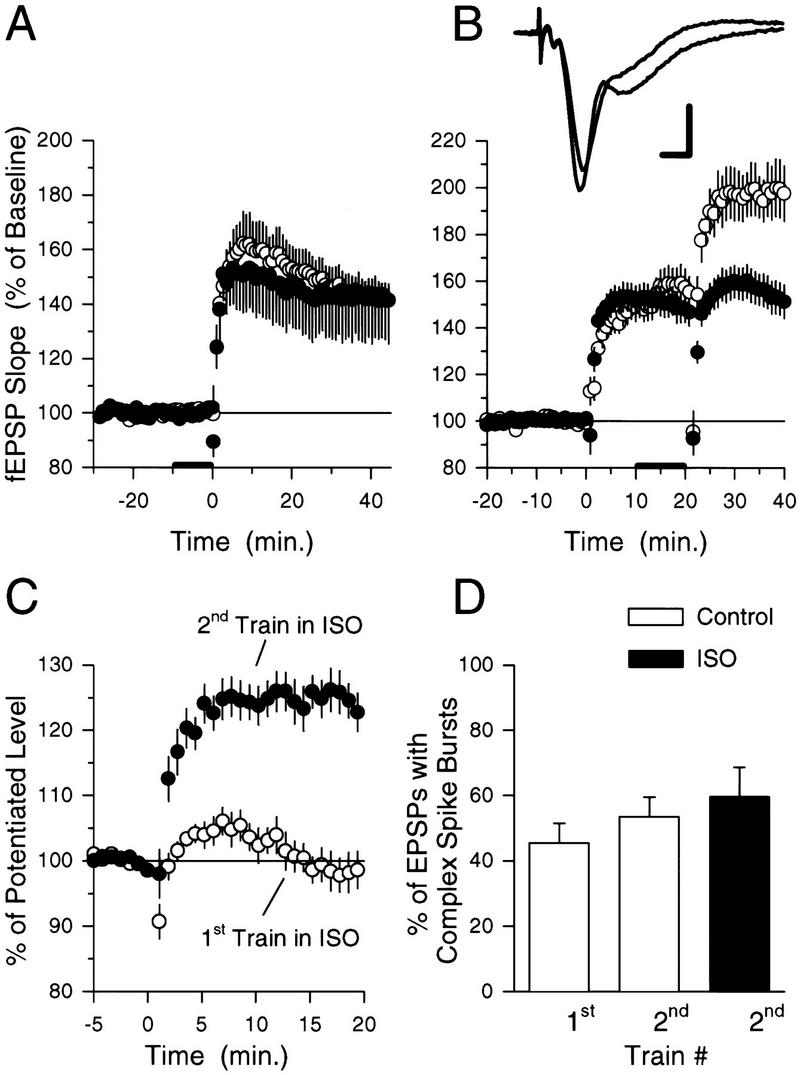
βAR activation enables additional LTP induction at potentiated synapses. (A) Seventy-five pulses of 5-Hz stimulation (delivered at time = 0) induced similar amounts of LTP in control slices (○, N = 13) and in slices to which 1.0 μm ISO was applied for 10 min prior to 5-Hz stimulation (●, N = 6). (B) Although ISO does not modulate the effects of 5-Hz stimulation at naive synapses, it enhances the induction of additional LTP at potentiated synapses. Ten minutes after the induction of LTP by 75 pulses of 5-Hz stimulation 1.0 μm ISO was bath-applied for 10 min before a second train of 5-Hz stimulation was delivered (○, N = 8). Note that the second train of 5-Hz stimulation now induces additional LTP. Results from control experiments (from Fig. 1B) showing the effects of two trains of 5-Hz stimulation delivered with an ITI of 20 min in the absence of ISO (●) are plotted for comparison. The traces show example fEPSPs recorded just before, and 20 min after (larger response), the second train of 5-Hz stimulation. Calibration bars, 2 mV and 2 mSec. (C) ISO enables the induction of additional LTP at potentiated synapses when present during the second, but not the first, train of 5-Hz stimulation. The plot shows the effects of a second train of 5-Hz stimulation delivered 20 min after LTP was initially induced by 75 pulses of 5-Hz stimulation (fEPSPs have been normalized to the size of the potentiated fEPSPs recorded over the last 5 min prior to the second 5-Hz stimulation train). The ●'s are from the experiment shown in B in which the second train of 5-Hz stimulation was delivered in the presence of ISO. The ○'s show the effects of the second train of 5-Hz stimulation in experiments in which the first train of 5-Hz stimulation was delivered at the end of a 10-min bath application of ISO (1.0 μm, N = 7). Note that βAR activation only enables the induction of additional LTP at potentiated synapses when present during the second train of 5-Hz stimulation. (D) Summary of the effects of ISO on complex spike bursting during 5-Hz stimulation. The open bars show the percentage of EPSPs during the 5-Hz stimulation trains that induce complex spike bursting in control experiments. The solid bar shows the percentage of EPSPs that evoke complex spike bursts in experiments in which the second train of 5-Hz stimulation was delivered in the presence of ISO. Note that ISO had no significant effect on the number of EPSP-evoked complex spike bursts elicited during the second train of 5-Hz stimulation. Complex spike bursting during 5-Hz stimulation was determined from the experiments shown in B.
Figure 6.
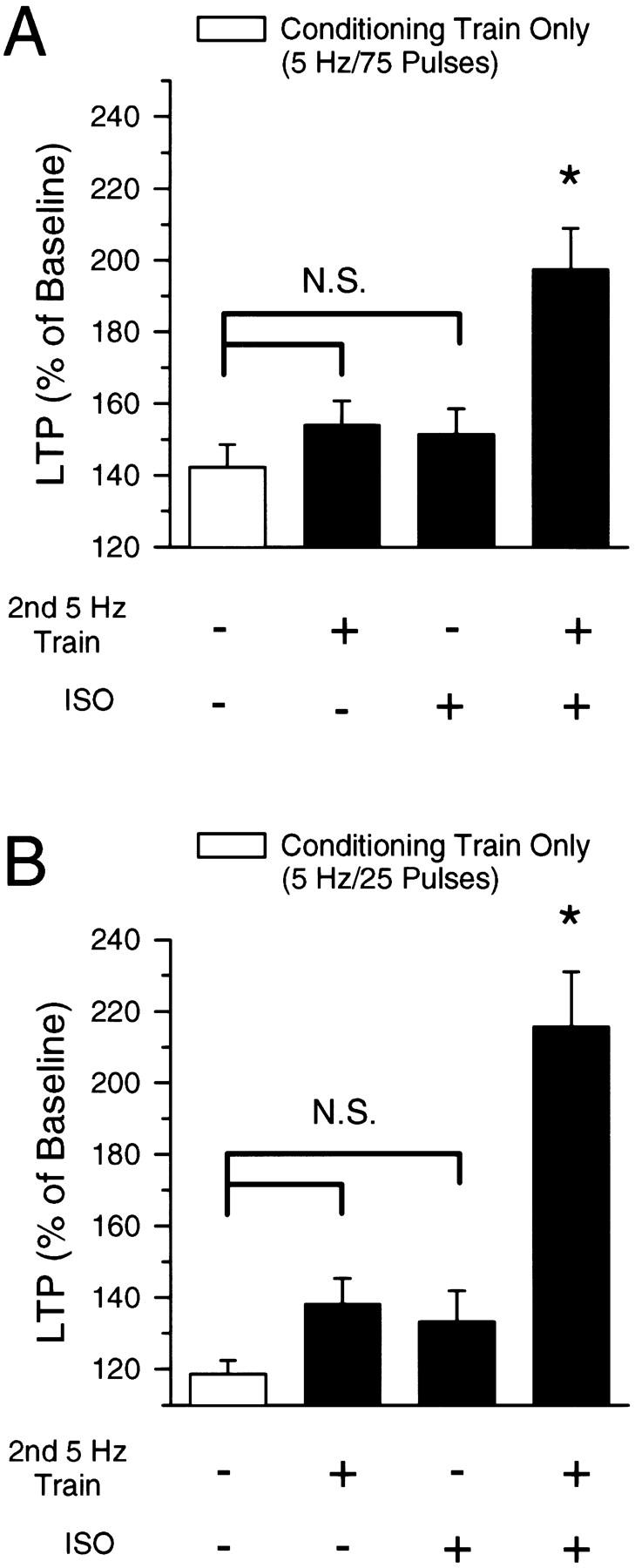
(A) Summary of the effects of 5-Hz stimulation on synaptic transmission. Values shown are the mean (±s.e.) amount of potentiation present 40–45 min after conditioning stimulation. The open bar shows the effects of a single, 75-pulse train of 5-Hz stimulation (conditioning stimulation only). Compared with the effects of conditioning stimulation alone, neither a second train of 5-Hz stimulation nor a 10-min application of 1.0 μm ISO (each delivered 20 min after conditioning stimulation) significantly enhanced the amount of potentiation seen 40 min after conditioning stimulation. In contrast, a significantly greater amount of potentiation was observed when a second train of 5-Hz stimulation was delivered in the presence of ISO (last bar). (*) P < 0.05; (N.S.) not significant (P > 0.05) Bonferroni test comparisons to conditioning stimulation alone. (B) Same as in A but with a 25-pulse train of 5-Hz stimulation as the conditioning stimulation. Only 75 pulses of 5-Hz stimulation in the presence of ISO (5-Hz stimulation delivered 20 min after conditioning stimulation) induced a significantly greater amount of LTP compared with 25 pulses of 5-Hz stimulation alone (P < 0.05, Bonferroni test).
In a separate series of experiments, we bath-applied 1.0 μm ISO for 10 min prior to the first of two 75-pulse trains of 5-Hz stimulation (ITI = 20 min) and then washed ISO from the recording chamber prior to delivering the second train of 5-Hz stimulation. As shown in Figure 5C, the presence of ISO during the first train of 5-Hz stimulation did not enable the induction of additional LTP by the second train of 5-Hz stimulation. Thus, although βAR activation opposes the metaplastic inhibition of LTP induction at potentiated synapses when applied during the second train of 5-Hz stimulation, it does not appear to prevent the induction of this form of metaplasticity.
The ability of EPSPs to evoke complex spike-like bursts of action potentials in CA1 pyramidal cells has a crucial role in the induction of LTP by 5-Hz stimulation (Thomas et al. 1998). Because the metaplastic inhibition of LTP induction at potentiated synapses appears to arise from an increase in the threshold for LTP induction, βAR activation might enable the induction of additional LTP by 5-Hz stimulation of potentiated synapses by producing changes in excitability that increase EPSP-evoked complex spike bursting during 5-Hz stimulation. We thus determined the effects of ISO on the amount of EPSP-evoked complex spike bursting induced during 5-Hz stimulation of potentiated synapses. In the experiments shown in Figure 5B, 59.7 ± 9% of the EPSPs evoked complex spike bursting when the second train of 5-Hz stimulation was delivered in ISO compared with 53.5 ± 6% evoked by the second train of 5-Hz stimulation in control experiments (see Fig. 5D). βAR activation thus does not appear to enable the induction of additional LTP at potentiated synapses by enhancing EPSP-evoked complex spike bursting during 5-Hz stimulation.
Discussion
THE BCM MODEL
A common observation in studies of LTP in the hippocampal CA1 region is that repeated trains of high-frequency synaptic stimulation induce incremental levels of potentiation until a maximal, or saturated, level of LTP is induced. However, a recent study by Frey et al. (1995) suggests that the saturation level of potentiation induced by multiple trains of high-frequency stimulation may not represent a true upper limit. Instead, LTP saturation may reflect the presence of a time-dependent metaplastic process that limits the ability of potentiated synapses to undergo additional LTP. Our experiments show that even patterns of synaptic stimulation that produce modest, “submaximal” levels of LTP can recruit processes that limit the ability of synaptic transmission to undergo additional potentiation. Even a very brief train of 5-Hz stimulation (25 pulses) that induced a marginal level of synaptic potentiation (20%) inhibited the induction of additional potentiation by 75 pulses of 5-Hz stimulation delivered 20 min later (see Fig. 6B).
In many ways the metaplastic inhibition of LTP induction by prior synaptic stimulation observed in our experiments, as well as in those of others (Huang et al. 1992; Fujii et al. 1996; for review, see Abraham and Tate 1997), is reminiscent of the sliding modification threshold regulating LTP induction in the Bienenstock, Cooper, Munroe (BCM) model of synaptic plasticity (Bienenstock et al. 1982; Bear et al. 1987). In the BCM model, low frequencies of synaptic stimulation that produce levels of coincident pre- and postsynaptic activity that fall below the modification threshold (υm) induce LTD, whereas higher frequencies of synaptic stimulation that produce levels of coincident synaptic activity that are above υm induce LTP. Importantly, υm is not a fixed property of the synapse but instead is modified in an activity-dependent manner. In the model, low frequencies of synaptic activation that fall below υm not only can induce LTD (or no change in synaptic strength) but also can shift υm towards lower frequencies, thus facilitating the induction of LTP by subsequent synaptic activity. Conversely, frequencies of synaptic activation that lie above υm not only induce LTP but also shift υm to higher frequencies and thus raise the threshold for LTP induction. According to the BCM model, a pattern of synaptic activation that lies just above υm will induce LTP of naive synapses but will subsequently fail to further influence synaptic strength (or even induce a depression, i.e., depotentiation) because of the shift in υm towards higher frequencies. Consistent with this, we found that a brief train of 5-Hz stimulation that elicited modest LTP at naive synapses, had little effect on synaptic strength at potentiated synapses, whereas additional LTP could be induced at potentiated synapses by stronger, 100-Hz stimulation trains. Our results, as well as previous findings (for reviews, see Deisseroth et al. 1995; Stanton 1996; Abraham and Tate 1997), thus indicate that metaplasticity may be the physiological manifestation of the “sliding” υm incorporated into the BCM model. However, as originally envisioned in the model, shifts in υm produce a cell-wide (i.e., heterosynaptic) change in the amounts of coincident synaptic activity needed for LTP or LTD induction (Bienenstock et al. 1982; Bear et al. 1987); yet in our experiments we observed that the inhibition of LTP by prior 5-Hz stimulation was homosynaptic (see also Huang et al. 1992). Because other studies have found evidence for heterosynaptic changes in υm (Holland and Wagner 1998), it seems likely that excitatory synapses onto pyramidal cells in the CA1 region of the hippocampus express both homosynaptic and heterosynaptic forms of metaplasticity.
MECHANISMS OF METAPLASTICITY
How might prior synaptic stimulation increase the threshold for LTP induction? One possibility is that an NMDA receptor-dependent activation of NOS produces a NO-mediated negative feedback of NMDA receptor activity that increases the threshold for LTP induction. Consistent with the idea that NMDA receptor activation has a crucial role, we observed that the effects of prior 5-Hz stimulation on LTP induction could be prevented if NMDA receptors were blocked with APV during the first 5-Hz stimulation train. The inhibition of additional LTP induction by 5-Hz stimulation delivered to potentiated synapses was not prevented by a high concentration of the NOS inhibitor L-NAME, however, suggesting that NO signaling is not involved. Moreover, using two different approaches we found no evidence for LTD of NMDA receptor-mediated synaptic responses following a 75-pulse train of 5-Hz stimulation (Fig. 4). Thus, neither NO signaling nor decreases in NMDA receptor activity seem to contribute to the metaplastic inhibition of LTP induction at potentiated synapses observed in our experiments. Our results do not rule out the possibility, however, that a NO-dependent down-regulation of NMDA receptor activity may be responsible for the inhibition of LTP induction following other types of synaptic stimulation.
An alternative mechanism that may underlie the activity-dependent increase in the threshold for LTP induction seen in our experiments is Ca2+/calmodulin trapping by autophosphorylated forms of the Ca2+/calmodulin-dependent protein kinase αCaMKII (Deisseroth et al. 1995; Mayford et al. 1995). Following activation by increases in intracellular Ca2+, αCaMKII can become persistently activated by autophosphorylation at a threonine residue (Thr-286) that resides within the inhibitory domain of αCaMKII (Miller and Kennedy 1986; Miller et al. 1988; Schworer et al. 1988; Thiel et al. 1988; Lou and Schulman 1989). Although this autophosphorylation event is thought to be important for the induction of LTP (Giese et al. 1998), perhaps by producing a persistent biochemical change that enhances postsynaptic AMPA receptor function (Lisman 1994; Barria et al. 1997; but see Otmakhov et al. 1997), autophosphorylation at this site also greatly (400-fold) enhances the affinity of αCaMKII for Ca2+/calmodulin (Meyer et al. 1992). Autophosphorylated αCaMKII can thus act as a calmodulin sink that limits calmodulin availability. Because the strength of synaptic transmission at excitatory synapses onto hippocampal CA1 pyramidal cells is regulated by opposing, Ca2+/calmodulin-activated protein kinase- and protein phosphatase-dependent signaling pathways (Malenka and Nicoll 1993; Malenka 1994; Coussens and Teyler 1996; Wang and Kelly 1996; Wang et al. 1997), changes in calmodulin availability could strongly influence how synaptic strength is modified by a given pattern of synaptic stimulation. Although the high calmodulin levels present at excitatory synapses in the hippocampus might limit the impact of Ca2+/calmodulin trapping by autophosphorylated CaMKII, levels of Ca2+/calmodulin may be a limiting factor in LTP induction under some experimental conditions (Wang and Kelly 1995). In the case of the 5-Hz stimulation trains used in our experiments, αCaMKII autophosphorylation induced during the first train of 5-Hz stimulation may inhibit the activation of the kinase-dependent mechanisms responsible for LTP or enhance the activation of the protein phosphatases that inhibit LTP induction during the second train of 5-Hz stimulation (or both). Consistent with the hypothesis that calmodulin-trapping by autophosphorylated αCaMKII contributes to the inhibition of additional LTP induction by 5-Hz stimulation of potentiated synapses, 5-Hz stimulation trains that induce low levels of LTP in the CA1 region of hippocampal slices from wild-type animals induce LTD in slices from transgenic animals expressing a mutant, persistently activated form of αCaMKII that traps calmodulin (Mayford et al. 1995). The calmodulin trapping by αCaMKII hypothesis also suggests that, with time, dephosphorylation of αCaMKII by protein phosphatases will reset (i.e., lower) the threshold level of coincident pre- and postsynaptic activity needed for LTP induction and re-enable the induction of LTP by 5-Hz stimulation. We found that the metaplastic inhibition of additional LTP induction at potentiated synapses fades with time following the initial induction of LTP (Fig. 2; see also Frey et al. 1995).
Although our results suggest that the metaplastic inhibition of LTP at potentiated synapses arises from changes in the activity of signaling molecules that lie downstream of NMDA receptor activation, rather than from a direct modification of NMDA receptor activity, they do not specifically implicate CaMKII in this process. Because activation of protein kinase C (PKC) with phorbol esters produces a persistent inhibition of LTP induction in the CA1 region of the hippocampus (Stanton 1995), PKC activation by the first train of 5-Hz stimulation could account for the inhibition of additional LTP induction at potentiated synapses. Unfortunately, the use of selective inhibitors of CaMKII and PKC to test the potential role of these two kinases in metaplasticity is not possible because both PKC and CaMKII inhibitors block LTP induction (for reviews, see Roberson et al. 1996; Wang et al. 1997).
βAR ACTIVATION AND METAPLASTICITY
The mechanisms responsible for metaplasticity may represent an important target through which modulatory transmitters can act to regulate LTP and LTD induction. For example, transmitters that up-regulate NMDA receptor activity could potently modulate forms of metaplasticity in which changes in NMDA receptor function underlie the shift in υmm that inhibits LTP induction. Alternatively, transmitters could regulate the effects of metaplasticity by activating signaling pathways that directly interact with the protein kinase- and protein phosphatase-dependent signaling pathways involved in LTP and LTD. Transmitters acting through the cAMP/protein kinase A (PKA) pathway might modulate metaplasticity via both sorts of mechanisms because PKA activation can directly oppose the effects of protein phosphatases on NMDA receptor activity (Raman et al. 1996) and may facilitate the induction of LTP by inhibiting the activity of protein phosphatase 1 (PP1) through a PKA-dependent phosphorylation of inhibitor-1, a PP1 regulatory protein (Blitzer et al. 1995, 1998; Thomas et al. 1996; Makhinson et al. 1999). Consistent with the idea that modulatory neurotransmitters can influence synaptic plasticity by regulating metaplasticity, we observed that βAR activation had no effect on the amount of LTP induced by 75 pulses of 5-Hz stimulation at naive synapses but enabled the induction of additional LTP by 75 pulses of 5-Hz stimulation at potentiated synapses (Figs. 5 and 6).
Previously, it was found that α-adrenergic receptor, but not βAR, activation potently inhibits the metaplastic inhibition of LTP induction produced by low levels of NMDA receptor activation (Izumi et al. 1992b). Although this may seem inconsistent with the effects of ISO observed in our experiments, we also found that the metaplastic inhibition of LTP induction at potentiated synapses is not prevented by NOS inhibitors, whereas previous studies have found that the inhibition of LTP induction produced by bath application of NMDA is blocked by NOS inhibitors (Izumi et al. 1992a). It thus seems likely that multiple metaplastic processes can regulate LTP induction at excitatory synapses onto CA1 pyramidal cells, and therefore, it may not be too surprising that these different forms of metaplasticity are differentially regulated by βAR activation.
How does βAR activation relieve the metaplastic inhibition of LTP induction at potentiated synapses? One possibility is that βAR activation overcomes the suppression of LTP induction at potentiated synapses by producing changes in postsynaptic excitability that facilitate NMDA receptor activation during 5-Hz synaptic stimulation. Compared with a second train of 5-Hz stimulation delivered to potentiated synapses in control experiments, ISO had no effect on the amount of EPSP-evoked complex spike bursting elicited by a second train of 5-Hz stimulation delivered to potentiated synapses. Changes in excitability that affect postsynaptic bursting thus do not account for the effects of βAR activation on metaplasticity. This observation does not rule out the possibility that more subtle changes in excitability might contribute to the effects of ISO, however. βAR activation has a number of effects on pyramidal cell excitability that could facilitate LTP induction (Heginbotham and Dunwiddie 1991; Dunwiddie et al. 1992; Sah and Bekkers 1996; Hoffman and Johnston 1999).
A second possibility is that βAR activation opposes the inhibition of additional LTP induction at potentiated synapses by regulating PP1 activity via a PKA-dependent phosphorylation of inhibitor-1 (Thomas et al. 1996; Makhinson et al. 1999). The induction of LTP by some patterns of synaptic stimulation, including 5-Hz stimulation, requires activation of Ca2+/calmodulin-dependent forms of adenylyl cyclase and a subsequent PKA-mediated suppression of PP1 (Blitzer et al. 1995, 1998; Thomas et al. 1996; Winder et al. 1998). Changes in Ca2+ signaling that occur as a result of LTP induction (e.g., because of calmodulin trapping by autophosphorylated αCaMKII) might thus hinder activation of Ca2+/calmodulin-dependent forms of adenylyl cyclase during 5-Hz stimulation of potentiated synapses, resulting in insufficient levels of PKA activation to inhibit the activity of protein phosphatases that oppose the induction of additional LTP. If changes in the ability of Ca2+ to activate adenylyl cyclase contributes to the inhibition of LTP induction at potentiated synapses, then stimulation of adenylyl cyclase following βAR activation may provide an alternative mechanism to achieve the PKA-mediated suppression of protein phosphatase activity needed to enable LTP induction. Although such a mechanism is consistent with our observations, our findings do not rule out other potential signaling mechanisms. One alternative possibility is that activation of the mitogen-associated protein (MAP) kinase signaling pathway following βAR activation (Daaka et al. 1997; Roberson et al. 1999) is involved because MAP kinase activation is required for LTP induction (English and Sweatt 1997).
In summary, our results show that brief trains of 5-Hz stimulation not only induce LTP of excitatory synapses in the CA1 region of the hippocampus but also induce a time-dependent inhibition of additional LTP induction by low-frequency synaptic stimulation. Although some patterns of synaptic stimulation may induce a metaplastic inhibition of LTP through NO-dependent processes and/or modifications of NMDA receptor activity, we find that the inhibition of LTP induction at potentiated synapses is not affected by NOS inhibitors and does not appear to arise from a depression in NMDA receptor activity. Thus, mechanistically distinct forms of metaplasticity may regulate synaptic plasticity in the CA1 region of the hippocampus. Although the signaling pathways responsible for the form of metaplasticity observed in our experiments remain to be determined, our results suggest that these pathways may represent an important target where norepinephrine acting through βARs can regulate how different patterns of synaptic activity modify synaptic strength in the hippocampus.
Acknowledgments
We are grateful to M. Makhinson for comments on an earlier version of this manuscript. This work was supported by grants from the National Institute of Mental Health, the PEW Charitable Trusts, and by the UCLA Center on Aging and Dr. P. Gail Mahoney. T.J.O. is a member of the UCLA Brain Research Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
References
- Abraham WC, Bear MF. Metaplasticity: The plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- Abraham WC, Huggett A. Induction and reversal of long-term potentiation by repeated high-frequency stimulation in rat hippocampal slices. Hippocampus. 1997;7:137–145. doi: 10.1002/(SICI)1098-1063(1997)7:2<137::AID-HIPO3>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Abraham WC, Tate WP. Metaplasticity: A new vista across the field of synaptic plasticity. Prog Neurobiol. 1997;52:303–323. doi: 10.1016/s0301-0082(97)00018-x. [DOI] [PubMed] [Google Scholar]
- Asztely F, Wigstrom H, Gustafsson B. The relative contribution of NMDA receptor channels in the expression of long-term potentiation in the hippocampal CA1 region. Eur J Neurosci. 1992;4:681–690. doi: 10.1111/j.1460-9568.1992.tb00177.x. [DOI] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- Barrionuevo G, Schottler F, Lynch G. The effects of repetitive low frequency stimulation on control and “potentiated” synaptic responses in the hippocampus. Life Sci. 1980;27:2385–2391. doi: 10.1016/0024-3205(80)90509-3. [DOI] [PubMed] [Google Scholar]
- Bashir ZI, Alford S, Davies SN, Randall AD, Collingridge GL. Long-term potentiation of NMDA receptor-mediated synaptic transmission in the hippocampus. Nature. 1991;349:156–158. doi: 10.1038/349156a0. [DOI] [PubMed] [Google Scholar]
- Bear MF, Cooper LN, Ebner FF. A physiological basis for a theory of synapse modification. Science. 1987;237:42–48. doi: 10.1126/science.3037696. [DOI] [PubMed] [Google Scholar]
- Bernard CL, Wheal HV. Simultaneous expression of long-term depression of NMDA and long-term potentiation of AMPA receptor-mediated synaptic responses in the CA1 area of the kainic acid-lesioned hippocampus. Eur J Neurosci. 1995;7:1651–1655. doi: 10.1111/j.1460-9568.1995.tb01160.x. [DOI] [PubMed] [Google Scholar]
- Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: Orientation specificity and binocular interaction in visual cortex. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in the hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Connor JH, Brown CP, Wong T, Shenolikar S, Iyengar R, Landau EM. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998;280:1940–1943. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- Böhme GA, Bon C, Stutzmann J-M, Doble A, Blanchard J-C. Possible involvement of nitric oxide in long-term potentiation. Eur J Pharmacol. 1991;199:379–381. doi: 10.1016/0014-2999(91)90505-k. [DOI] [PubMed] [Google Scholar]
- Chetkovich DM, Klann E, Sweatt JD. Nitric oxide synthase-independent long-term potentiation in area CA1 of hippocampus. NeuroReport. 1993;5:919–922. doi: 10.1097/00001756-199307000-00020. [DOI] [PubMed] [Google Scholar]
- Christie BR, Stellwagen D, Abraham WC. Reduction of the threshold for long-term potentiation by prior theta-frequency synaptic activity. Hippocampus. 1995;5:52–59. doi: 10.1002/hipo.450050107. [DOI] [PubMed] [Google Scholar]
- Clark KA, Collingridge GL. Synaptic potentiation of dual-component excitatory postsynaptic currents in the rat hippocampus. J Physiol (Lond) 1995;482:39–52. doi: 10.1113/jphysiol.1995.sp020498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coan EJ, Irving AJ, Collingridge GL. Low-frequency activation of the NMDA receptor system can prevent the induction of LTP. Neurosci Lett. 1989;105:205–210. doi: 10.1016/0304-3940(89)90038-4. [DOI] [PubMed] [Google Scholar]
- Coussens CM, Teyler TJ. Protein kinase and phosphatase activity regulate the form of synaptic plasticity expressed. Synapse. 1996;24:97–103. doi: 10.1002/(SICI)1098-2396(199610)24:2<97::AID-SYN1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Bito H, Schulman H, Tsien RW. A molecular mechanism for metaplasticity. Curr Biol. 1995;5:1334–1338. doi: 10.1016/s0960-9822(95)00262-4. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Taylor M, Heginbotham LR, Proctor WR. Long-term increases in excitability in the CA1 region of rat hippocampus induced by β-adrenergic stimulation: Possible mediation by cAMP. J Neurosci. 1992;2:506–517. doi: 10.1523/JNEUROSCI.12-02-00506.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long-term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Frey U, Schollmeier K, Reymann KG, Seidenbecher T. Asymptotic hippocampal long-term potentiation in rats does not preclude additional potentiation at later phases. Neuroscience. 1995;67:799–807. doi: 10.1016/0306-4522(95)00117-2. [DOI] [PubMed] [Google Scholar]
- Fujii S, Saito K, Miyakawa H, Ito KI, Kato H. Reversal of long-term potentiation (depotentiation) induced by tetanus stimulation of the input to CA1 neurons of guinea pig hippocampal slices. Brain Res. 1991;555:112–122. doi: 10.1016/0006-8993(91)90867-u. [DOI] [PubMed] [Google Scholar]
- Fujii S, Kuroda Y, Miura M, Hidekazu F, Sasaki H, Kaneko K, Ito K-I, Chen Z, Kato H. The long-term suppressive effect of prior activation of synaptic inputs by low-frequency stimulation on induction of long-term potentiation in CA1 neurons of guinea pig hippocampal slices. Exp Brain Res. 1996;111:305–312. [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the α calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- Gribkoff VK, Lum-Ragan JT. Evidence for nitric oxide synthase inhibitor-sensitive and insensitive hippocampal synaptic potentiation. J Neurophysiol. 1992;68:639–642. doi: 10.1152/jn.1992.68.2.639. [DOI] [PubMed] [Google Scholar]
- Haley JE, Wilcox GL, Chapman PF. The role of nitric oxide in hippocampal long-term potentiation. Neuron. 1992;8:211–216. doi: 10.1016/0896-6273(92)90288-o. [DOI] [PubMed] [Google Scholar]
- Haley JE, Malen PL, Chapman PF. Nitric oxide synthase inhibitors block long-term potentiation induced by weak but not strong tetanic stimulation at physiological brain temperatures in rat hippocampal slices. Neurosci Lett. 1993;160:85–88. doi: 10.1016/0304-3940(93)90919-c. [DOI] [PubMed] [Google Scholar]
- Heginbotham LR, Dunwiddie TV. Long-term increases in the evoked population spike in the CA1 region of rat hippocampus induced by β-adrenergic receptor activation. J Neurosci. 1991;11:2519–2527. doi: 10.1523/JNEUROSCI.11-08-02519.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Johnston D. Neuromodulation of dendritic action potentials. J Neurophysiol. 1999;81:408–411. doi: 10.1152/jn.1999.81.1.408. [DOI] [PubMed] [Google Scholar]
- Holland LL, Wagner JJ. Primed facilitation of homosynaptic long-term depression and depotentiation in rat hippocampus. J Neurosci. 1998;18:887–894. doi: 10.1523/JNEUROSCI.18-03-00887.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-Y, Colino A, Selig DK, Malenka RC. The influence of prior synaptic activity on the induction of long-term potentiation. Science. 1992;255:730–733. doi: 10.1126/science.1346729. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Clifford DB, Zorumski CF. Inhibition of long-term potentiation by NMDA-mediated nitric oxide release. Science. 1992a;257:1273–1276. doi: 10.1126/science.1519065. [DOI] [PubMed] [Google Scholar]
- ————— Norepinephrine reverses N-methly-D-aspartate-mediated inhibition of long-term potentiation in rat hippocampal slices. Neurosci Lett. 1992b;142:163–166. doi: 10.1016/0304-3940(92)90364-d. [DOI] [PubMed] [Google Scholar]
- Kato K, Zorumski CF. Nitric oxide inhibitors facilitate the induction of hippocampal long-term potentiation by modulating NMDA responses. J Neurophysiol. 1993;70:1260–1263. doi: 10.1152/jn.1993.70.3.1260. [DOI] [PubMed] [Google Scholar]
- Katsuki H, Izumi Y, Zorumski CF. Noradrenergic regulation of synaptic plasticity in the hippocampal CA1 region. J Neurophysiol. 1997;77:3013–3020. doi: 10.1152/jn.1997.77.6.3013. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC, Nicoll RA. A persistent postsynaptic modification mediates long-term potentiation in the hippocampus. Neuron. 1988;1:911–917. doi: 10.1016/0896-6273(88)90148-1. [DOI] [PubMed] [Google Scholar]
- Ko GY, Kelly PT. Nitric oxide acts as a postsynaptic signaling molecule in calcium/calmodulin-induced synaptic potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 1999;19:6784–6794. doi: 10.1523/JNEUROSCI.19-16-06784.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE. The CaM kinase II hypothesis for the storage of synaptic memory. Trends Neurosci. 1994;17:406–412. doi: 10.1016/0166-2236(94)90014-0. [DOI] [PubMed] [Google Scholar]
- Lou LL, Schulman H. Distinct autophosphorylation sites sequentially produce autonomy and inhibition of the multifunctional Ca2+/calmodulin-dependent protein kinase. J Neurosci. 1989;9:2020–2032. doi: 10.1523/JNEUROSCI.09-06-02020.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhinson M, Chotiner JK, Watson JB, O'Dell TJ. Adenylyl cyclase activation modulates activity-dependent changes in synaptic strength and Ca2+/calmodulin-dependent kinase II autophosphorylation. J Neurosci. 1999;19:2500–2510. doi: 10.1523/JNEUROSCI.19-07-02500.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malen P, Chapman PF. Nitric oxide facilitates long-term potentiation, but not long-term depression. J Neurosci. 1997;17:2645–2651. doi: 10.1523/JNEUROSCI.17-07-02645.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC. Synaptic plasticity in the hippocampus: LTP and LTD. Cell. 1994;78:535–538. doi: 10.1016/0092-8674(94)90517-7. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: Multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- Mayford M, Wang J, Kandel ER, O'Dell TJ. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell. 1995;81:891–904. doi: 10.1016/0092-8674(95)90009-8. [DOI] [PubMed] [Google Scholar]
- Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 1992;256:1199–1201. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- Miller SG, Kennedy MB. Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation: A Ca2+-triggered molecular switch. Cell. 1986;44:861–870. doi: 10.1016/0092-8674(86)90008-5. [DOI] [PubMed] [Google Scholar]
- Miller SG, Patton BL, Kennedy MB. Sequences of autophosphorylation sites in neuronal type II CaM kinase that control Ca2+-independent activity. Neuron. 1988;1:593–604. doi: 10.1016/0896-6273(88)90109-2. [DOI] [PubMed] [Google Scholar]
- Moody TD, Thomas MJ, Makhinson M, O'Dell TJ. 5 Hz stimulation of CA3 pyramidal cell axons induces a β-adrenergic modulated potentiation at synapses on CA1, but not CA3, pyramidal cells. Brain Res. 1998;794:75–79. doi: 10.1016/s0006-8993(98)00217-0. [DOI] [PubMed] [Google Scholar]
- Muller D, Lynch G. Synaptic modulation of N-methyl-D-aspartate receptor mediated responses in hippocampus. Synapse. 1990;5:94–103. doi: 10.1002/syn.890050203. [DOI] [PubMed] [Google Scholar]
- Muller D, Joly M, Lynch G. Contributions of quisqualate and NMDA receptors to the induction and expression of LTP. Science. 1988;242:1694–1697. doi: 10.1126/science.2904701. [DOI] [PubMed] [Google Scholar]
- Murphy KPSJ, Bliss TVP. Photolytically released nitric oxide produces a delayed but persistent suppression of LTP in area CA1 of the rat hippocampal slice. J Physiol (Lond) 1999;515:453–462. doi: 10.1111/j.1469-7793.1999.453ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Conner JJ, Rowan MJ, Anwyl RA. Tetanically induced LTP involves a similar increase in the AMPA and NMDA receptor components of the excitatory postsynaptic current: Investigations of the involvement of mGlu receptors. J Neurosci. 1995;15:2013–2020. doi: 10.1523/JNEUROSCI.15-03-02013.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dell TJ, Kandel ER. Low-frequency stimulation erases LTP through an NMDA receptor-mediated activation of protein phosphatases. Learn & Mem. 1994;1:129–139. [PubMed] [Google Scholar]
- O'Dell TJ, Huang PL, Dawson TM, Dinerman JL, Snyder SH, Kandel ER, Fishman MC. Endothelial NOS and blockade of LTP by NOS inhibitors in mice lacking neuronal NOS. Science. 1994;265:542–546. doi: 10.1126/science.7518615. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Griffith LC, Lisman JE. Postsynaptic inhibitors of calcium/calmodulin-dependent protein kinase type II block induction but not maintenance of pairing-induced long-term potentiation. J Neurosci. 1997;17:5357–5365. doi: 10.1523/JNEUROSCI.17-14-05357.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkel DJ, Nicoll RA. Evidence for all-or-none regulation of neurotransmitter release: Implications for long-term potentiation. J Physiol (Lond) 1993;471:481–500. doi: 10.1113/jphysiol.1993.sp019911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Tong G, Jahr CE. β-Adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron. 1996;16:415–421. doi: 10.1016/s0896-6273(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Roberson ED, English JD, Sweatt JD. A biochemist's view of long-term potentiation. Learn & Mem. 1996;3:1–24. doi: 10.1101/lm.3.1.1. [DOI] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Bekkers JM. Apical dendritic location of slow afterhyperpolarization current in hippocampal pyramidal neurons: Implications for the integration of long-term potentiation. J Neurosci. 1996;16:4537–4542. doi: 10.1523/JNEUROSCI.16-15-04537.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selig DK, Hjelmstad GO, Herron C, Nicoll RA, Malenka RC. Independent mechanisms for long-term depression of AMPA and NMDA responses. Neuron. 1995;15:417–426. doi: 10.1016/0896-6273(95)90045-4. [DOI] [PubMed] [Google Scholar]
- Schworer CM, Colbran RJ, Keefer JR, Soderling TR. Ca2+/calmodulin-dependent protein kinase II: Identification of a regulatory autophosphorylation site adjacent to the inhibitory and calmodulin-binding domains. J Biol Chem. 1988;263:13486–13489. [PubMed] [Google Scholar]
- Stanton PK. Transient protein kinase C activation primes long-term depression and suppresses long-term potentiation of synaptic transmission in hippocampus. Proc Natl Acad Sci. 1995;92:1724–1728. doi: 10.1073/pnas.92.5.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— LTD, LTP, and the sliding threshold for long-term synaptic plasticity. Hippocampus. 1996;6:35–42. doi: 10.1002/(SICI)1098-1063(1996)6:1<35::AID-HIPO7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Staubli U, Lynch G. Stable depression of potentiated synaptic responses in the hippocampus with 1-5 Hz stimulation. Brain Res. 1990;513:113–118. doi: 10.1016/0006-8993(90)91096-y. [DOI] [PubMed] [Google Scholar]
- Thiel G, Czernik AJ, Gorelick F, Nairn AC, Greengard P. Ca2+/calmodulin-dependent protein kinase II: Identification of threonine-286 as the autophosphorylation site in the α subunit associated with the generation of Ca2+-independent activity. Proc Natl Acad Sci. 1988;85:6337–6341. doi: 10.1073/pnas.85.17.6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Moody T, Makhinson M, O'Dell TJ. Activity dependent β-adrenergic modulation of low-frequency stimulation induced LTP in the hippocampal CA1 region. Neuron. 1996;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Watabe AM, Moody TD, Makhinson M, O'Dell TJ. Postsynaptic complex spike bursting enables the induction of LTP by theta frequency synaptic stimulation. J Neurosci. 1998;18:7118–7126. doi: 10.1523/JNEUROSCI.18-18-07118.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JJ, Alger BE. GABAergic and developmental influences on homosynaptic LTD and depotentiation in rat hippocampus. J Neurosci. 1995;15:1577–1586. doi: 10.1523/JNEUROSCI.15-02-01577.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Kelly PT. Postsynaptic injection of Ca2+/CaM induces synaptic potentiation requiring CaMKII and PKC activity. Neuron. 1995;15:443–452. doi: 10.1016/0896-6273(95)90048-9. [DOI] [PubMed] [Google Scholar]
- ————— The balance between postsynaptic Ca2+-dependent protein kinase and phosphatase activities controlling synaptic strength. Learn & Mem. 1996;3:170–181. doi: 10.1101/lm.3.2-3.170. [DOI] [PubMed] [Google Scholar]
- Wang JH, Ko GY, Kelly PT. Cellular and molecular bases of memory: Synaptic and neuronal plasticity. J Clin Neurophysiol. 1997;14:264–293. doi: 10.1097/00004691-199707000-00002. [DOI] [PubMed] [Google Scholar]
- Wexler EM, Stanton PK. Priming of homosynaptic long-term depression in hippocampus by previous synaptic activity. NeuroReport. 1993;4:591–594. doi: 10.1097/00001756-199305000-00034. [DOI] [PubMed] [Google Scholar]
- Winder DG, Mansuy IM, Osman M, Moallem TM, Kandel ER. Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell. 1998;92:25–37. doi: 10.1016/s0092-8674(00)80896-x. [DOI] [PubMed] [Google Scholar]
- Zorumski CF, Izumi Y. Modulation of LTP induction by NMDA receptor activation and nitric oxide release. Prog Brain Res. 1998;118:173–182. doi: 10.1016/s0079-6123(08)63207-0. [DOI] [PubMed] [Google Scholar]