

Abstract
Extreme corneal fragility and thinning, which have a high risk of catastrophic spontaneous rupture, are the cardinal features of brittle cornea syndrome (BCS), an autosomal-recessive generalized connective tissue disorder. Enucleation is frequently the only management option for this condition, resulting in blindness and psychosocial distress. Even when the cornea remains grossly intact, visual function could also be impaired by a high degree of myopia and keratoconus. Deafness is another common feature and results in combined sensory deprivation. Using autozygosity mapping, we identified mutations in PRDM5 in families with BCS. We demonstrate that regulation of expression of extracellular matrix components, particularly fibrillar collagens, by PRDM5 is a key molecular mechanism that underlies corneal fragility in BCS and controls normal corneal development and maintenance. ZNF469, encoding a zinc finger protein of hitherto undefined function, has been identified as a quantitative trait locus for central corneal thickness, and mutations in this gene have been demonstrated in Tunisian Jewish and Palestinian kindreds with BCS. We show that ZNF469 and PRDM5, two genes that when mutated cause BCS, participate in the same regulatory pathway.
Introduction
Brittle cornea syndrome (BCS) is an autosomal-recessive generalized connective tissue disorder.1 Individuals with this condition have a high risk of corneal rupture, which could occur either spontaneously2 or because of minor injury.1 It is known that deafness affects many but not all patients with BCS and results in combined sensory deprivation when present.1 The etiology of this is unclear because both sensorineural and conductive deafness have been identified in affected patients.1 Other common clinical features include joint hypermobility and other features of connective tissue disorder such as scoliosis,1,3 but for many patients these are mild in comparison to the dramatic ocular phenotype.1 Abu et al.4 demonstrated mutations in ZNF469, a 13 kb open reading frame at 16q24 (NM_001127464.1) that is probably a single exon gene, as a cause of BCS. Two frameshift mutations were identified, a founder mutation, c.5943delA, in patients of Tunisian Jewish origin, and c.9527delG in a single large Palestinian family. Both of these mutations would be predicted to result in premature termination codons, p.Gly1983AlafsX16 and p.Gln3178ArgfsX23, respectively. Homozygous missense and truncating mutations in ZNF469 have subsequently been described in two further families of Norwegian3 and Syrian5 origins with BCS, c.10016G>A, predicting p.Cys3339Tyr, and c.4174G>T, predicting p.Gln1392X. Subsequently, a role for ZNF469 in normal corneal development has also been confirmed: genome-wide association studies have each identified this locus as a determinant of the highly heritable quantitative trait of corneal thickness.6–8 ZNF469 encodes a evolutionarily poorly conserved zinc finger protein of unknown function, and the mechanisms by which it could influence corneal structure and function and result in the other features of BCS remain unknown. We demonstrate that BCS is genetically heterogeneous and can also result from mutations in the gene encoding the transcription factor PRDM5, a determinant of corneal thickness that is critical for extracellular matrix development and maintenance.
Subjects and Methods
A family of Pakistani origin (BCS-001, Figure 1D and Table 1) presented when individual IV:6 suffered an expulsive hemorrhage (Figures 1A and 1B), necessitating emergency enucleation, after a minor impact when a small child's shoe, thrown accidentally, hit her in the face. Three of her sisters had previously lost eyes after similarly minor trauma, suggesting BCS as the likely diagnosis.1 Another family of Pakistani origin, BCS-002, was also identified for analysis by the presence of thin corneas, blue sclera, and mixed sensorineural/conductive deafness (Figure 2). Data on the clinical features of both affected and unaffected members of these two families was collected for analysis of the phenotypic spectrum associated with homozygous and heterozygous mutations. Clinical information was also collated about further individuals with BCS from six additional affected consanguineous families whose DNA was available for analysis and in whom mutations in ZNF469 had previously been excluded. Each of these families included either a single individual or pair of siblings affected with BCS (as shown in Table 2).
Figure 1.
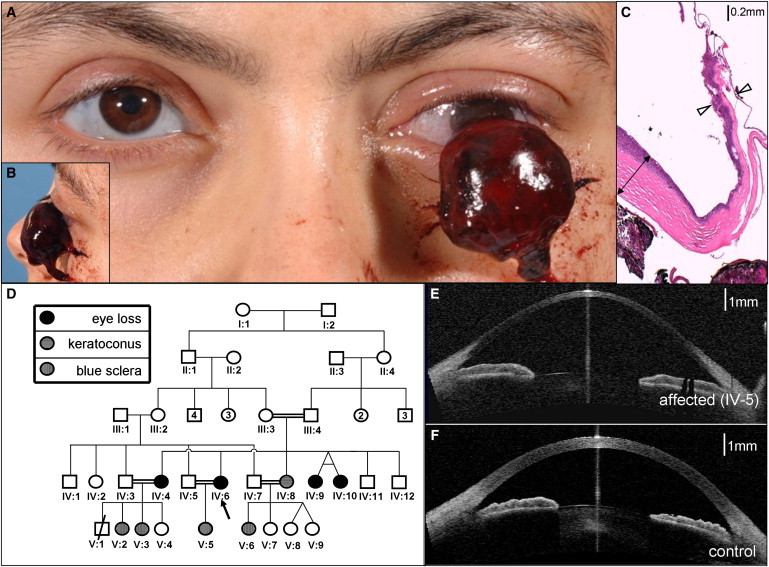
Extreme Corneal Fragility in BCS-001
(A and B) Expulsive hemorrhage affecting BCS-001 IV:6. Note also the blue-gray sclera in the remaining eye.
(C) 40× magnification of hematoxylin and eosin-stained section through the eviscerated cornea. Note the relative preservation of peripheral corneal thickness (indicated by double-headed arrow) in contrast to extreme thinning of central cornea in the region of rupture (indicated by open arrowheads). The stroma (stained pink) is almost absent here: much of the remaining corneal thickness derives from epithelial and endothelial layers.
(D) The pedigree of family BCS-001. The circles indicate females, and the squares indicate males. Filled shapes indicate affected individuals, as assessed by history of previous enucleation after minor trauma or phthisis. Blue sclerae and keratoconus in individuals without corneal rupture are also as indicated.
(E) Optical coherence tomography demonstrating cross-sectional appearance of remaining eye of individual IV:6 from family BCS-001; note the extreme thinning of central cornea and keratoglobus compared to that in the control image (F).
Table 1.
Phenotypic Characteristics of Individuals with PRDM5 Mutations from Families BCS-001 and BCS-002
|
BCS-001 |
BCS-002 |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IV:4 | IV:6 | IV:8 | IV:9 | IV:7 | V:2 | V:3 | V:5 | V:6 | IV:6 | V:1 | V:4 | V:5 | IV:3 | |
| Homozygous/heterozygous | hom | hom | hom | hom | het | het | het | het | het | hom | hom | hom | hom | het |
| Corneal rupture | + | + | + | + | ||||||||||
| myopia | + | + | + | + | + | + | + | + | ||||||
| Blue sclera | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| Keratoconus | + | + | + | + | + | + | ||||||||
| Keratoglobus | + | + | + | + | ||||||||||
| Megalocornea | ||||||||||||||
| Poor healing/abnormal scarring | + | |||||||||||||
| Soft skin/easy bruising | + | + | + | + | ||||||||||
| Treatment for DDH | + | + | + | |||||||||||
| Femoral epiphyseal changes | + | + | + | |||||||||||
| Scoliosis | + | |||||||||||||
| Small joint hypermobility | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| Fractures | + | + | + | |||||||||||
| Myalgia | + | + | + | |||||||||||
| Abnormal gait | + | + | + | + | + | + | ||||||||
| Deafness | + | + | + | + | + | + | + | + | ||||||
| Hypercompliant TMs | + | + | + | + | + | + | + | |||||||
| Other features | P | LD | H | LD | LD CLP PKU | |||||||||
| CCT <400 μm | + | + | + | + | + | + | + | + | ||||||
| CCT 400–550 μm | + | + | + | + | + | + | ||||||||
Affected, homozygous, individuals in each family are indicated. + indicates present; and empty box indicates not present. N/A indicates data not available.
The following abbreviations are used: DDH, developmental dysplasia of the hip; TM, tympanic membrane; CCT, central corneal thickness. P, primiparous cervical incompetence; LD, learning disability; H, hernia (inguinal, umbilical or epigastric); CLP, cleft lip and palate; PKU, phenylketonuria.
Figure 2.
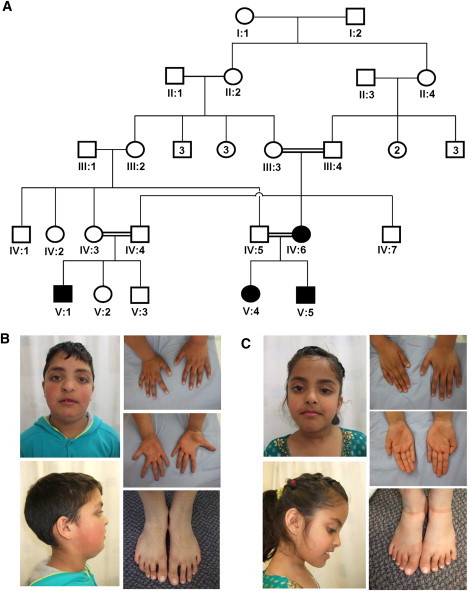
Corneal Thinning without Rupture in BCS-002
(A) the pedigree of family BCS-002. The circles indicate females, and the squares indicate males. Filled shapes indicate affected individuals as assessed by the presence of thin corneas, deafness, and hypercompliant tympanic membranes. Keratoconus and developmental dysplasia of the hip were present in individual V:1, developmental dysplasia of the hip in individual V:4, and cleft lip and palate in individual V:5
(B) Hypertelorism, downslanting palpebral fissures, a short nose with anteverted nares, long fingers and toes, and flat feet of individual V:5. are evident. A repaired left-sided orofacial cleft, blue sclera, and single palmar creases are also present.
(C). Note similar facial features and hands and feet to those of her brother (individual V.5) but no orofacial clefting in individual V:4.
Table 2.
Phenotypic Characteristics of Individuals with PRDM5 Mutations from Families 914, K921, and K923
|
901 |
902 |
914 |
K921 |
K923 |
|||
|---|---|---|---|---|---|---|---|
| 04a | 03a | 05a | 04 | 06 | 03 | 03 | |
| Homozygous/heterozygous | hom | hom | hom | hom | hom | hom | hom |
| Corneal rupture | + | + | + + | + | |||
| myopia | + | + | + | N/A | N/A | + | + |
| Blue sclera | + | + | + | + | + | + | + |
| Keratoconus | + | + | |||||
| Keratoglobus | + | + | |||||
| Megalocornea | + | N/A | N/A | + | |||
| Poor healing/abnormal scarring | |||||||
| Soft skin/easy bruising | + | ||||||
| Treatment for DDH | + | ||||||
| Femoral epiphyseal changes | |||||||
| Scoliosis | + | + | |||||
| Small joint hypermobility | + | + | + | + | + | ||
| Fractures | |||||||
| Myalgia | + | ||||||
| Abnormal gait | + | + | |||||
| Deafness | + | ||||||
| Hypercompliant TMs | |||||||
| Other features | SF SH | S CP | S CP | CO H JH | H SH GV JH PP TB | ||
| CCT <400 μm | N/A | + | N/A | N/A | N/A | N/A | + |
| CCT 400–550 μm | N/A | N/A | N/A | N/A | N/A | ||
Affected, homozygous, individuals in each family are indicated. + indicates present; and empty box indicates not present. N/A indicates data not available.
The following abbreviations are used: DDH, developmental dysplasia of the hip; TM, tympanic membrane; CCT, central corneal thickness. H, hernia (inguinal, umbilical or epigastric); SF, severe scleral fragility; SH, skin hyperelasticity; S, sclerocornea; CP, cornea plana; CO, corneal opacity; JH, generalized joint hypermobility; GV, genu vara; PP, pes planus; TB, tibial bowing.
Patients described initially in Cameron et al. as Cases 2, 10 and 11.20
Clinical Assessment
Individuals were examined by clinical geneticists, ophthalmologists, and audiologists. Central corneal thickness was assessed by pachymetry and optical coherence tomography with the Oculus Pentacam (Oculus, Lynnwood, WA) and Visante optical coherence tomography (Carl Zeiss Meditech, Oberkochen) systems. Pure tone audiometry and tympanometry were carried out with standard equipment.
Clinical Diagnostic Samples
Urinary pyridinoline crosslinks were analyzed as previously described.9 Histological analysis of the eviscerated cornea was carried out in accordance with standard diagnostic protocols: the tissue was fixed and sectioned for hematoxylin and eosin staining.
Genetic and Genomic Analysis
Ethical approval was granted by a National Health Service Research Ethics Committee (reference 06/Q1406/52), and informed written consent was obtained from participants. The open reading frame of ZNF469 was sequenced as described in Abu et al.4 Autozygosity mapping was then undertaken by SNP analysis on the SNP6.0 platform (Affymetrix, Santa Clara, CA), as previously described.10 From the results of SNP copy number analysis within the autozygous region, PRDM5 was identified as a candidate gene for BCS. PCR and Sanger sequencing of exons 1–16 of this gene was therefore undertaken with primers and reaction conditions as listed in Table S1, available online.
For analysis of variants identified in PRDM5, the coding sequence of this gene was evaluated from exome sequencing data in a control set of 401 subjects enrolled in the ClinSeq cohort.11 DNA was captured with the SureSelect system and sequenced as described.12
Cell Culture
Primary dermal fibroblast cultures from individuals with BCS (individuals IV:4 of family BCS-001 and V:1 of family BCS-002) were established from skin biopsies by routine procedures. Control samples were obtained from normal skin of age- and sex-matched patients undergoing dermatologic procedures. Fibroblasts were routinely maintained in DMEM (PAA, Somerset) supplemented with 10% fetal calf serum (PAA, Somerset) at 37°C in 5% CO2. Fibroblasts were expanded until full confluency was achieved, and then harvested by trypsin treatment at the same passage number.
Expression Microarray
Total RNA was extracted from cell lines with RNeasy Mini Kit and QIA Shredder (QIAGEN, USA). We conducted microarrays on skin-derived fibroblasts from patients and controls described above by using the Affymetrix Human Genome U133 Plus 2.0 array according to the manufacturer's instructions (Affymetrix Inc. High Wycombe, UK). RNA was quantified with a Nanodrop ultra-low-volume spectrophotometer (Nanodrop Technologies, Ringmer, UK), and RNA quality was checked with the RNA 6000 Nano Assay and analyzed on an Agilent 2100 Bioanalyzer (Agilent Technologies, UK). Background correction, quantile normalization, and gene-expression analysis were performed with RMA in Bioconductor,13 and the full data set was submitted to ArrayExpress. Results for mutant cell lines and their respective controls were paired, allowing calculation of fold change in expression and assessment of q values (q < 0.01). The 500 genes with the greatest fold changes were analyzed with the Database for Annotation, Visualization and Integrated Discovery (DAVID) version 6.7 software for gene ontology and molecular interactions.14 Molecular components of pathways highlighted in this analysis were further validated with quantitative PCR.
Quantitative PCR
Extracted total RNA was reverse-transcribed into single-stranded cDNA with a High Capacity RNA-to-cDNA Kit (Applied Biosystems, USA). The RT-PCR was performed with first-strand cDNA with TaqMan Fast Universal PCR Master Mix (Applied Biosystems). The assay numbers for the mRNA endogenous control (GAPDH) and target genes were as follows: GAPDH (Hs03929097_g1∗), HAPLN1 (Hs00157103_m1), TGFB2 (Hs00234244_m1), COL4A1 (Hs00266237_m1), COL11A1 (Hs01097664_m1), and EDIL3 (Hs00174781_m1). Quantitative PCR was performed on an Applied Biosystems StepOnePlus Real-Time PCR system (Applied Biosystems) with the following conditions: 50°C incubation for 2 min, 95°C for 10 min, 40 cycles of PCR at 95°C for 15 s, and 60°C for 1 min. All reactions were performed in a 10 μl reaction volume in triplicate. mRNA expression levels were determined by the 2−ΔCt method with Applied Biosystem's StepOne software. Seven technical replicates were performed for each cell line and control.
We used one-way ANOVA and Dunnett's multiple comparison posttest by using mean values and standard error to compare results from mutant cells and to those from the wild-type cells. These were performed on all the fold change means in all groups assessed (F(5,18) = 9.447, p < 0.0001), allowing the significance of multiple means to be assessed.
Indirect Immunofluorescent Microscopy
Fibroblasts used for indirect immunofluorescence (IF) microscopy were maintained as described above and the media additionally supplemented with 100 IU/ml penicillin and 100 μg/ml streptomycin. All assays were performed on cells at the eighth passage.
Antibodies against type I, type III, and type V collagens and α2β1 and α5β1 integrins were sourced from Millipore Chemicon International (Billerica, MA). Fibronectin antibody, ascorbic acid, and tetramethylrhodamine isothiocyanate (TRITC)-conjugated rabbit anti-goat antibody were from Sigma Aldrich, and fluoroscein isothiocyanate (FITC)-conjugated goat anti-rabbit and TRITC-conjugated goat anti-mouse secondary antibodies were from Calbiochem-Novabiochem. A total of 1.0 × 105 cells were grown for 48 hr on glass coverslips in complete MEM supplemented with 10% fetal calf serum and then fixed in methanol and incubated with the specific antibodies, as previously described.15,16 For analysis of integrins, cells were fixed in 3% paraformaldehyde and 60 mM sucrose and permeabilized in 0.5% Triton X-100. Cells were subsequently incubated for 45 min with 10 μg/ml FITC- or TRITC-conjugated anti-rabbit, anti-goat, or anti-mouse IgG. IF signals were acquired by a charge-coupled device black and white TV camera (SensiCam-PCO Computer Optics GmbH, Germany) mounted on a Zeiss fluorescence-Axiovert microscope and digitalized by Image-Pro Plus program (Media Cybernetics, Silver Spring, MD).
Results
Clinical Findings
Phenotypic details of affected individuals from families BCS-001 and BCS-002 and the five other families in whom mutations were subsequently identified are summarized in Tables 1 and 2.
Ocular Phenotype
Corneal pachymetry and optical coherence tomography revealed profoundly reduced central corneal thickness (CCT) in affected individuals (Figure 1E, compared to that in the control in Figure 1F; see also Table 1). CCT in the remaining eyes of affected individuals from family BCS-001 was measured at 220 μm (IV:6) and 270 μm (IV:4) (the normal range is 515–575 μm17). Affected individuals from family BCS-002 showed corneal thicknesses of 275–370 μm in the six eyes in which this could be quantified. A paucity of corneal stroma, and almost total absence in the central cornea, was confirmed on histological examination of the eviscerated cornea of IV:6 of BCS-001 (Figure 1C).
Several affected individuals were also noted to have keratoconus. Individuals IV:4 and IV:9 from family BCS-001 were each noted in early childhood, prior to ocular rupture, to have this condition, whereas their sister who did not have BCS, individual IV:8, developed keratoconus as a young adult. In family BCS-002, affected individual V:1 developed severe keratoconus by the age of 6 and required operative intervention before the age of 10; keratoconus was not found in other individuals (with or without very thin corneae) in this family.
A high degree of myopia was present in all affected individuals of BCS-001. Blue sclera was present in all affected individuals (Table 1, Figures 1 and 2).
Audiological Phenotype
The affected individuals in families BCS-001 and BCS-002 had mixed sensorineural and conductive hearing loss. This was the primary indication for referral to genetic services for family BCS-002. Conductive losses appeared to predominate in the early years for both families; a progressive sensorineural hearing loss was apparent through childhood and became severe in adulthood in affected members of BCS-001. Importantly, highly characteristic tympanometry, with hypercompliant tympanic membranes, was seen in both families among all affected patients in whom this could be assessed (several affected children were not assessable because of the presence of tympanostomy tubes). No audiological phenotype was apparent in heterozygous mutation carriers.
Generalized Connective Tissue Phenotype
In family BCS-001, several affected individuals had experienced hip problems in early childhood: individual IV:4 was diagnosed with bilateral developmental dysplasia of the hip, whereas twins IV:9 and IV:10 had femoral capital epiphyseal changes on X-radiography ascribed to Perthes disease. Similarly, individuals V:1 and V:4 of BCS-002 had both been affected with developmental dysplasia of the hip. Small joint hypermobility was marked in affected individuals and present to a milder degree in many of their relatives who did not have BCS. Easy bruising had been noted in most affected individuals, but skin healing appeared normal except for mildly hypotrophic scars in individual V:1 of BCS-002. Obstetric and perinatal problems had been present in several affected individuals: individual V:1 of BCS-002 was born prematurely after premature rupture of membranes, whereas individual IV:4 of BCS-001 suffered a second trimester pregnancy loss due to primary primiparous cervical incompetence.
Molecular Analysis
A normal lysylpyridinoline/hydroxylysylpyridinoline (LP/HP) urinary excretion ratio, characteristic of BCS,1 was confirmed in affected members of families BCS-001 (0.14 in individual IV:4; 0.20 in individual IV:6) and BCS-002 (0.23 in individual V:1) (the normal range is 0.10–0.35). This was in contrast to the markedly increased LP/HP ratio present in Ehlers-Danlos syndrome (EDS) type VIA (MIM 225400),18,19 thereby excluding this diagnosis.
Mutations in ZNF469 were not present in individuals who were affected members of families BCS-001 and BCS-002 and who were also heterozygous for SNPs within the coding sequence of the transcript, despite the history of consanguinity. Autozygosity mapping undertaken with the Affymetrix SNP 6.0 platform identified a single 18 Mb autozygous region on chromosome 4 (114,039,300–131,838,000 bp) that was shared by affected individuals across both families (Figure 3A). This region encompasses 53 known or hypothetical genes. There was no evidence for a shared haplotype between the two families. However, within this autozygous region a 52.46 kb homozygous deletion, resulting in deletion of exons 9–14 of PRDM5 (NM_018669.2), was identified in all affected members of BCS-001 (Figure 3B). The presence of exons 1–8 and 15 and 16 was demonstrated by PCR in these individuals (data not shown). The deletion was absent from 454 control chromosomes, including 60 chromosomes of matched ethnic origin. It is predicted that the in-frame deletion of exons 9–14 would result in the loss of zinc fingers 6–13 of the protein (Figure 3C). In family BCS-002, a homozygous nonsense mutation in exon 16, c.1768C>T, encoding p.Arg590X, was identified in all affected individuals. Because this mutation is in the last exon, it would be predicted to escape nonsense-mediated decay and to result in production of a truncated protein with absence of the two most C-terminal zinc fingers (Figure 3C). Further screening of patient samples identified homozygous mutations in PRDM5 in five additional BCS families (Figures 3B and 3C; Tables 2 and 3). Two of these individuals had previously been published in the premolecular era as cases of EDS VI,20 underlining the clinical similarities between EDS VIA and BCS. Other mutations included a splice-site mutation, c.93+1G>A, and three frameshift mutations resulting in premature termination codons, all likely to be consistent with null alleles as a result of nonsense-mediated decay. One missense variant, c.320A>G, encoding p.Tyr107Cys, was also identified. This substitution lies in the PR-SET domain, within a region where 34 of 36 sequential residues are conserved down to zebrafish and for which in silico modeling by PolyPhen2 and SIFT21 analyses predicts pathogenicity. All variants described are not included in online databases of normal genomic variation such as dbSNP and were absent from 401 control individuals in the ClinSeq cohort.11,12 The coverage of this gene was >95% of its coding base pairs with a metric of sufficient coverage to generate a most probable genotype (MPG)12 base-calling score of 10 or more. Six novel SNPs in PRDM5 were identified in this cohort (Table S2).
Figure 3.
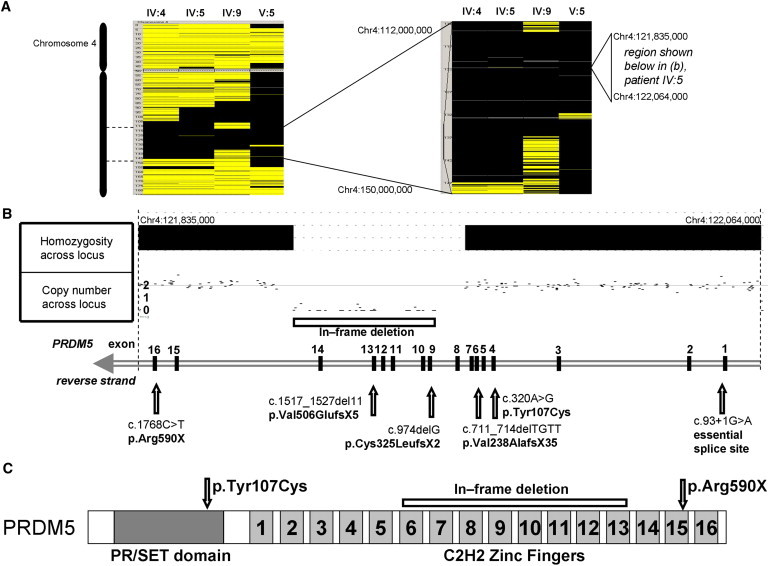
Mutations in PRDM5 Cause Brittle Cornea Syndrome
(A) Autozygosity mapping by SNP6.0 array demonstrates homozygosity at 4q28 in affected members of families BCS-001 (individuals IV:4, IV:6, and IV:9) and BCS-002 (individual V:5).
(B) Nullizygosity for 34 adjacent SNPs within the autozygous region demonstrates a homozygous 52.46 kb deletion encompassing exons 9–14 of PRDM5 in individual IV:5 of family BCS-001. This in-frame deletion and further mutations in PRDM5 identified are shown.
(C) Schematic of PRDM5 protein showing N-terminal PR-SET domain and 16 zinc fingers toward the C terminus. Missense mutation p.Tyr107Cys is within the PR-SET domain in a sequence of highly conserved amino acid residues. Nonsense mutation p.Arg590X predicts a truncated protein, resulting in loss of zinc fingers 15 and 16, whereas deletion of exons 9–14 predicts loss of zinc fingers 6–13.
Table 3.
Description of Observed Mutations
| Family | Mutation | Protein Prediction | |
|---|---|---|---|
| BCS-001 | deletion of exons 9–14 | in-frame deletion | internally deleted protein |
| 901 | c.93+1G>A | splice site | splice error |
| 902 | c.320A>G | missense | p.Tyr107Cys |
| K921 | c.711_714delTGTT | frameshift | p.Val238AlafsX35 |
| K923 | c.974delG | frameshift | p.Cys325LeufsX2 |
| 914 | c.1517_1527del TTCATATCCGG | frameshift | p.Val506GlufsX5 |
| BCS-002 | c.1768C>T | nonsense | p.Arg590X |
PRDM5 Mutation Results in Reduced Expression of Extracellular Matrix Macromolecules
Expression microarray analysis on dermal fibroblasts from two patients with PRDM5 mutations (deletion of exons 9–14 (family BCS-001, individual IV:4), c.1768C>T (BCS-002 V:1), compared to those from age- and sex-matched controls, demonstrated 47 upregulated and 49 downregulated transcripts with a fold change greater than 10. Large, significant differences from controls were identified for key genes that encode molecules involved in extracellular matrix (ECM) development and maintenance and are of particular relevance to the multisystemic connective tissue phenotype of BCS (Figure 4A). No direct effect on ZNF469 expression was apparent in PRDM5 mutant fibroblasts. We used the DAVID Microarray Analysis Tool14 to identify enriched biological pathways common to both of the PRDM5 mutant fibroblast lines. In particular, genes encoding fibrillar collagens (e.g., COL4A1 and COL11A1), connective tissue components (e.g., HAPLN1), and molecules regulating cell migration and adhesion (e.g., EDIL3 and TGFB2) were all significantly downregulated on microarray analysis. EDIL3, HAPLN1, and COL11A1 each demonstrated a greater than 30-fold decrease in mutant lines relative to controls. These findings were subsequently tested and validated by quantitative PCR (Figure 4B).
Figure 4.
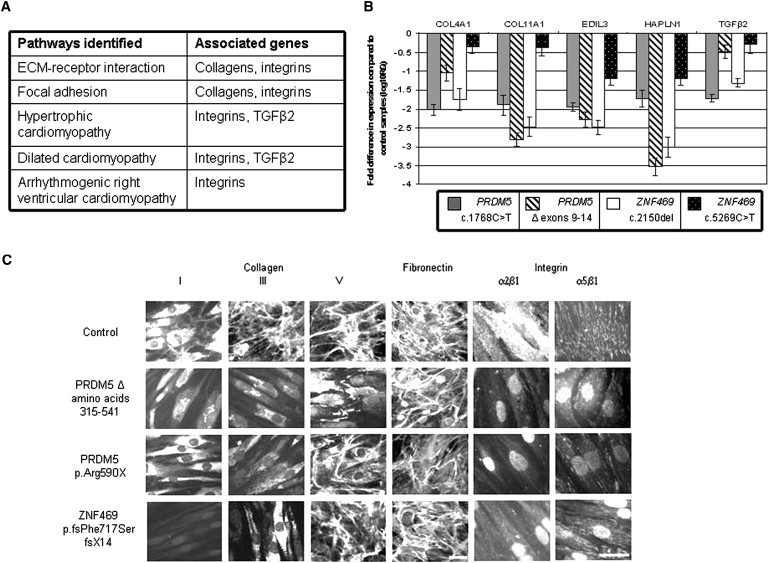
Functional Consequences of Mutation in PRDM5
(A) Results of KEGG pathway analysis. Pathways up- and downregulated in fibroblasts from IV:4 from family individual BCS-001, compared to those in an age- and sex-matched control, were analyzed with the DAVID functional annotation bioinformatics microarray analysis tool. Pathways shown here were identified by altered expression of a number of molecular components as a result of mutation in PRDM5. Selected genes associated with these pathways whose expression was altered in the microarray are specified in the right hand column.
(B) Quantitative PCR assessment of target genes identified in microarray analysis. Fold changes in mRNA expression of genes highlighted in the microarray were assessed in dermal fibroblasts isolated from four BCS patients with different mutations: PRDM5 deletion of exons 9–14, PRDM5 c.1768C>T, ZNF469 c.2150del, and ZNF469 c.5269C>T. mRNA levels were normalized to GAPDH expression, and the fold change is displayed relative to a control fibroblast line. The y axis represents fold changes (log10) in gene expression, whereas the x axis shows the target assessed. Error bars represent the standard error of the mean. For each of the five genes assessed, transcript levels were significantly reduced and assessed by one-way ANOVA and Dunnett's multiple comparison test (p < 0.0001).
(C) Immunofluorescence of fibroblasts from BCS patients. Disarray of fibrillar collagens I, III, and V, fibronectin, and their integrin receptors, the α2β1 and α5β1 integrins is demonstrated (the scale bars represent 10 μm). In control cells, collagen I was synthesized and mainly detected in the cytoplasm, and only a few fibrils were assembled in the extracellular compartment. Collagen I staining was strongly reduced in PRDM5 mutant cells and was detected mainly associated with the perinuclear endoplasmic reticulum; in ZNF469 mutant cells it was not detectable in the cytoplasm and was not assembled in the ECM. Collagen III was organized in the ECM by control fibroblasts; this network was absent from both PRDM5 and ZNF469 mutant fibroblasts, which showed only diffuse cytoplasmic staining. Collagen V was organized in the ECM by control fibroblasts; in BCS cells this structure was almost completely disorganized, with cytoplasmic accumulation in PRDM5 but not in ZNF469 mutant cells.
We hypothesized that the clinical phenotype of BCS, whether it arises from mutation in PRDM5 or ZNF469, has a single molecular etiology, and therefore we evaluated expression of the same five genes in ZNF469 mutant fibroblasts from previously described individuals with BCS (915-07, ZNF469 p.Phe717SerfsX14 and 909-06, ZNF469 p.Gln1757X, clinically described1). Highly significant downregulation was confirmed for all transcripts examined. The similarity in the gene-expression profiles for ECM genes in fibroblasts with PRDM5 and ZNF469 mutations (Figure 4B) supports our hypothesis of a shared molecular pathogenesis.
To examine the effects of mutations on the organization of the ECM in vitro, indirect immunofluorescence microscopy of PRDM5- and ZNF469 mutant fibroblasts was undertaken (Figure 4C). Disarray of collagens I and III, fibronectin, and their receptor α2β1 and α5β1 integrins was more evident in both mutants than in control cells. Results for collagen V showed some differences, with cytoplasmic accumulation and loss of expression in the ECM in PRDM5 mutant cells, whereas the appearance of ZNF469 mutant cells was closer to that of controls.
Discussion
PRDM5 is widely expressed and encodes a transcriptional regulator that modulates many aspects of tissue development and maintenance, including cell fate and cell adhesion, via mechanisms that include Wnt signaling.22 We have demonstrated that one form of brittle cornea syndrome is associated with PRDM5 mutations, some of which are predicted to result in nonsense-mediated decay and hence absence of the protein. PRDM5 is therefore the second gene in which mutations could cause BCS, along with ZNF469. The identification of only a single affected family that has BCS but no mutations in PRDM5 or ZNF469 suggests that, although there might be a third gene involved in the pathogenesis of this condition, the large majority of cases could be attributable to mutation in one of these two genes.
The ocular phenotype is the most dramatic and potentially distressing aspect of BCS. We found affected individuals to have dramatically reduced corneal thickness; in patients from families BCS-001 and BCS-002 this was in the range 220–370 μm, whereas in other individuals with homozygous PRDM5 mutations, CCT was reported to range from 300–455 μm. The presence of greatly reduced CCT is, therefore, likely to be a sensitive diagnostic discriminator for BCS. Reduced CCT has been reported in other connective tissue disorders, specifically classical EDS,23 but to a much lesser extent than confirmed here in BCS. The presence of a distinctive auditory phenotype with hypercompliant tympanic membranes could also be a useful diagnostic indicator for this condition.
The confirmation of the existence of a carrier phenotype in the majority of heterozygous individuals examined in this study is an important observation and is in keeping with findings in previous studies.5 Blue sclera is present in the large majority of those identified with heterozygous PRDM5 mutations as is small joint hypermobility. Individuals with heterozygous mutations in PRDM5 were also noted to have mildly reduced central corneal thickness, range 480–505 μm. However, the auditory and certain connective tissue aspects of the phenotype, such as easy bruising and laxity of the large joints, appeared to be limited to individuals with biallelic mutations. Nonetheless, the possibility that heterozygote status might confer higher than baseline risk for features such as developmental dysplasia of the hip, visual impairment, or deafness, should be considered and appropriate screening employed.
The phenotypic spectrum of BCS appears extremely similar, if not identical, in patients with mutations in either ZNF469 or PRDM5, suggesting that the two genes act within the same developmental pathway. To investigate this, we undertook microarray analyses of fibroblasts from two patients with mutations in PRDM5. Fibroblasts with mutations encoding either internally deleted or truncated PRDM5 showed highly similar results, and there was disruption in the expression of several key extracellular matrix components. The significance of these observed alterations to the molecular pathogenesis of BCS was confirmed by the demonstration of similar downregulation of these ECM genes in skin-derived fibroblasts from patients with BCS because of mutations in ZNF469. These mutations are likely to account for the generalized connective tissue phenotype observed in individuals with BCS. Data for gene expression in corneal stroma from such patients are not currently available, and it is possible that additional genes might also be important in the dramatic phenotype observed in this highly specialized connective tissue.
Fibroblasts from BCS patients with both PRDM5 and ZNF469 mutations also showed similar cellular phenotypes, with disruption in the deposition of several collagens, fibronectin and integrins, further supporting the hypothesis that PRDM5 and ZNF469 regulate ECM organization through similar biochemical mechanisms. The sole difference observed between PRDM5 and ZNF469 mutant fibroblasts was with respect to the disarray of collagen V in the former, but not in the latter. This suggests that, although the overall effects of mutations in either gene are similar at both the molecular and macroscopic level, differences might exist in the regulation of individual gene expression by the two transcription factors.
The demonstration of disarray of many collagens in BCS is strongly reminiscent of other connective tissue disorders, in particular those within the EDS spectrum.15,24 The clinical features of BCS are also reminiscent of those of EDS, as is apparent from the overlapping phenotype with EDS VIA. Another condition that shows considerable overlap with the clinical features of BCS is Stickler syndrome. Deafness, epiphyseal dysplasia, joint hypermobility, and orofacial clefting are classical features of Stickler syndrome, which is caused by mutations in type II and XI collagen genes.25,26 Because mutation of PRDM5 results in greatly reduced expression of type XI collagen, it is possible that mutations in PRDM5 could also be responsible for Stickler-like phenotypes in individuals without collagen gene mutations.
Reduced central corneal thickness (CCT) in individuals with homozygous and heterozygous mutations in PRDM5 suggests that this gene and the biochemical pathways it controls are significant contributors to this trait. A dosage effect is suggested by the more mildly reduced CCT seen in heterozygous than homozygous mutation carriers.
Similarly, mutated PRDM5 appears to be a strong predisposing factor for the development of keratoconus in the families described here. In family BCS-001, keratoconus in childhood was noted prior to ocular ruptures in affected homozygous individuals (IV:4 and IV:9), whereas an individual heterozygous for the family mutation (IV:8), developed keratoconus as a young adult. In family BCS-002, a homozygous affected individual (V:1) also had early-onset keratoconus, but this was not found in other individuals (with either homozygous or heterozygous mutations) in this family.
ZNF469 has been associated with CCT in several genome-wide association studies (GWAS),6–8 and there are concomitant implications for risks for development of glaucoma or keratoconus. Although PRDM5 has not been identified by GWAS, this does not exclude rarer variants in this gene as significant contributors to CCT, as such alleles would not necessarily be identified by these approaches. Additional genes recently associated with CCT by GWAS include COL8A27 and COL5A1,8 which is notable given the disarray of collagen V on immunofluorescent staining of BCS fibroblasts with mutations in PRDM5 (but not ZNF469) (Figure 4C).
Appropriate management of patients with BCS requires a multidisciplinary approach, including primary prevention of corneal rupture by provision of protective polycarbonate eyeglasses and careful screening of vision and hearing. The distinctive syndromic features of BCS identified here include developmental dysplasia of the hip and hypercompliant tympanic membranes and serve as important diagnostic clues in the early recognition of patients with this condition, particularly where they are the only affected individual in their family.
Now that the molecular basis of BCS in the large majority of affected families ascertained to date has been clarified, effective genetic testing and further assessment for genotype-phenotype correlations in this condition become possible. Significant differences exist between the phenotypes of families BCS-001 and BCS-002 in this study, and although the presentation of all affected individuals in family BCS-001 appears similar, for members of BCS-002 there is much greater phenotypic variability. These observations of inter- and intrafamilial variability emphasize the difficulty in making a firm clinical diagnosis of BCS and hence the importance of molecular diagnostic testing of individuals at risk for this condition. Early diagnosis by presymptomatic genetic testing will help to avert the devastating physical and psychological sequelae of corneal rupture, as well as identifying individuals at risk for the associated features of this condition, whereas carrier testing will permit effective genetic counseling and identification of individuals at risk of late-onset complications, such as keratoconus.
In identifying that mutations in PRDM5 cause extreme corneal fragility, as well as the related auditory and musculoskeletal connective tissue phenotypes seen in BCS, we have identified a pathway that is central to corneal development and maintenance and that, as a result, profoundly influences CCT. Further work to determine the role of the genes mutated in BCS in common ocular disorders associated with a low CCT, such as glaucoma and keratoconus, will lead to improved understanding of these disorders and potentially to improved management. The pathways in which PRDM5 exerts its effects could also present therapeutic targets for BCS or other connective tissue disorders in the future.
Acknowledgments
We thank all healthcare professionals contributing to the care of the families described here, particularly R.W. Paton (Royal Blackburn Hospital), and J. Teer of the National Human Genome Research Institute, for sequence coverage analysis in the ClinSeq cohort. We thank B. Steinmann for useful discussion and support, H. Al-Hussein for referral of some BCS families, and A. Schwarze and C. Bürer for expert technical assistance. The support of the Manchester Biomedical Research Centre, funded by the UK National Institute for Health Research, is gratefully acknowledged. E.B.W. is funded by a Clinical Research Training Fellowship from the Wellcome Trust, and F.D.C.M. by Fight for Sight. The NISC comparative sequencing program and the ClinSeq study are funded by the National Human Genome Research Institute of the National Institutes of Health.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
NCBI's Single Nucleotide Polymorphism, http://www.ncbi.nlm.nih.gov/projects/SNP/
Accession Numbers
The ArrayExpress accession number for the PRDM5 sequence reported in this paper is E-MEXP-3077.
References
- 1.Al-Hussain H., Zeisberger S.M., Huber P.R., Giunta C., Steinmann B. Brittle cornea syndrome and its delineation from the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VI): Report on 23 patients and review of the literature. Am. J. Med. Genet. A. 2004;124A:28–34. doi: 10.1002/ajmg.a.20326. [DOI] [PubMed] [Google Scholar]
- 2.Izquierdo L., Jr., Mannis M.J., Marsh P.B., Yang S.P., McCarthy J.M. Bilateral spontaneous corneal rupture in brittle cornea syndrome: A case report. Cornea. 1999;18:621–624. doi: 10.1097/00003226-199909000-00019. [DOI] [PubMed] [Google Scholar]
- 3.Christensen A.E., Knappskog P.M., Midtbø M., Gjesdal C.G., Mengel-From J., Morling N., Rødahl E., Boman H. Brittle cornea syndrome associated with a missense mutation in the zinc-finger 469 gene. Invest. Ophthalmol. Vis. Sci. 2010;51:47–52. doi: 10.1167/iovs.09-4251. [DOI] [PubMed] [Google Scholar]
- 4.Abu A., Frydman M., Marek D., Pras E., Nir U., Reznik-Wolf H., Pras E. Deleterious mutations in the Zinc-Finger 469 gene cause brittle cornea syndrome. Am. J. Hum. Genet. 2008;82:1217–1222. doi: 10.1016/j.ajhg.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan A.O., Aldahmesh M.A., Mohamed J.N., Alkuraya F.S. Blue sclera with and without corneal fragility (brittle cornea syndrome) in a consanguineous family harboring ZNF469 mutation (p.E1392X) Arch. Ophthalmol. 2010;128:1376–1379. doi: 10.1001/archophthalmol.2010.238. [DOI] [PubMed] [Google Scholar]
- 6.Lu Y., Dimasi D.P., Hysi P.G., Hewitt A.W., Burdon K.P., Toh T., Ruddle J.B., Li Y.J., Mitchell P., Healey P.R. Common genetic variants near the Brittle Cornea Syndrome locus ZNF469 influence the blinding disease risk factor central corneal thickness. PLoS Genet. 2010;6:e1000947. doi: 10.1371/journal.pgen.1000947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vithana E.N., Aung T., Khor C.C., Cornes B.K., Tay W.T., Sim X., Lavanya R., Wu R., Zheng Y., Hibberd M.L. Collagen-related genes influence the glaucoma risk factor, central corneal thickness. Hum. Mol. Genet. 2011;20:649–658. doi: 10.1093/hmg/ddq511. [DOI] [PubMed] [Google Scholar]
- 8.Vitart V., Bencić G., Hayward C., Skunca Herman J., Huffman J., Campbell S., Bućan K., Navarro P., Gunjaca G., Marin J. New loci associated with central cornea thickness include COL5A1, AKAP13 and AVGR8. Hum. Mol. Genet. 2010;19:4304–4311. doi: 10.1093/hmg/ddq349. [DOI] [PubMed] [Google Scholar]
- 9.Kraenzlin M.E., Kraenzlin C.A., Meier C., Giunta C., Steinmann B. Automated HPLC assay for urinary collagen cross-links: effect of age, menopause, and metabolic bone diseases. Clin. Chem. 2008;54:1546–1553. doi: 10.1373/clinchem.2008.105262. [DOI] [PubMed] [Google Scholar]
- 10.Daly S.B., Urquhart J.E., Hilton E., McKenzie E.A., Kammerer R.A., Lewis M., Kerr B., Stuart H., Donnai D., Long D.A. Mutations in HPSE2 cause urofacial syndrome. Am. J. Hum. Genet. 2010;86:963–969. doi: 10.1016/j.ajhg.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biesecker L.G., Mullikin J.C., Facio F.M., Turner C., Cherukuri P.F., Blakesley R.W., Bouffard G.G., Chines P.S., Cruz P., Hansen N.F. Instrumenting the health care enterprise for discovery research in the genomic era. Genome Res. 2009;19:1675–1681. doi: 10.1101/gr.094615.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teer J.K., Bonnycastle L.L., Chines P.S., Hansen N.F., Aoyama N., Swift A.J., Abaan H.O., Albert T.J., Margulies E.H., Green E.D., NISC Comparative Sequencing Program Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res. 2010;20:1420–1431. doi: 10.1101/gr.106716.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolstad B.M., Irizarry R.A., Astrand M., Speed T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 14.Dennis G., Jr., Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- 15.Zoppi N., Gardella R., De Paepe A., Barlati S., Colombi M. Human fibroblasts carrying mutations in COL5A1 and COL3A1 genes do not organize collagens and fibronectin in the extracellular matrix, down-regulate α2β1 integrin, and recruit α5β3 instead of α5β1 integrin. J. Biol. Chem. 2004;279:18157–18168. doi: 10.1074/jbc.M312609200. [DOI] [PubMed] [Google Scholar]
- 16.Zoppi N., Barlati S., Colombi M. FAK-independent α5β3 integrin-paxillin-EGFR complexes rescue from anoikis matrix-defective fibroblasts. Biochim. Biophys. Acta. 2008;1783:1177–1188. doi: 10.1016/j.bbamcr.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Jahadi Hosseini H.R., Katbab A., Khalili M.R., Abtahi M.B. Comparison of corneal thickness measurements using Galilei, HR Pentacam, and ultrasound. Cornea. 2010;29:1091–1095. doi: 10.1097/ICO.0b013e3181cf98e5. [DOI] [PubMed] [Google Scholar]
- 18.Steinmann B., Eyre D.R., Shao P. Urinary pyridinoline cross-links in Ehlers-Danlos syndrome type VI. Am. J. Hum. Genet. 1995;57:1505–1508. [PMC free article] [PubMed] [Google Scholar]
- 19.Giunta C., Randolph A., Al-Gazali L.I., Brunner H.G., Kraenzlin M.E., Steinmann B. Nevo syndrome is allelic to the kyphoscoliotic type of the Ehlers-Danlos syndrome (EDS VIA) Am. J. Med. Genet. A. 2005;133A:158–164. doi: 10.1002/ajmg.a.30529. [DOI] [PubMed] [Google Scholar]
- 20.Cameron J.A. Corneal abnormalities in Ehlers-Danlos syndrome type VI. Cornea. 1993;12:54–59. doi: 10.1097/00003226-199301000-00009. [DOI] [PubMed] [Google Scholar]
- 21.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 22.Meani N., Pezzimenti F., Deflorian G., Mione M., Alcalay M. The tumor suppressor PRDM5 regulates Wnt signaling at early stages of zebrafish development. PLoS ONE. 2009;4:e4273. doi: 10.1371/journal.pone.0004273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Segev F., Héon E., Cole W.G., Wenstrup R.J., Young F., Slomovic A.R., Rootman D.S., Whitaker-Menezes D., Chervoneva I., Birk D.E. Structural abnormalities of the cornea and lid resulting from collagen V mutations. Invest. Ophthalmol. Vis. Sci. 2006;47:565–573. doi: 10.1167/iovs.05-0771. [DOI] [PubMed] [Google Scholar]
- 24.Steinmann B., Royce P.M., Superti-Furga A. The Ehlers-Danlos syndrome. In: Royce P.M., Steinmann B., editors. Connective Tissue and its Heritable Disorders. 2nd ed. Wiley-Liss; New York: 2002. pp. 431–523. [Google Scholar]
- 25.Ahmad N.N., Ala-Kokko L., Knowlton R.G., Jimenez S.A., Weaver E.J., Maguire J.I., Tasman W., Prockop D.J. Stop codon in the procollagen II gene (COL2A1) in a family with the Stickler syndrome (arthro-ophthalmopathy) Proc. Natl. Acad. Sci. USA. 1991;88:6624–6627. doi: 10.1073/pnas.88.15.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richards A.J., Yates J.R., Williams R., Payne S.J., Pope F.M., Scott J.D., Snead M.P. A family with Stickler syndrome type 2 has a mutation in the COL11A1 gene resulting in the substitution of glycine 97 by valine in alpha 1 (XI) collagen. Hum. Mol. Genet. 1996;5:1339–1343. doi: 10.1093/hmg/5.9.1339. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.