

Abstract
Hereditary leukonychia (porcelain nails or white nails) is a rare nail disorder with an unknown genetic basis. To identify variants in a gene underlying this phenotype, we identified four families of Pakistani origin showing features of hereditary leukonychia. All 20 nails of each affected individual were chalky and white in appearance, consistent with total leukonychia, with no other cutaneous, appendageal, or systemic findings. By using Affymetrix 10K chip, we established linkage to chromosome 3p21.3-p22 with a LOD score (Z) of 5.1. We identified pathogenic mutations in PLCD1 in all four families, which encodes phosphoinositide-specific phospholipase C delta 1 subunit, a key enzyme in phosphoinositide metabolism. We then identified localization of PLCD1 in the nail matrix. It was recently shown that PLCD1 is a component of the human nail plate by proteomic analysis and is localized in the matrix of human nails. Furthermore, mutations detected in PLCD1 resulted in reduced enzymatic activity in vitro. Our data show that mutations in PLCD1 underlie hereditary leukonychia, revealing a gene involved in molecular control of nail growth.
Main Text
The nail is a specialized skin appendage with a complex structure involving a fully keratinized nail plate produced by the germinative epithelium of the nail matrix. Significant advances have been made in the diagnosis and management of nail diseases, yet our knowledge of the underlying molecular controls of nail growth and development is relatively scant.1 A white appearance of nails can result from whitening of the nail plate (true leukonychia), the nail bed (pseudoleukonychia), or neither (apparent leukonychia) and can be due to a variety of factors including infectious, metabolic, or systemic diseases, trauma, or drugs.2 One of the rare causes of whitening of the nail plate is hereditary leukonychia (MIM 151600), a rare autosomal-dominant or -recessive nail disorder with an unknown genetic basis. In hereditary leukonychia, whitening of the nails may exist as an isolated feature or be associated with other cutaneous or systemic abnormalities, including keratosis palmoplantaris, pilar and sebaceous cysts, severe keratosis pilaris, pili torti, hypotrichosis with keratoderma, onychorrhexis, koilonychia, renal calculi, and hair dysplasia with acanthosis-nigricans-like lesions.2–14
We undertook this study to identify a gene involved in hereditary leukonychia, with the aim of defining a new genetic determinant of nail growth in humans. We identified four families of Pakistani origin showing features of hereditary leukonychia that were present since birth. Two of the families included consanguineous marriages or other features consistent with recessively inherited leukonychia (Figure 1C), whereas the other two families demonstrated dominant inheritance (Figure 1C). All 20 nails of each affected individual were chalky white, consistent with total leukonychia (Figure 1A), although some displayed incomplete leukonychia with translucency and yellowish coloration in the distal parts of the nail plate. There was a negative family history for malignancy. No other skin, hair, dermatologic, or systemic findings were detected. Informed consent was obtained from all subjects and approval for this study was provided by the Institutional Review Board of Columbia University. The study was conducted in adherence to the Declaration of Helsinki Principles. Peripheral blood samples were collected from members of four Pakistani families as well as 130 unrelated healthy control individuals of Pakistani origin. Genomic DNA was isolated from these samples according to standard techniques.15 We performed a genome-wide scan on eight members of family A with the low-density Affymetrix 10K SNP array. Genespring GT (Agilent Software) was used for quality control measures and to perform a number of analyses. SNPs showing Mendelian inheritance errors were removed, and the remaining analyzed data set contained 9887 SNPs. Haplotypes, or clusters of SNPs that tend to be inherited together, were inferred from the data by Genespring GT. Using haplotypes minimizes the effect of linkage disequilibrium on multipoint linkage analysis, reducing type II error. Initial analysis included genome-wide autozygosity mapping to identify regions identical by descent that were shared among affected individuals. We performed parametric linkage analysis twice, once with SNP genotypes and once with haplotypes. All tests assumed a recessive mode of inheritance with 100% penetrance and a disease allele frequency of 0.001. We identified a region of excess homozygosity shared among affected individuals on chromosome 3p21.3-p22 (Figure 1B) with a LOD score (Z) of 5.1. Microsatellite markers spanning the region of autozygosity were then used to genotype all families, revealing linkage to the same location in the other three families (Figure 1C). Key recombination events were detected between markers D3S1483 and D3S4134 in the affected individuals V-1 and V-3 of family A (Figure 1C) and between markers D3S1298 and D3S1260 in the unaffected individual V-6 (Figure 1C), which allowed the interval of linkage to be narrowed to 2.0 Mb flanked by markers D3S1483 and D3S1260.
Figure 1.
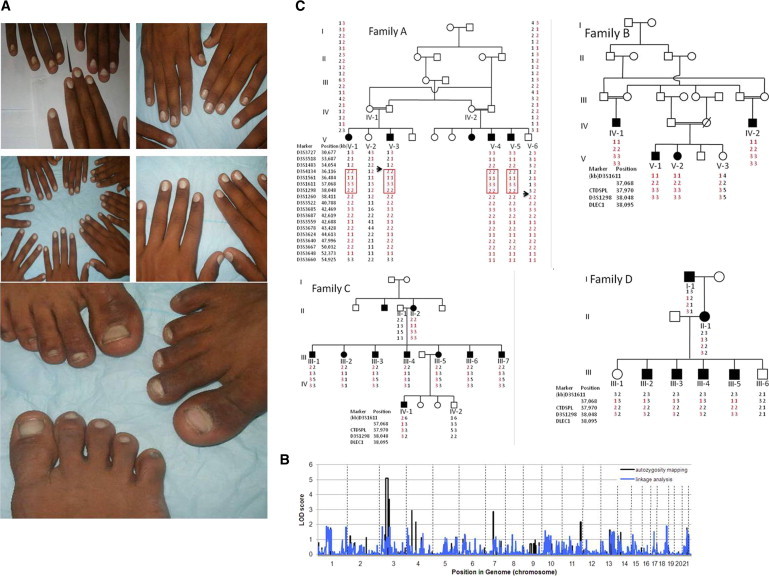
Clinical Features of Affected Individuals and Linkage to Chromosome 3 and Identification of Mutations in PLCD1
(A) Complete nail plate leukonychia involving all the finger and toe nails in affected individuals from both recessive families.
(B) Results of autozygosity mapping in family A. The maximum LOD score was obtained for a region on chromosome 3.
(C) Haplotype analysis in families A, B, C, and D. The linked haplotype is indicated in red, and key recombination events are indicated by arrows.
The region of interest harbored 18 known genes, 15 pseudogenes, and 3 predicted transcripts (Figure 2A). After excluding genes associated with other inherited disorders and sequencing seven known genes in the region (STAC [MIM 602317], ITGA9 [MIM 603963], DCLK3 [MIM 613167], SLC22A13 [MIM 604047], GOLGA4 [MIM 602509], SLC22A14 [MIM 604048], and PLCD1 [MIM 602142] [NM_001130964.1]), we identified predicted mutations in PLCD1 in all four families (Figures 2B and 2C). All exons of PLCD1 were amplified by PCR with gene-specific primers (see Table S1 available online). The PCR products were directly sequenced in an ABI Prism 310 Automated Sequencer, with the ABI Prism Big Dye Terminator Cycle Sequencing Ready Reaction Kit (PE Applied Biosystems). We screened for the PLCD1 mutations in genomic DNA from 130 unrelated healthy control individuals (260 chromosomes) of Pakistani origin. Affected individuals of autosomal-recessive family A displayed a homozygous mutation c.1309C>T leading to a premature termination codon p.Arg437X (Figure 2B). The affected individuals in the second family with autosomal-recessive inheritance, family B, also showed a homozygous protein-truncating mutation consisting of a 10 bp deletion spanning the splice junction of intron 11 and exon 12 (c.1792-10delTGTAGTGGCC) resulting in a premature termination codon because of either a frameshift or missplicing (Figure 2C). By contrast, affected individuals of families C and D with autosomal-dominant inheritance displayed heterozygous missense mutations c.1720C>T (antisense strand) and c.625T>C in PLCD1 leading to the amino acid substitutions p.Ala574Thr and p.Cys209Arg, respectively (Figures 2D and 2E). None of the mutations were found in 130 population-matched, unrelated, unaffected control individuals screened by restriction enzymes (data not shown) or direct sequencing (for p.Ala574Thr and p.Cys209Arg, data not shown). p.Ala574Thr and p.Cys209Arg are both highly conserved among several organisms (Figure S1A). The identification of protein-truncating mutations indicated that an absence or reduction of PLCD1 activity might underlie the leukonychia phenotype in the affected individuals. To confirm this hypothesis, we assayed the function of the p.Arg437X mutant PLCD1 in vitro. The wild-type PLCD1-cDNA was amplified by PCR from human foreskin total mRNA with the following primers: forward 5′-AAACTCGAGGCCACCATGCAGTGCCTGGGGATC-3′ and reverse 5′-AAAGGTACCGGCCTAGTCCTGGAGGGAGATC-3′. The PCR product was cloned into the mammalian expression vector pCXN2.116 with XhoI and KpnI sites. The expression construct for the p.Arg437X mutant PLCD1-p.Arg437X-pCXN2.1 was generated with the QuikChange II XL Site-Directed Mutagenesis Kit (QIAGEN) with the PLCD1-pCXN2.1 construct as a template. To measure PLCD1 function, accumulation of its downstream metabolite, inositol monophosphate (IP1), was quantified with IP-One ELISA competitive immunoassay kit (Cisbio). We plated 1.5 × 105 HEK293 cells on 24-well plates and transfected with pCXN2.1 (vector only), the wild-type PLCD1 containing construct (PLCD1-pCXN2.1), or the mutant (p.Arg437X) PLCD1 containing construct (PLCD1 p.Arg437X-pCXN2.1) with Fugene transfection reagent (Invitrogen). Forty-eight hours after transfection, cells were stimulated in the presence of LiCl causing the accumulation of IP1 upon receptor activation and cell lysates were collected. Competitive immunoassay with IP1-HRP and a IP1 monoclonal antibody was then carried out according to manufacturer's instructions. The optical density (OD) was read at 450 nm with BioRad microplate reader model 680.
Figure 2.
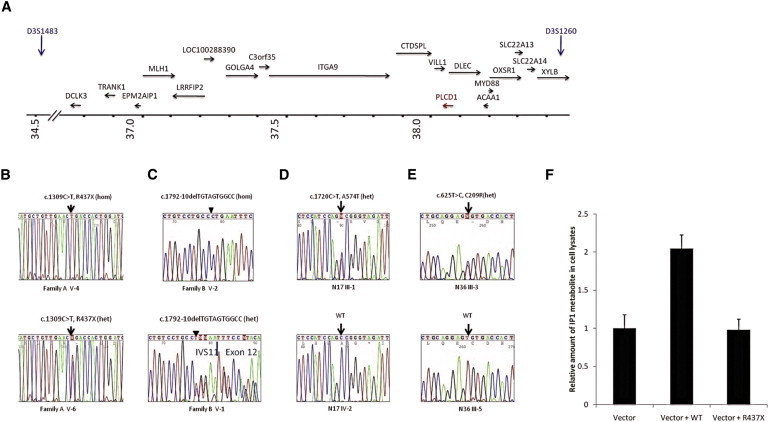
Identification of Mutations in PLCD1 and Function of Mutant PLCD1
(A) Schematic representation of the candidate region harboring PLCD1. Arrows indicate the position and the direction of transcription of genes in the region. FCHP, LOC100130503, ARPP21, and STAC are located upstream of the region shown where the axis has been truncated.
(B and C) Mutation in PLCD1 in families A and B.
(B) Family A; homozygous mutation c.1309C>T, p.Arg437X in an affected individual V-4 (top); heterozygous mutation in an unaffected carrier V-6 (bottom).
(C) Family B; homozygous mutation c.1792-10delTGTAGTGGCC in affected member V-2 and heterozygous in carrier member V-1 of family B.
(D) Heterozygous c.1720C>T (antisense strand), p.Ala574Thr in family C.
(E) Heterozygous c.625T>C, p.Cys209Arg in family D.
(F) Function of p.Arg437X mutant PLCD1 protein in vitro by detection of phospholipase C downstream metabolite IP1. The quantity of IP1 was measured by ELISA 48 hr after transfection of HEK293T cells with vector only (negative control), vector and wild-type PLCD1 construct (positive control), and vector and mutant PLCD1 construct. The test was repeated five times and difference was statistically significant (p < 0.01).
We found a significant reduction (p < 0.01) in IP1, a downstream metabolite of inositol 1,4,5-trisphosphate (IP3) that accumulates in cells after activation of phospholipase C-coupled receptors, PLCD1 in 293 cells transfected with the mutant construct compared with the wild-type (Figure 2F).
To further implicate PLCD1 in the pathogenesis of hereditary leukonychia, we examined its localization in the nail. For PLCD1 immunohistochemistry, paraffin-embedded sections of human fetal (IRB-AAAB2447) nail were used. Antigen unmasking was carried out with Antigen Unmasking Solution (Vector Laboratories). After blocking with normal goat serum, samples were incubated with a rabbit polyclonal antibody raised against human PLCD1 (1:50 dilution; HPA020107; Sigma Prestige) at 4°C overnight in a humidified chamber. Primary antibody was omitted from the negative control. To visualize the staining, the Vectastain Elite rabbit ABC-AP kit (Vector Laboratories) and alkaline phosphatase (AP) staining with 5-Bromo-4-chloro-3-indolyl phosphate and 4-Nitro Blue Tetrazolium Chloride (Roche Applied Science) in AP staining buffer (100 mM Tris [pH 9.5], 50 mM MgCl2, 10 mM NaCl, and 1% Tween 20 in H2O) were used. By immunohistochemistry analysis, prominent localization was detected in the nail matrix and the nail bed (Figures 3A–3D). We then isolated total RNA from plucked human scalp hair, human scalp, human foreskin, and foreskin-derived keratinocytes and fibroblasts with the RNeasy Minikit (QIAGEN). 2 μg of total RNA was reverse transcribed with random primers and SuperScript III (Invitrogen). The cDNAs were then amplified by PCR with PLCD1-specific primers (forward 5′-CAGCGTCAGAAGCTACAGCA-3′, reverse 5′-TCTGTCTGGGAGTGGTCACA-3′) and SYBR Green master mix. The PCR reactions were run in an Applied Biosystems Sequence Detection System 7700. Relative expression levels were calculated with the ΔΔCt method (Applied Biosystems), with GAPDH as the internal control. The expression level of human scalp was set as the calibrator. Although PLCD1 mRNA was expressed in human skin and hair follicle as evidenced by real-time quantitative RT-PCR analysis (Figure S1B), we found no evidence of hair or skin abnormalities in the affected individuals.
Figure 3.
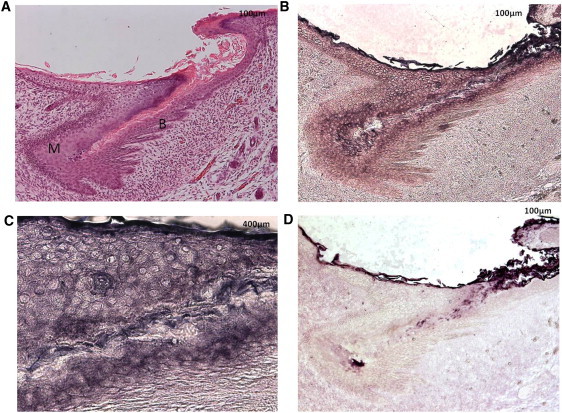
PLCD1 Localization in Human Embryo Nail
(A–C) Anatomy of human embryo nail by (A) hematoxylin and eosin staining (M, nail matrix; B, nail bed) and by (B and C) PLCD1 staining (purple indicates positive staining) revealing cytoplasmic and membranous localization of PLCD1.
(D) Negative control.
Although many hereditary disorders with isolated or combined nail defects have been described, the underlying gene defect in most of these diseases is yet unknown.1 Some of the genes implicated in human nail diseases include LMX1B (MIM 602575), a transcription factor that regulates collagen IV expression in the nail-patella syndrome (NPS [MIM 161200]),17 K6 (MIM 148041), K16 (MIM 148067), and K17 (MIM 148069) in pachyonychia congenita type 1 (PC1 [MIM 167200]) and pachyonychia congenita type 2 (PC2 [MIM 167210]), a group of rare autosomal-dominant ectodermal dysplasias,18 and RSPO4 (MIM 610573), a Wnt signaling ligand we found to be mutated in congenital anonychia (MIM 206800).19 Previously, Norgett et al.20 showed genetic linkage in a family with autosomal-dominant hereditary leukonychia to the type II cytokeratin locus on 12q13 and detected abundant intracellular vacuoles and a lesser compactness of keratins in the affected nails, postulating that the white appearance of nails seemed to be due to an abnormal keratinization of cells originating from the proximal nail matrix.
The finding of PLCD1 mutations in four families with autosomal-recessive and autosomal-dominant hereditary leukonychia (Table 1) broaden our knowledge of genetic control of nail growth in humans and emphasize the involvement of nonkeratin genes in these processes.
Table 1.
Summary of the Four Families and Their Mutations
| Family | Pattern of Inheritance | Mutation | Consequence |
|---|---|---|---|
| A | autosomal recessive | c.1309C>T, p.Arg437X | suppresses IP1, a downstream metabolite of PLCD1 |
| B | autosomal recessive | c.1792-10delTGTAGTGGCC | not tested |
| C | autosomal dominant | c.1720C>T (antisense strand), p.Ala574Thr | not tested |
| D | autosomal dominant | c.625T>C, p.Cys209Arg | not tested |
Phosphoinositide-specific phospholipase C (PLC) is a key enzyme in phosphoinositide turnover by hydrolyzing phosphatidylinositol 4,5-bisphosphate (PIP2) to generate two second messengers, diacylglycerol (DG) and IP3.20 DG further mediates the activation of protein kinase C, and IP3 releases calcium from intracellular stores playing a role in a variety of physiological functions.21 PLCs are divided into six isotypes: four β, two γ, four δ, one ɛ, one ζ, and one η types. The catalytic domains of all PLCs share 40%–60% similarity. These isotypes are regulated by different mechanisms. For instance, PLC-β isoenzymes are activated by the Gαq/11 family of heterotrimeric G-proteins and βγ subunits are released from the Gαi/o family while PLC-γ isoenzymes are activated by tyrosine kinases through tyrosine phosphorylation. PLC-ɛ is activated by Gα12 and ras through membrane translocation. These isotypes are expressed in various tissues where they are believed to play fundamental roles in triggering cellular responses.22 Recently, it was found that PLCD1 acts as a tumor-suppressor gene in cell lines of gastric and breast cancer.23,24 None of our families in this study reported any history of malignancies.
Interestingly, Plcd1-deficient mice undergo progressive hair loss because of epidermal hyperplasia, occlusion of the hair canal, sebaceous gland hyperplasia, and formation of abnormal cysts, though a nail phenotype was not described.25 Furthermore, Plcd1-knockout mice share striking phenotypic similarity to the Foxn1-deficient nude mice, whose skin defects are caused in part by insufficient expression of Plcd1.26 Further studies are needed to examine the link between Plcd1 and the nail dystrophy in nude mice. Nude mice also have nail dystrophy with shorter and abnormally shaped nails, severe parakeratosis of the dorsal nail plate because of dysplasia and impaired cornification of the nail matrix, and lower nail plate sulfur concentrations.27 Although PLCD1 mRNA was expressed in human skin and hair follicle as shown by real-time quantitative RT-PCR analysis, we found no evidence of hair or skin abnormalities in the affected individuals, suggesting that other members of the phosphoinositide-specific phospholipase C family may compensate for the PLCD1 defect in the skin and the hair follicle.
Most recently, it was shown through proteomic studies that PLCD1 is abundant in human nails.28 The mechanism through which it leads to the formation of leukonychia remains to be determined. The phenomenon that recessive and dominant mutations in the same gene result in a similar phenotype is very rare. Ataxia telangiectasia (AT [MIM 208900]) is an autosomal-recessive condition caused by mutations in ATM (MIM 607585). In 2008, Smirnov and Cheung investigated the effect of heterozygous and homozygous ATM mutations on gene and microRNA expression and found that some genes and/or pathways require only one wild copy of ATM to function normally whereas others require two copies. In summary, they demonstrated that mutations in ATM result in both recessive and dominant expression phenotypes of genes and microRNAs. Moreover, recessive and dominant mutations in keratins 5 and 14 have been reported in epidermolysis bullosa simplex (EBS).29,30 This is one possibility that can explain the occurrence of white nails in the setting of recessive or dominant mutations. A second possibility would be that the missense mutations exert a dominant-negative effect on the wild-type allele with complete loss of function.31
In conclusion, our findings indicate that mutations in PLCD1 underlie hereditary leukonychia and, more broadly, reveal a gene involved in controlling nail growth in humans.
Acknowledgments
We are grateful to the family members for their participation in this study. We would like to thank Dr. Robert H. Rice for his valuable discussion, comments, and ideas and H. Lam and M. Zhang for technical assistance. We appreciate the collaboration with K. Fantauzzo, C. Higgins, K. Inoue, and other members of the A.M.C. laboratory. Supported in part by USPHS/NIH grant RO1AR44924 from NIAMS (to A.M.C.) and the skin Disease Research Center at Columbia University (P30AR44535). M.K. was a trainee on NIH Institutional Research Training Grant T32AR007605 (P.I. D. Bickers, Department of Dermatology, Columbia University).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Agilent, http://www.chem.agilent.com/en-US/Products/software/lifesciencesinformatics/Pages/gp63308.aspx
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Paus R., Peker S., Sundberg J.P. Biology of hair and nails. In: Bolognia J.L., Jorizzo J.L., Rapini R.P., editors. Dermatology. Second Edition. Elsevier; St. Louis, MO: 2007. pp. 965–986. [Google Scholar]
- 2.Grossman M., Scher R.K. Leukonychia. Review and classification. Int. J. Dermatol. 1990;29:535–541. doi: 10.1111/j.1365-4362.1990.tb03463.x. [DOI] [PubMed] [Google Scholar]
- 3.Albright S.D., 3rd, Wheeler C.E., Jr. Leukonychia. Total and partial leukonychia in a single family with a review of the literature. Arch. Dermatol. 1964;90:392–399. doi: 10.1001/archderm.1964.01600040020003. [DOI] [PubMed] [Google Scholar]
- 4.Bart R.S., Pumphrey R.E. Knuckle pads, leukonychia and deafness. A dominantly inherited syndrome. N. Engl. J. Med. 1967;276:202–207. doi: 10.1056/NEJM196701262760403. [DOI] [PubMed] [Google Scholar]
- 5.Başaran E., Yilmaz E., Alpsoy E., Yilmaz G.G. Keratoderma, hypotrichosis and leukonychia totalis: A new syndrome? Br. J. Dermatol. 1995;133:636–638. doi: 10.1111/j.1365-2133.1995.tb02720.x. [DOI] [PubMed] [Google Scholar]
- 6.Bushkell L.L., Gorlin R.J. Leukonychia totalis, multiple sebaceous cysts, and renal calculi. A syndrome. Arch. Dermatol. 1975;111:899–901. [PubMed] [Google Scholar]
- 7.Crosby E.F., Vidurrizaga R.H. Knuckle pads, leukonychia, deafness, and keratosis palmoplantaris: Report of a family. Johns Hopkins Med. J. 1976;139(SUPPL):90–92. [PubMed] [Google Scholar]
- 8.Galadari I., Mohsen S. Leukonychia totalis associated with keratosis pilaris and hyperhidrosis. Int. J. Dermatol. 1993;32:524–525. doi: 10.1111/j.1365-4362.1993.tb02841.x. [DOI] [PubMed] [Google Scholar]
- 9.Giustina T.A., Woo T.Y., Campbell J.P., Ellis C.N. Association of pili torti and leukonychia. Cutis. 1985;35:533–534. [PubMed] [Google Scholar]
- 10.Juhlin L. Hereditary leukonychia. Acta Derm. Venereol. 1963;43:136–141. [PubMed] [Google Scholar]
- 11.Slee J.J., Wallman I.S., Goldblatt J. A syndrome of leukonychia totalis and multiple sebaceous cysts. Clin. Dysmorphol. 1997;6:229–231. doi: 10.1097/00019605-199707000-00005. [DOI] [PubMed] [Google Scholar]
- 12.Stevens K.R., Leis P.F., Peters S., Baer S., Orengo I. Congenital leukonychia. J. Am. Acad. Dermatol. 1998;39:509–512. doi: 10.1016/s0190-9622(98)70341-x. [DOI] [PubMed] [Google Scholar]
- 13.Lee Y.B., Kim J.E., Park H.J., Cho B.K. A case of hereditary leukonychia totalis and partialis. Int. J. Dermatol. 2011;50:233–234. doi: 10.1111/j.1365-4632.2010.04306.x. [DOI] [PubMed] [Google Scholar]
- 14.Le Corre Y., Steff M., Croue A., Filmon R., Verret J.L., Le Clech C. Hereditary leukonychia totalis, acanthosis-nigricans-like lesions and hair dysplasia: A new syndrome? Eur. J. Med. Genet. 2009;52:229–233. doi: 10.1016/j.ejmg.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 15.Sambrook J., Fritsch E.F., Maniatis T. Second Edition. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- 16.Niwa H., Yamamura K., Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 17.Morello R., Zhou G., Dreyer S.D., Harvey S.J., Ninomiya Y., Thorner P.S., Miner J.H., Cole W., Winterpacht A., Zabel B. Regulation of glomerular basement membrane collagen expression by LMX1B contributes to renal disease in nail patella syndrome. Nat. Genet. 2001;27:205–208. doi: 10.1038/84853. [DOI] [PubMed] [Google Scholar]
- 18.Terrinoni A., Smith F.J., Didona B., Canzona F., Paradisi M., Huber M., Hohl D., David A., Verloes A., Leigh I.M. Novel and recurrent mutations in the genes encoding keratins K6a, K16 and K17 in 13 cases of pachyonychia congenita. J. Invest. Dermatol. 2001;117:1391–1396. doi: 10.1046/j.0022-202x.2001.01565.x. [DOI] [PubMed] [Google Scholar]
- 19.Bergmann C., Senderek J., Anhuf D., Thiel C.T., Ekici A.B., Poblete-Gutierrez P., van Steensel M., Seelow D., Nürnberg G., Schild H.H. Mutations in the gene encoding the Wnt-signaling component R-spondin 4 (RSPO4) cause autosomal recessive anonychia. Am. J. Hum. Genet. 2006;79:1105–1109. doi: 10.1086/509789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Norgett E.E., Wolf F., Balme B., Leigh I.M., Perrot H., Kelsell D.P., Haftek M. Hereditary ‘white nails’: a genetic and structural study. Br. J. Dermatol. 2004;151:65–72. doi: 10.1111/j.1365-2133.2004.05994.x. [DOI] [PubMed] [Google Scholar]
- 21.Nishizuka Y. The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature. 1988;334:661–665. doi: 10.1038/334661a0. [DOI] [PubMed] [Google Scholar]
- 22.Stewart A.J., Mukherjee J., Roberts S.J., Lester D., Farquharson C. Identification of a novel class of mammalian phosphoinositol-specific phospholipase C enzymes. Int. J. Mol. Med. 2005;15:117–121. [PubMed] [Google Scholar]
- 23.Hu X.T., Zhang F.B., Fan Y.C., Shu X.S., Wong A.H., Zhou W., Shi Q.L., Tang H.M., Fu L., Guan X.Y. Phospholipase C delta 1 is a novel 3p22.3 tumor suppressor involved in cytoskeleton organization, with its epigenetic silencing correlated with high-stage gastric cancer. Oncogene. 2009;28:2466–2475. doi: 10.1038/onc.2009.92. [DOI] [PubMed] [Google Scholar]
- 24.Xiang T., Li L., Fan Y., Jiang Y., Ying Y., Putti T.C., Tao Q., Ren G. PLCD1 is a functional tumor suppressor inducing G(2)/M arrest and frequently methylated in breast cancer. Cancer Biol. Ther. 2010;10:520–527. doi: 10.4161/cbt.10.5.12726. [DOI] [PubMed] [Google Scholar]
- 25.Nakamura Y., Fukami K., Yu H., Takenaka K., Kataoka Y., Shirakata Y., Nishikawa S., Hashimoto K., Yoshida N., Takenawa T. Phospholipase Cdelta1 is required for skin stem cell lineage commitment. EMBO J. 2003;22:2981–2991. doi: 10.1093/emboj/cdg302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura Y., Ichinohe M., Hirata M., Matsuura H., Fujiwara T., Igarashi T., Nakahara M., Yamaguchi H., Yasugi S., Takenawa T., Fukami K. Phospholipase C-delta1 is an essential molecule downstream of Foxn1, the gene responsible for the nude mutation, in normal hair development. FASEB J. 2008;22:841–849. doi: 10.1096/fj.07-9239com. [DOI] [PubMed] [Google Scholar]
- 27.Mecklenburg L., Paus R., Halata Z., Bechtold L.S., Fleckman P., Sundberg J.P. FOXN1 is critical for onycholemmal terminal differentiation in nude (Foxn1) mice. J. Invest. Dermatol. 2004;123:1001–1011. doi: 10.1111/j.0022-202X.2004.23442.x. [DOI] [PubMed] [Google Scholar]
- 28.Rice R.H., Xia Y., Alvarado R.J., Phinney B.S. Proteomic analysis of human nail plate. J. Proteome Res. 2010;9:6752–6758. doi: 10.1021/pr1009349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yasukawa K., Sawamura D., McMillan J.R., Nakamura H., Shimizu H. Dominant and recessive compound heterozygous mutations in epidermolysis bullosa simplex demonstrate the role of stutter region in keratin intermediate filament assembly. J. Biol. Chem. 2002;277:23670–23674. doi: 10.1074/jbc.M200974200. [DOI] [PubMed] [Google Scholar]
- 30.Has C., Chang Y.R., Volz A., Hoeping D., Kohlhase J., Bruckner-Tuderman L. Novel keratin 14 mutations in patients with severe recessive epidermolysis bullosa simplex. J. Invest. Dermatol. 2006;126:1912–1914. doi: 10.1038/sj.jid.5700312. [DOI] [PubMed] [Google Scholar]
- 31.Smirnov D., Cheung V. ATM gene mutations result in both recessive and dominant expression phenotypes of genes and micrRNAs. Am. J. Hum. Genet. 2008;83:243–253. doi: 10.1016/j.ajhg.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.