

Abstract
The low prevalence rate of orphan diseases (OD) requires special combined efforts to improve diagnosis, prevention, and discovery of novel therapeutic strategies. To identify and investigate relationships based on shared genes or shared functional features, we have conducted a bioinformatic-based global analysis of all orphan diseases with known disease-causing mutant genes. Starting with a bipartite network of known OD and OD-causing mutant genes and using the human protein interactome, we first construct and topologically analyze three networks: the orphan disease network, the orphan disease-causing mutant gene network, and the orphan disease-causing mutant gene interactome. Our results demonstrate that in contrast to the common disease-causing mutant genes that are predominantly nonessential, a majority of orphan disease-causing mutant genes are essential. In confirmation of this finding, we found that OD-causing mutant genes are topologically important in the protein interactome and are ubiquitously expressed. Additionally, functional enrichment analysis of those genes in which mutations cause ODs shows that a majority result in premature death or are lethal in the orthologous mouse gene knockout models. To address the limitations of traditional gene-based disease networks, we also construct and analyze OD networks on the basis of shared enriched features (biological processes, cellular components, pathways, phenotypes, and literature citations). Analyzing these functionally-linked OD networks, we identified several additional OD-OD relations that are both phenotypically similar and phenotypically diverse. Surprisingly, we observed that the wiring of the gene-based and other feature-based OD networks are largely different; this suggests that the relationship between ODs cannot be fully captured by the gene-based network alone.
Introduction
The US Rare Disease Act of 2002 defined a rare or orphan disease (OD) as a disease that affects fewer than 200,000 inhabitants, equivalent to approximately 6.5 patients per 10,000 inhabitants.1 There are an estimated 8000 ODs, many of which are known to be of genetic origin, affect children at a very early age, and be life-threatening and/or chronically debilitating.2,3 Orphan diseases exist in all disease classes and range from exceptionally rare to more prevalent. Because there are so many ODs and because each has such a low prevalence, it is difficult to develop a public health policy specific to each disease. It is possible, however, to have a global rather than a piecemeal approach in the areas of OD and orphan drug research and development, information, and training.4 In the decade before the US Orphan Drug Act in 1983, only ten drugs for rare diseases had received FDA marketing approval, compared with more than 300 orphan drugs that were subsequently approved.5 Most of these orphan drugs, however, are for rare cancers or metabolic diseases, and very few are for ODs of other classes. Furthermore, most of these are symptomatic therapies rather than curative or able to modify fundamental pathophysiology. Additionally, the prices of such approved drugs are in many cases high and hence are a burden for health insurers or patients.6
A recent study reports that ODs featured in a high number of scientific publications are more likely to obtain a therapeutic product than those with a low number of publications.7 Previous studies indicate that many human diseases are interrelated or grouped together due to perturbation of the same gene. Disease networks, disease-causing mutant gene networks, and drug target networks8–10 are increasingly explored as a complement to networks centered on protein or gene interactions. However, the quality of these networks is heavily dependent on the quantity and quality of information that supports their creation11 and is also constrained by the number of known disease-causing mutant genes. One way to overcome this is to use functional linkages based on features other than genes alone. Linghu et al.12 used such functional linkages to identify associations between genes involved in different diseases and to identify relationships that might be associated functionally related sets of genes rather than with the same genes.
Elucidating the mechanisms and interconnectivity of most of the 1700 ODs with known disease-causing mutant genes is important not only for ODs and orphan drug development but also for the understanding of normal biological pathways and common diseases. For example, some of the most effective treatments for coronary artery disease, a very common disease, were first established during the study of familial hypercholesterolemia, an orphan disease. In the current study, using ODs and their known disease-causing mutant genes,13–15 we built a bipartite graph of the human orphan diseasome to investigate the ODs and OD-causing mutant genes (ODMG) in the context of shared gene networks, protein interactions, functional linkages, and literature-based connectivity.
Material and Methods
Data Resources and Analysis
The orphan diseases and mutant gene information was downloaded from Orphanet13 with the Uniprot Knowledgebase15 interface. We then parsed these 2092 orphan disease terms into 1772 distinct orphan diseases by merging some of the disease subtypes of a single disease based on their given disorder names as described previously.9 To compare the ODMGs with common disease genes, we extracted the current curated list of all known disorder-gene associations from the Morbid Map of the Online Mendelian Inheritance in Man (OMIM).14 The human protein interactome used in this study was compiled from several resources16–21 with both redundant interactions and self-loops removed. We defined essential genes (n = 2481) as previously described9 by retrieving a list of human orthologs of mouse genes that resulted in lethal phenotype in embryonic and postnatal stages upon knockout (cataloged in the Mouse Genome database22). The list of ubiquitously expressed human genes was compiled from Ramskold et al.23 and Tu et al.24 The mitochondrial genes were downloaded from the MitoCarta database,25 an inventory of mammalian mitochondrial genes. To identify enriched features (Biological Process, Cellular Component, Mammalian Phenotype, and pathways), we used the ToppGene Suite.26 The feature-based OD networks were constructed with the shared enriched feature (p cut-off 0.05; Bonferroni correction) as an edge. Literature-based orphan disease networks (ODNs) were generated with the shared cited literature in the corresponding OMIM14 disease records of the ODs from the Orphanet. The mappings of OD to OMIM were obtained from the Orphanet. Two ODs are connected if they have a same article cited in their respective OMIM disease records (Figure 1).
Figure 1.
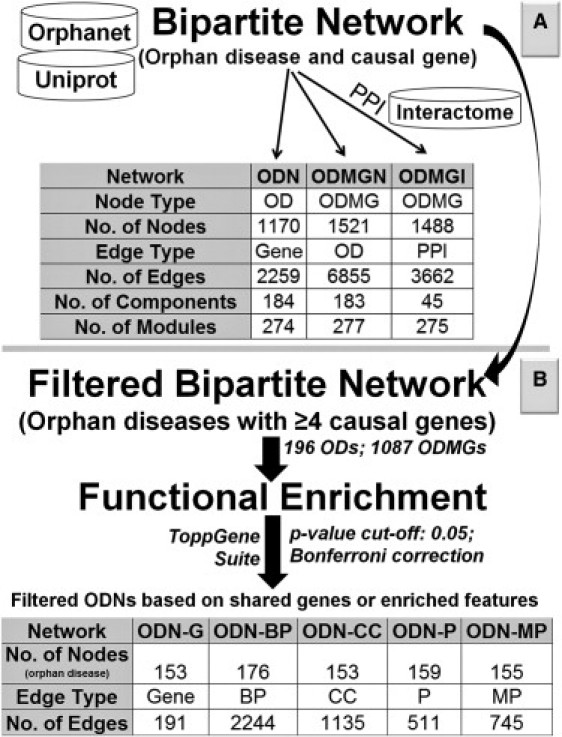
Workflow for Generating the OD Networks on the Basis of Shared Genes or Enriched Functional Features
(A) The details of the ODN, ODMGN, and ODMGI that are generated with the orphan disease and OD-causing mutant gene bipartite network and the human protein interactome.
(B) Outlines of the method and results of the functionally connected ODs.
Analysis of the Orphan Disease Network and OD-Causing Mutant Gene Network
We defined hubs as all nodes that are in the top 20% of the degree distribution (i.e., ODs or OD-causing mutant genes that have the 20% highest number of neighbors), whereas bottlenecks are defined as the nodes that are in the top 20% in terms of betweenness.27 Betweenness measures the total number of nonredundant shortest paths going through a certain node or edge in a network, and, combined with the degree, it is used to assess the relevance of the location of nodes in a network.28 The degree and betweenness centrality values are calculated with TopNet-like Yale Network Analyzer (tYNA).29 We used three well-known centrality measures, namely betweenness centrality, closeness centrality, and eigenvector centrality (available as part of the Gephi package30) to analyze the ODN and orphan disease-causing mutant gene network (ODMGN). In brief, eigenvector centrality is a measure of node importance in a network based on a node's connections, and closeness centrality is the average distance from a given starting node to all other nodes in the network. We define a subnetwork or a connected component as a portion of a network that consists of nodes that are only reachable from nodes in the same network. For all the networks constructed in this study, we determined the number of connected components and their respective sizes by using Gephi.30 Community or modularity, on the other, hand represents the tightness of coupling among a specific group of nodes in comparison to other nodes in the entire network. The community detection algorithm (Louvain Method31) in Gephi was used to identify the modules in each of the networks generated in the study.
Visualization of Orphan Disease Networks
All the networks and related analyses in the current study were performed with Gephi,30 and the results are made available as a browseable web-based resource (Orphan Diseasome). Users can interactively query the different networks for genes or ODs of interest.
Results
Generating and Analyzing Networks of Orphan Diseases and Mutant Genes
We start our analysis with a curated list of 1772 ODs that have at least one implicated gene mutation (2124 OD-causing mutant genes or ODMGs) (Table S1, available online). A gene and OD are considered connected if a known mutation in that gene is implicated as a causal mutation for the OD. Of the 1772 ODs analyzed, 1223 (∼69%) have only one known gene implicated, whereas the remaining 549 have more than one disease-causing mutant gene. On the basis of the mutant genes currently known to cause OD, this finding indicates that the majority of ODs are monogenic. There are only 39 ODs that have 10 or more known disease-causing mutant genes. Of the 2124 OD-causing mutant genes, 1393 are implicated in only one OD, whereas the remaining 731 genes are causative for two or more ODs. For example, mutations of LMNA are implicated in 17 ODs, whereas the OD nonsyndromic genetic deafness has the most number (43) of known disease-causing mutant genes. The average degree of 1772 ODs is 1.94 (number of disease-causing mutant genes per OD), whereas it is 1.62 (number of ODs per gene) for 2124 ODMGs.
The global bipartite network of ODs and ODMGs comprises 3896 nodes (1772 ODs plus 2124 ODMGs) and 3437 edges (gene mutations connecting ODs with ODMGs). There are a total of 786 connected components or subnetworks (nodes that are only reachable from nodes in the same network), ranging in size from 734 genes and 530 ODs to just one gene and one OD. A large number (602; ∼77%) of these subnetworks comprise only one OD and one gene. The number of communities or modules (the tightness of coupling among a specific group of nodes in comparison to other nodes in the network) is 1254 (Louvain modularity31 = 0.81). From this OD-gene bipartite network, we first built and analyzed two types of networks: (1) the ODN and (2) the ODMGN. Second, we selected a subset of all ODs with four or more disease-causing mutant genes and performed functional enrichment analysis to identify enriched biological processes, cellular components, pathways, and mammalian phenotype terms. We then generated a functional feature-based orphan disease network by using shared enriched features (Figure 1). Thus, in this network two ODs are connected (by a shared functional feature) even if they do not share an OD-causing mutant gene. Lastly, using the cited literature in the OD records, we constructed a document-based OD network to analyze and compared it with the traditional gene-based OD network.
The gene-based ODN contains 1170 nodes and 2259 edges (Figure 1). In this network, each node represents an OD, and an edge represents at least one shared ODMG. There are 184 connected components (maximally connected subgraphs) in the ODN with the largest connected component (or subnetwork) of 530 nodes and 1396 edges (Figure 2 and Table S2). On the other hand, the ODMGN contains 1521 nodes and 6855 edges. In this network, each node represents an OD-causing mutant gene, and an edge represents at least one OD shared between two genes (Figure 1). In case of the ODMGN, there are 183 connected components, and the largest connected component has 734 nodes and 4817 edges (Figure 3 and Table S2). There are 274 closely connected modules or communities (modularity score 0.85) in the ODN, whereas there are 277 communities (modularity score 0.87) in the ODMGN, suggesting important pathophysiological relatedness between different orphan diseases and OD-causing mutant genes (see Table S2 for a complete list of subnetworks and communities in the orphan disease and orphan disease-causing mutant gene networks).
Figure 2.
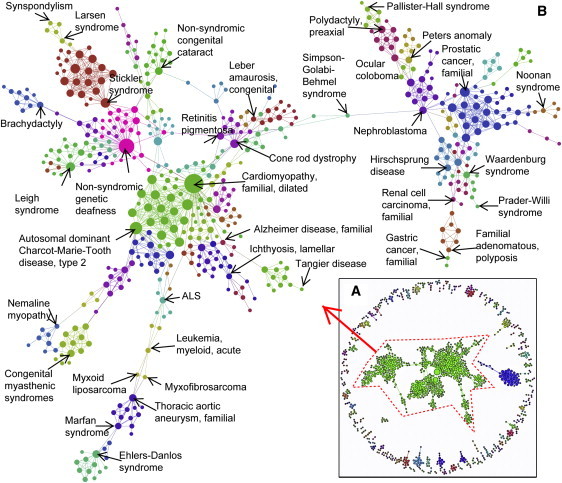
Network of OD Based on Shared Genes
(A) The loosely connected 184 components (subnetworks) of the ODN.
(B) One of the largest subnetworks of the ODN and the 76 modules within it. Modularity indicates the tightness of coupling among a specific group of nodes in comparison to others in the entire network. For simplicity and clarity, only some of the nodes are labeled. The size of the nodes is proportional to the number of other ODs that are connected to it.
Figure 3.
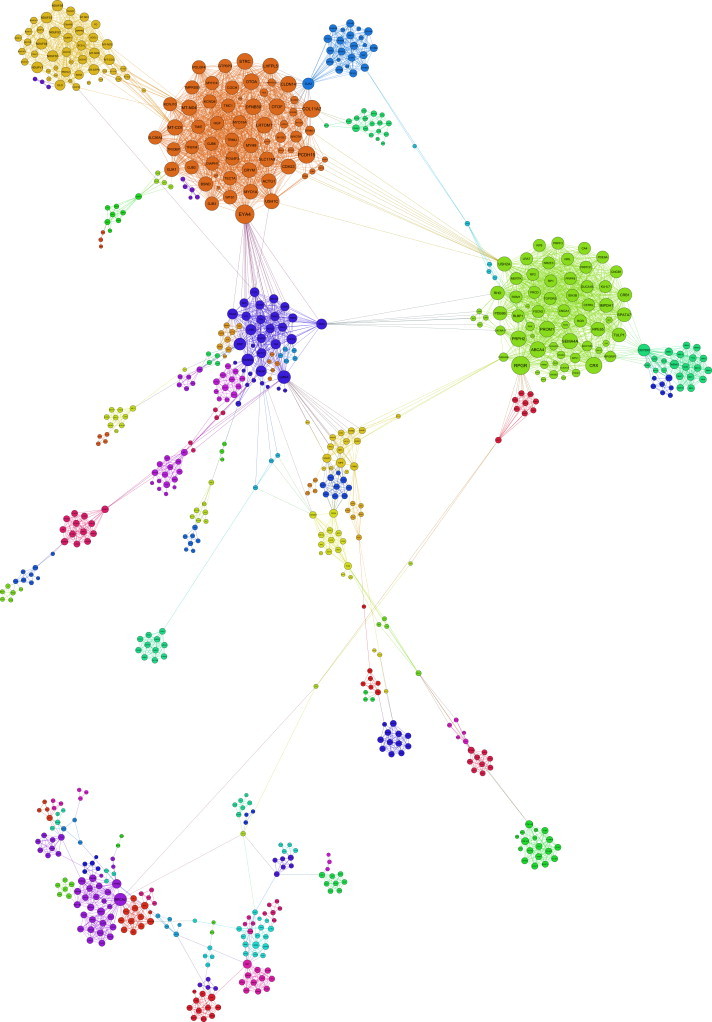
Largest Subnetwork of ODMGN Based on Shared OD
This network represents the largest connected component of the ODMGN and has 734 nodes (OD-causing mutant genes) and 4817 edges (representing at least one shared OD between two OD-causing mutant genes) divided into various modules (indicated by various colors). The size of the node is proportional to the number of other OD-causing mutant genes it is connected to.
To estimate the significance of connectivity in the ODN and ODMGN, we randomly shuffled the relations between orphan diseases and orphan disease-causing mutant genes in the bipartite graph, whereas keeping the number of links or edges per node (OD or ODMG) unchanged. The average sizes of the largest connected component in the randomized ODN and ODMGN were 954 ± 18 and 1305 ± 19, respectively. Both of these were significantly larger than those of actual ODN (530) and ODMGN (734) with both p values being less than 1.0 × 10−5 (one-sample t test), respectively. Thus, clustering of ODs and ODMGs deviates significantly from a random distribution and is consistent with a previous study on the human disease network9 that attributed such clustering to important pathophysiological relatedness between different diseases and disease genes.
Orphan Disease-Causing Mutant Gene Interactome
Genes, whose mutations cause disease, tend to be nonessential and show no tendency to encode hub proteins.9 To check whether genes whose mutations cause ODs are similar to or different from common disease-causing mutant genes, we next built the OD-causing mutant gene interactome by using an assembled human protein interactome. The human protein interactome used in our study contains protein-protein interactions (PPI) from large-scale yeast two-hybrid experiments,16,17 computational predictions,18 and curation of the literature,19–21 with both redundant interactions and self-loops removed. The assembled PPI network consists of 12,260 proteins and 70,576 interactions. Although 1811 out of 2124 ODMGs encode proteins that are part of human PPI network, 1488 of them interact with another protein encoded by an ODMG and 559 interactions overlap between the ODMGN and the orphan disease-causing mutant gene interactome (ODMGI) (Table S3). Additionally, this network of 1488 proteins of ODMGs has 3662 interactions, much more than the expected number of 1539 interactions. The expected number is calculated by dividing the number of all PPIs in the PPI network (70,576) by the number of all possible PPIs between all protein pairs (75,147,670) and then multiplying by all possible PPIs between ODMG pairs (1,638,955). The 559 PPIs (representing 590 OD-causing mutant genes for 266 ODs) that not only interact physically but also share an OD are organized as 145 connected clusters of size 3 and larger (at least two interacting ODMGs and an OD) of proteins implicated with the same or a related disorder (Figure 4 and Table S3). The findings and conclusions drawn from the ODMGI analyses are presented in the following three sections.
Figure 4.
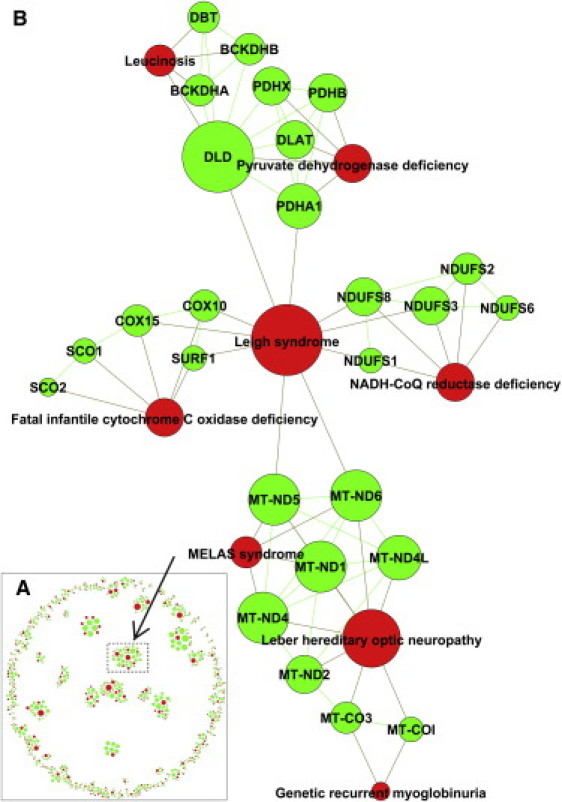
Interacting Genes Associated with Same or Related OD
(A) The 559 protein-protein interactions (representing 590 orphan disease-causing mutant genes for 266 orphan diseases) that not only interact physically but share an OD also and are organized as 145 connected clusters (of size 3 and larger).
(B) One of the connected clusters. The red-colored nodes are ODs, whereas the green ones represent ODMGs. An edge between an OD and ODMG represents known orphan disease-gene relationships, whereas an edge between two genes represents a protein-protein interaction.
OD-Causing Mutant Genes Have High Connectivity or Serve as Bridges between ODMG Communities
We found that proteins encoded by OD-causing mutant genes in the human PPI network tend to have a higher-than-average degree (the number of edges of a node) and betweenness centrality (the number of shortest paths between all pairs of nodes that go through a node) when compared to all other proteins in the network. On one hand, about 28% (507 out of 1811) of ODMGs are hubs in the PPI network, which is a higher percentage than the 20% cutoff definition for all hubs (Table S4). On the other hand, the average degree of proteins encoded by 1811 ODMGs in the PPI network (15.40) is also significantly higher than that of other proteins in the network (10.84) (p < 1.0 × 10−5; Wilcoxon rank sum test). Similarly, the percentage and the average betweenness values for ODMGs are both higher than those of all proteins in the network (Table S4). This is in contrast to previous studies (based on all diseases in the OMIM database) that reported a weak correlation between hubs and disease genes9 and that the majority of disease genes are nonessential and show no tendency to encode hub proteins.9
We next checked whether the opposite is true, that is, whether ODMGs that are highly connected in the human PPI network are responsible for multiple ODs. We found that ODMGs encoding protein hubs (or bottlenecks) in the PPI network tend to be implicated in more ODs than nonhubs (or nonbottlenecks). The average number of implicated ODs (the OD degree) for the 1811 ODMGs is 1.65, and there are significant differences between the average OD degree of hubs (1.85) and nonhubs (1.58) (p = 0.0167 by one-sided Wilcoxon rank sum test). Similar results are observed in comparisons of bottlenecks and nonbottlenecks (1.87 versus 1.56; p = 4.32 × 10−4) (Table S5).
Protein Products of ODMGs Are More Likely to Physically Interact with Those of Other ODMGs
For all 1811 ODMGs for which encoded protein products are in the PPI network, the average number of interacting partners also known to be OD-causing mutant genes is 4.04 (Table S6), and the average ratio between the ODMG-interactant degree and the PPI degree is 0.358, which is significantly higher than the expected ratio of 0.148 (1,810 out of 12,259) (p < 1.0 × 10−5; one-sample t test). This suggests that protein products of ODMGs tend to physically interact with other protein products of ODMGs. Although PPIs alone might not be capable of detecting every novel OD protein, the relatively high proportion of other OD proteins localized within the immediate ODMG-protein interactome space is promising. Indeed, previous studies have shown that the systematic use of PPI data improves positional candidate gene prediction by 10-fold.32
Hubs in the ODMGN Do Not Tend to Be Hubs/Bottlenecks in the Human PPI Network
To address the question of whether an ODMG encoded protein is a hub both in the OD and PPI network, we next compared the ODMGN (an edge is a shared OD) with ODMGI (an edge is PPI). Of the 1521 genes in the ODMGN, 1302 have known protein interactions. Among these 1302 ODMGs, 375 are hubs (the top 20% of nodes with the highest degree values) in the human interactome, whereas the remaining 927 nodes are nonhubs; 388 are bottlenecks (the top 20% of nodes with highest betweenness centrality values), whereas the remaining 914 are nonbottlenecks. The degree and betweenness centrality values are calculated by tYNA.29 We found that hubs in the ODMGN do not tend to be hubs or bottlenecks in the human PPI network (or ODMGI). The average number of OD-causing mutant genes that share the same ODs (the ODMG degree) with other ODMGs in the ODMGN and are hubs (8.28) is not significantly different from the number that are nonhubs (9.00) in the PPI network (p = 0.404; one-sided Wilcoxon rank sum test). Furthermore, the average ODMG degree in the ODMGN for bottlenecks in the PPI network (8.48) is not significantly different from that of nonbottlenecks (8.92) (p = 0.544; one-sided Wilcoxon rank sum test) (Table S7). There were 220 ODMGs for which the encoded proteins do not have any known protein interactions. These ODMGs in the ODMGN have a higher average degree (10.34) compared to ODMGs in the PPI network (8.79), although not statistically significant (p = 0.173) (Table S7). This implies that hub ODMGs in ODMGN tend to be important irrespective of their status in the PPI network. However, it should be noted that the knowledge of the interactome remains incomplete and that many conclusions about global measures (e.g., network topology) should be viewed with some skepticism.33
Orphan Disease-Causing Mutant Genes Encode Proteins that Tend to Be Essential
To confirm our findings that OD genes tend to encode hub or bottleneck proteins, and therefore most of them could be essential genes, we performed a direct comparison with essential genes as described earlier.9 About 36% (765/2124) of the ODMGs are essential genes whose ortholog gene knockout in mice is lethal; this is much higher than the 22% (398/1777) of essential genes in the disease network reported by Goh et al.9 Additionally, we have also observed that 376 ODMGs (∼18%) cause premature deaths in mouse ortholog gene knockout models. Thus, altogether 907 genes (∼43%) from the 2124 ODMGs result in either premature death and/or lethality in mouse gene knockout models (Table S8). We believe that this is even more significant and specific to ODs because Goh et al.'s diseasome9 comprised several ODs, and the reported 22% is probably due to the presence of some of the ODs and genes in their dataset. To test whether this is indeed true, we separated all ODMGs from the entire set of OMIM disease genes (Morbid Map of the OMIM14 database), resulting in two classes of disease genes: 2124 ODMGs and 1901 non-ODMGs (NODMG) or common disease genes (Figure 5 and Table S9). Although ODMGs, as defined earlier, are genes that when mutated caused an orphan disease, NODMGs are genes whose mutant forms are not associated with any orphan disease (based on current orphan disease and gene relationships in the Orphanet database). Compared to NODMGs, ODMGs are significantly enriched for lethality and mitochondrion, as well as premature death (p < 1.0 × 10−5; Fisher's exact test) (Figure 4). A total of 765 (∼36% of 2124) of ODMGs are essential, whereas only 10% (192/1901) of NODMGs are essential. Interestingly, when we checked the extent to which essential genes overlapped with the entire set of disease genes from OMIM Morbid Map (as in Goh et al.9 but with updated disease and essential gene lists), there were 920 (24%) essential disease genes, which is similar to the 22% reported by Goh et al.9 This confirms the original findings of Goh et al.,9 whose study was based on all disease genes, still hold good despite the increase in the database sizes of human disease genes (from 1776 to 3864) and the essential genes (from 1267 to 2481). It also strengthens our conclusion that the enrichment of essential genes is something specific to ODMGs because the percentage of essential ODMGs is higher when compared to either NODGMs or all disease genes from OMIM. Additionally, these results suggest the robustness of our conclusions as well as previous conclusions,9 and we do not expect them to change significantly even if the resource databases are updated with additional genes and annotations. To confirm our findings further, we also repeated this analysis by focusing only on those genes (from each category) that are in the human PPI, and we found similar results (see Table S9 for additional details). Of the 2124 ODMGs, 1811 (∼85%) were also present in the human protein interactome (i.e., they have at least one interacting protein), whereas only 619 (∼33%) of 1901 NODMGs had at least one known interacting protein. There was no significant difference in degree and betweenness of ODMGs when compared to NODMGs. Although this is surprising, one of the reasons could be the relatively low representation (∼33%) of NODMGs in the protein interactome when compared to the representation of ODMGs (∼85%). We also intersected the ODMGs and NODMGs with the ubiquitously expressed human genes (UEHG)23,24 and found that ODMGs are significantly enriched with UEHGs when compared to NODMGs (p < 1.0 × 10−5; Fisher's exact test). Of the 2124 ODMGs, 863 (∼41%) are UEHGs, whereas only about 13% (247/1901) of NODMGs are UEHGs. Together, about 62% (1314/2124) of ODMGs are essential, ubiquitously expressed, or both, whereas in the case of NODMGs, this figure is only ∼18% (348/1901) (see Table S9 for details).
Figure 5.
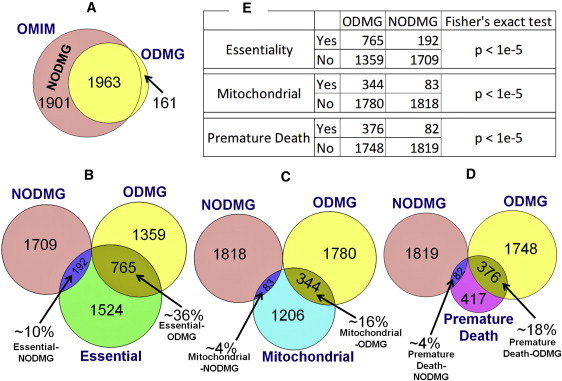
Venn Diagrams Showing the Relationships between ODMG and NODMG with Different Categories of Genes
(A) The overlap between OMIM disease genes and ODMG.
(B) The intersection of ODMG and NODMG with essential genes, whereas (C and D) show the intersections with mitochondrial genes and genes whose knockout in mouse causes premature death.
The table in (E) shows that, compared to NODMGs, ODMGs are enriched for essentiality, mitochondrial genes, and genes associated with premature death in the mouse knockouts.
Function-Based Orphan Disease Networks
In the current study, to obtain a statistically significant and representative functional signature from the 1772 ODs, we first extracted all those ODs with four or more mutant genes from our original data set. Starting with this filtered subbipartite network of 196 ODs and 1087 genes (1283 total nodes and 1395 total edges), we built OD-OD networks based on shared genes and shared functions. The enriched functions (p < 0.05) for each of the 196 ODs were determined with the ToppFun application.26 The shared functions we considered for enrichment analysis included biological processes (BP) and cellular components (CC) from Gene Ontology, KEGG pathways, and mammalian phenotype (MP) (Figure 1). Using the enriched features for each of the orphan diseases (see Table S10 for a complete list of functional enrichment results of the 196 ODs), we rebuilt the orphan disease networks. However, this time the edge between two ODs represents an enriched shared function (BP, CC, Pathway, or MP) and not necessarily a shared gene. After generating these function-based OD networks, we compared them with the gene-based orphan disease networks to find the overlapping nodes and edges. The results were surprising.
The gene-based OD network (153 OD nodes and 191 edges; an edge indicates shared ODMG) is largely different from various function-based OD networks, including a BP-based OD network (176 OD nodes and 2244 edges; edges are shared BP terms), a CC-based OD network (153 OD nodes and 1135 edges; edges are shared CC terms), a MP-based OD network (155 OD nodes and 745 edges; edges are shared MP terms), and a pathway-based OD network (159 OD nodes and 511 edges; edges are shared pathways) (Figure 1 and Table S10). Although the node agreement between the gene-based ODN and function-based ODNs was higher and corresponding Jaccard indices ranged from 0.647 to 0.732, the edge agreement was much lower, and Jaccard indices ranged from 0.0592 to 0.162 (p < 1.0 × 10−5 compared with p for random expectations, one-sample t test; we assessed random expectations by calculating the overlap between the gene-based network and randomized function-based networks with shuffled edges and unchanged node degrees). To address the effect of data incompleteness, we added up to 20% random edges into the gene-based and term-based networks to approximate uncovered associations and compared the overlap of edges with what would be expected as a result of chance, and the results are consistent (Table S10).
Literature-Based ODNs
To test the effectiveness of literature-based networks versus traditional gene-centric approaches in identifying OD-OD relationships, we regenerated the ODN with the edge as a shared published article instead of a shared gene. To avoid potential false positives, we used the corresponding OMIM records of ODs, which summarize results from publications about gene-disease relationships, instead of mining literature. Specifically, we used the cited literature (the links to PubMed records for the references cited in an OMIM entry) in the OMIM records. For 1461 ODs there is a corresponding OMIM record (obtained from Orphanet). Of the 1475 mapped OMIM records, 1370 had at least one cited article (indicated by presence of at least one PubMed ID). We used this subset of 1370 ODs to compare the gene-based OD network with the literature-based OD network.
The gene-based OD network contained 811 ODs as nodes and 1277 edges, indicating common ODMGs shared by a pair of ODs. The literature-based OD network contained 747 ODs as nodes and 927 edges, representing shared literature (PubMed IDs) for a pair of ODs. To estimate the significance of connectivity, we randomly shuffled the relationships between ODs and PubMed IDs in the bipartite graph while keeping the number of links per OD or PubMed ID unchanged. The average size of the largest components in the randomized ODN is 823 ± 12, significantly larger than that of the actual PubMed-based ODN (432) with a p < 1.0 × 10−5 by one-sample Student's t test. This indicates pathophysiological clustering of ODs that deviates from a random distribution as was seen in case of ODN and ODMGNs also.
Although a large number of common nodes exist between the gene- and literature-based networks (517 ODs, 0.5 by Jaccard index), common edges are fewer (255 common edges, 0.13 by Jaccard index; p < 1.0 × 10−5 than would be expected as a result of chance, one-sample t test; random expectations were assessed by calculating the overlap between the gene-based network and randomized PubMed-based networks with shuffled edges and unchanged node degrees). In addition, among the 517 common ODs, less than one fourth of the hubs (31 out of 166 and 143 hubs in the gene-based and the PubMed-based networks, respectively) are conserved. To address the effect of data incompleteness, we randomly added up to 20% edges into the gene-based and PubMed-based networks to approximate uncovered associations and compared the overlap of edges with what would have been expected as a result of chance, and the results are consistent (Table S11). These results indicate that the wirings of these two networks are largely different, which suggests that many ODs with no shared mutant genes might still be related. We also observed that the measures of topological importance differ significantly between the two networks with hardly any overlap. For instance, comparing the top 100 OD nodes (ranks are based on three centrality measures—betweenness centrality, closeness centrality, and eigenvector centrality) in gene-based and literature-based networks shows very little overlap (Table S11). Furthermore, the literature-based OD network was able to identify additional relationships for those diseases sharing no known disease genes but having potential functional links between their corresponding disease gene sets. Among the 927 potentially related OD pairs with literature support, 255 (∼28%) pairs also share known disease genes and are identified by both methods. However, a large number (672 edges; ∼72%) share no known disease genes, and their relationships are identified solely on the basis of literature-connectivity (Figure 6 and Table S11). For instance, Tay-Sachs disease (mutant HEXA and GM2A) and Sandhoff syndrome (mutant HEXB) do not share any disease genes and hence are not connected in shared-gene-based studies.8,9 However, Tay-Sachs disease and Sandhoff disease are connected in the literature-based OD network, which is not surprising because these two disorders arise because of the failure of the same metabolic pathway. Some other examples include Rubinstein-Taybi syndrome (CREBP and EP300 mutants) and ICF syndrome (mutant DNMT3B), which are both syndromes of chromatin modeling; ornithine transcarbamylase deficiency, arginosuccinic aciduria, and citrullinemia, which are all urea cycle disorders; Prader-Willi syndrome and Angelman syndrome, which are both genomic-imprinting disorders; and lathosterolosis, Smith-Lemli-Opitz syndrome, and Greenberg dysplasia, which are all inborn errors of cholesterol synthesis (see Table S11 for a complete list of subnetworks and communities in the literature-based ODN).
Figure 6.
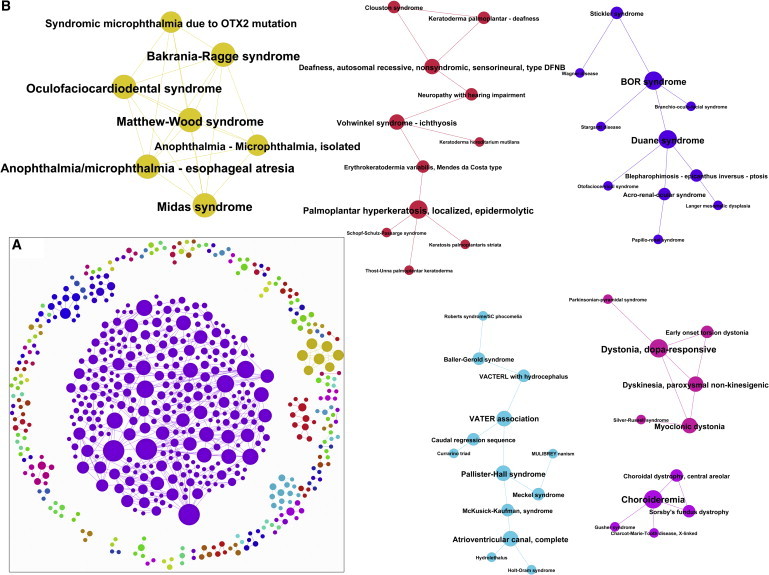
Orphan Disease Network Based on Literature Connectivity
(A) The 577 orphan diseases (672 edges) that are connected by a shared published article. Although these diseases do not share any common OD-causing mutant genes, they are still connected by virtue of a shared published article.
(B) A few of the literature-connected orphan diseases.
Discussion
Although opportunities now exist to accelerate progress toward understanding the basis of many more orphan diseases and for developing innovative medical approaches, relatively few efforts have successfully addressed scientific or technical questions across a spectrum of orphan diseases.34 Therefore, finding common genes, pathways, and targets is critical if we are going to make more than baby steps in orphan disease research. Constructing networks that underlie biological processes and pathways associated with orphan diseases facilitates identification of the functional units that respond to genetic perturbations and potentially affect disease risk or therapeutic response and can systematically move the field in a favorable direction. We believe that the decomposition of orphan disease networks can help us to understand the relationship between orphan diseases and their genetic mechanisms. Studies of biological networks can identify common pathways or processes for multiple orphan diseases that are biologically related and comprehensive understanding such molecular basis could provide opportunities for interventions that are beneficial for an array of related orphan diseases. This capability could open the door for the discovery of single therapies that can benefit multiple disorders and also, potentially, more common diseases.34
Previous studies focusing on all diseases (from OMIM) reported that there was a weak correlation between hubs and disease genes9 and that the majority of disease genes are nonessential and show no tendency to encode hub proteins.9 Our results, in contrast, have shown that genes whose mutations cause orphan diseases tend to encode proteins that are hubs, are ubiquitously expressed, and are essential. Although we acknowledge that the partition of disease genes into such groups (ODMG and NODMG) is a simplification, this partition helped in gaining insights into the relationship between the orphan disease characteristics (rare, lethal, and syndromic in nature) and the underlying causal mutant gene. First, by an evolutionary argument, the partition could explain the rarity of orphan diseases in a population because mutations in hubs might not be compatible with survival and are less likely to be maintained in a population. Second, the partition could explain the severity and lethality associated with most of the ODs because mutations in hubs could have wider repercussions and larger consequences on entire system than those in nonhubs. Additionally, functional enrichment analysis of ODMGs showed that a majority result in premature deaths or are lethal in the orthologous mouse gene knockout models. Third, because hubs through their multiple interacting proteins connect heterogeneous cellular processes, the partition might explain the complex phenotypic or syndromic nature of ODs that have an impact on multiple physiological systems. The ubiquitous nature of ODMGs might also explain this. At the same time, the paradox of ubiquitous expression and tissue-specific phenotypes seen in some of the orphan diseases (e.g., IMPDH and retinitis pigmentosa) is difficult to explain. Some of this has been explained by the existence of novel tissue-specific isoforms and relatively high levels of UEHGs in a particular tissue.35 Together, our results provide further evidence that the genetic and network properties of human genes are related and that some of the disease characteristics can be explained by the topological features of an individual or group of nodes in the network.
Biological networks are known to be modular, and their decomposition into modules or communities provides deep insight into living systems and human diseases.36 We found high connectivity among different orphan diseases or OD-causing mutant genes that can be used not only to infer the common mechanism and targeted pathways but also to find candidates for drug repositioning or drug repurposing (i.e., to extrapolate or suggest novel applications for already approved drugs), especially when one or more than one orphan disease in the community has an approved drug.
Because most of the previous studies elucidating relationships between diseases are genecentric, they are limited in their discovery of new and unknown disease relationships.37 To address this, three recent studies12,37,38 recommend using functional linkage maps. However, each of these approaches focuses on a limited number of features, such as gene expression and PPI data, biological processes, or pathways to connect diseases. Although the node agreement between the gene-based and function-based ODNs was relatively higher, the edge agreement was much lower and indicates that their wiring is significantly different. This suggests that the relationship between the ODs cannot be fully captured by the gene-based networks alone. Thus, by considering functional connectivity between causative genes involved in different orphan diseases, relationships between orphan diseases that are based on underlying molecular mechanisms can be revealed. Such associations can potentially be used to generate novel hypotheses on the molecular mechanisms of diseases, and can in turn guide the development of relevant therapy12 or potential drug repositioning candidates.
A literature-based discovery methodology39 was shown to be effective in identifying disease genes. Indeed, literature-based relations between orphan diseases could provide functional modularity and immediate insight into the underlying molecular mechanisms and thus generate novel hypotheses for therapeutic strategies (e.g., drug repositioning). In this study, we found about 670 related and diverse OD-OD relationships that are identified only by literature connectivity but not by shared genes. To overcome or limit the number of false positives typically associated with text-mining exercises, we focused in the current study only on cited literature in OD records. However, there are some potential limitations to this approach. For instance, we have seen examples of literature that list some of the ODs in a context other than those relating them mechanistically or functionally.
Apart from leading to new insights into the biological underpinnings of various ODs, we believe that our global analysis of orphan diseasome will encourage the development of new and innovative research on these rare conditions that have been hitherto understudied. The global analysis of all ODs can help in analyzing comorbidities and the underlying molecular basis apart from establishing potential networking opportunities. The functional feature-based OD networks, apart from partially addressing the limitations of the conventional gene-based connectivity maps of diseases, can have direct implications to drug discovery process. Physical protein-protein interactome-based ODMGI maps can be used to generate lists of genes potentially enriched for new candidate ODMGs. We have also used several different types of biological data to build functional interaction networks of ODs that are an advantage over gene-based disease networks. These functional interaction networks of ODs can provide a generic framework for integrating disparate data types into a common predictive network. Additionally, the shared functional features between different ODs can be mined for predicting specific OD genetic modifiers or drug targets. Indeed, integration of various interactome and functional relationship networks have been used previously to predict cancer and other types of disease susceptibility candidate and modifier genes.40,41 An important tool in the quest of orphan drug discovery is the ODN that represents a genome-wide roadmap for future studies on orphan diseasome and druggome. As such, it can be used to assess interactions between orphan diseases and the disease-causing mutant genes through the orphan diseasome web site that we have made available online and that offers global perspective and a rapid visual reference of the genetic links between orphan diseases and mutant genes. For instance, overlaying the network of ODs and/or genes with orphan drugs or common disease drugs can be used as a discovery platform for identifying potential drug repositioning candidates.
Acknowledgments
We thank the Orphanet team for providing us with the data. We thank Thomas Boat and John Hutton for useful comments and scientific discussions on the manuscript. We acknowledge the help of Ron Bryson for language editing. This work was supported in part by Cincinnati Digestive Health Sciences Center (Public Health Service Grant P30 DK078392) and Cincinnati Children's Hospital Medical Center.
Supplemental Data
This table shows the orphan disease gene associations extracted from Uniprot. The degree corresponds to the total number of known genes associated with an orphan disease. The networks were generated using the 1772 curated orphan disease terms and 2124 orphan disease-causing mutant genes (ODMGs). The table also includes the gene IDs and gene descriptions of the ODMGs.
These interactions form 145 connected clusters (of size 2 and larger) of genes and are associated with the same disorder (see also Figure 4 in the main text).
The first four worksheets have the details of the enriched (p = 0.05; Bonferroni correction) GO biological process (BP), cellular component (CC), mammalian phenotype (MP), and pathways for 196 orphan diseases (ODs; ODs with four or more causal genes) respectively. The next four worksheets show orphan disease (OD) connectivity based on shared GO-BP, GO-CC, MP, and pathways, respectively. The last worksheet has the results of the comparisons between gene-based and functional feature-based ODNs (node and edge overlaps) and also the results from randomization.
This table has six worksheets that include OD-OD networks based on literature, gene, or both; literature-based ODN versus gene-based ODN and comparison of the top 100 ranked nodes based on eigenvector centrality (EC) measure; literature-based ODN versus gene-based ODN and comparison of the top 100 ranked nodes based on betweenness centrality (BC) measure; literature-based ODN versus gene-based ODN and comparison of top 100 ranked nodes based on closeness centrality (CC) measure; literature-based ODN amd subnetworks and communities: This network of orphan disease is based exclusively on literature connectivity; comparison of node/edge overlaps of gene-based and literature-based ODNs and the effect of incompleteness in data.
Web Resources
The URLs for data presented herein are as follows:
Gene Ontology, http://www.geneontology.org
Gephi, http://gephi.org
HPRD, http://www.hprd.org
Mammalian Phenotype, http://www.informatics.jax.org/phenotypes.shtml
MitoCarta database, http://www.broadinstitute.org/pubs/MitoCarta
Orphan Diseasome, http://research.cchmc.org/od
Orphanet, http://www.orpha.net
PubMed, http://www.pubmed.org
ToppGene Suite, http://toppgene.cchmc.org
UniProt, http://www.uniprot.org
References
- 1.Dear J.W., Lilitkarntakul P., Webb D.J. Are rare diseases still orphans or happily adopted? The challenges of developing and using orphan medicinal products. Br. J. Clin. Pharmacol. 2006;62:264–271. doi: 10.1111/j.1365-2125.2006.02654.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stolk P., Willemen M.J., Leufkens H.G. Rare essentials: Drugs for rare diseases as essential medicines. Bull. World Health Organ. 2006;84:745–751. doi: 10.2471/blt.06.031518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schieppati A., Henter J.I., Daina E., Aperia A. Why rare diseases are an important medical and social issue. Lancet. 2008;371:2039–2041. doi: 10.1016/S0140-6736(08)60872-7. [DOI] [PubMed] [Google Scholar]
- 4.Aymé S., Schmidtke J. Networking for rare diseases: A necessity for Europe. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 2007;50:1477–1483. doi: 10.1007/s00103-007-0381-9. [DOI] [PubMed] [Google Scholar]
- 5.Haffner M.E. Adopting orphan drugs—two dozen years of treating rare diseases. N. Engl. J. Med. 2006;354:445–447. doi: 10.1056/NEJMp058317. [DOI] [PubMed] [Google Scholar]
- 6.Remuzzi G., Garattini S. Rare diseases: What's next? Lancet. 2008;371:1978–1979. doi: 10.1016/S0140-6736(08)60847-8. [DOI] [PubMed] [Google Scholar]
- 7.Heemstra H.E., van Weely S., Büller H.A., Leufkens H.G., de Vrueh R.L. Translation of rare disease research into orphan drug development: Disease matters. Drug Discov. Today. 2009;14:1166–1173. doi: 10.1016/j.drudis.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 8.Feldman I., Rzhetsky A., Vitkup D. Network properties of genes harboring inherited disease mutations. Proc. Natl. Acad. Sci. USA. 2008;105:4323–4328. doi: 10.1073/pnas.0701722105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goh K.I., Cusick M.E., Valle D., Childs B., Vidal M., Barabási A.L. The human disease network. Proc. Natl. Acad. Sci. USA. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yildirim M.A., Goh K.I., Cusick M.E., Barabási A.L., Vidal M. Drug-target network. Nat. Biotechnol. 2007;25:1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- 11.Oti M., Huynen M.A., Brunner H.G. The biological coherence of human phenome databases. Am. J. Hum. Genet. 2009;85:801–808. doi: 10.1016/j.ajhg.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linghu B., Snitkin E.S., Hu Z., Xia Y., Delisi C. Genome-wide prioritization of disease genes and identification of disease-disease associations from an integrated human functional linkage network. Genome Biol. 2009;10:R91. doi: 10.1186/gb-2009-10-9-r91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aymé S. Orphanet, an information site on rare diseases. Soins. 2003;672:46–47. [PubMed] [Google Scholar]
- 14.Hamosh A., Scott A.F., Amberger J., Valle D., McKusick V.A. Online Mendelian Inheritance in Man (OMIM) Hum. Mutat. 2000;15:57–61. doi: 10.1002/(SICI)1098-1004(200001)15:1<57::AID-HUMU12>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 15.Apweiler R., Bairoch A., Wu C.H., Barker W.C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004;32(Database issue):D115–D119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stelzl U., Worm U., Lalowski M., Haenig C., Brembeck F.H., Goehler H., Stroedicke M., Zenkner M., Schoenherr A., Koeppen S. A human protein-protein interaction network: A resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 17.Rual J.F., Venkatesan K., Hao T., Hirozane-Kishikawa T., Dricot A., Li N., Berriz G.F., Gibbons F.D., Dreze M., Ayivi-Guedehoussou N. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 18.Ramani A.K., Bunescu R.C., Mooney R.J., Marcotte E.M. Consolidating the set of known human protein-protein interactions in preparation for large-scale mapping of the human interactome. Genome Biol. 2005;6:R40. doi: 10.1186/gb-2005-6-5-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prasad T.S., Kandasamy K., Pandey A. Human Protein Reference Database and Human Proteinpedia as discovery tools for systems biology. Methods Mol. Biol. 2009;577:67–79. doi: 10.1007/978-1-60761-232-2_6. [DOI] [PubMed] [Google Scholar]
- 20.Joshi-Tope G., Gillespie M., Vastrik I., D'Eustachio P., Schmidt E., de Bono B., Jassal B., Gopinath G.R., Wu G.R., Matthews L. Reactome: A knowledgebase of biological pathways. Nucleic Acids Res. 2005;33(Database issue):D428–D432. doi: 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alfarano C., Andrade C.E., Anthony K., Bahroos N., Bajec M., Bantoft K., Betel D., Bobechko B., Boutilier K., Burgess E. The Biomolecular Interaction Network Database and related tools 2005 update. Nucleic Acids Res. 2005;33(Database issue):D418–D424. doi: 10.1093/nar/gki051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blake J.A., Richardson J.E., Bult C.J., Kadin J.A., Eppig J.T., Mouse Genome Database Group MGD: The Mouse Genome Database. Nucleic Acids Res. 2003;31:193–195. doi: 10.1093/nar/gkg047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramsköld D., Wang E.T., Burge C.B., Sandberg R. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput. Biol. 2009;5:e1000598. doi: 10.1371/journal.pcbi.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tu Z., Wang L., Xu M., Zhou X., Chen T., Sun F. Further understanding human disease genes by comparing with housekeeping genes and other genes. BMC Genomics. 2006;7:31. doi: 10.1186/1471-2164-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.E., Walford G.A., Sugiana C., Boneh A., Chen W.K. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J., Bardes E.E., Aronow B.J., Jegga A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37(Web Server issue):W305–W311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu H., Kim P.M., Sprecher E., Trifonov V., Gerstein M. The importance of bottlenecks in protein networks: Correlation with gene essentiality and expression dynamics. PLoS Comput. Biol. 2007;3:e59. doi: 10.1371/journal.pcbi.0030059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Girvan M., Newman M.E. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA. 2002;99:7821–7826. doi: 10.1073/pnas.122653799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yip K.Y., Yu H., Kim P.M., Schultz M., Gerstein M. The tYNA platform for comparative interactomics: A web tool for managing, comparing and mining multiple networks. Bioinformatics. 2006;22:2968–2970. doi: 10.1093/bioinformatics/btl488. [DOI] [PubMed] [Google Scholar]
- 30.Bastian, M., Heymann, S., Jacomy, M. (2009). Gephi: An open source software for exploring and manipulating networks. In International AAAI Conference on Weblogs and Social Media. (San Jose, CA: AAAI Publications) (http://gephi.org/publications/gephi-bastian-feb09.pdf).
- 31.Blondel V.D., Guillaume J., Lambiotte R., Lefebvre E. Fast unfolding of communities in large networks. J. Stat. Mech. 2008;2008:P10008. [Google Scholar]
- 32.Oti M., Snel B., Huynen M.A., Brunner H.G. Predicting disease genes using protein-protein interactions. J. Med. Genet. 2006;43:691–698. doi: 10.1136/jmg.2006.041376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hakes L., Pinney J.W., Robertson D.L., Lovell S.C. Protein-protein interaction networks and biology—what's the connection? Nat. Biotechnol. 2008;26:69–72. doi: 10.1038/nbt0108-69. [DOI] [PubMed] [Google Scholar]
- 34.Field, M.J., and Boat, T.F. (2010). Rare diseases and orphan products: Accelerating research and development. In Rare Diseases and Orphan Products: Accelerating Research and Development, M.J. Field and T.F. Boat, eds. Committee on Accelerating Rare Diseases Research and Orphan Product Development, Institute of Medicine (Washington DC: The National Academies Press). [PubMed]
- 35.Bowne S.J., Liu Q., Sullivan L.S., Zhu J., Spellicy C.J., Rickman C.B., Pierce E.A., Daiger S.P. Why do mutations in the ubiquitously expressed housekeeping gene IMPDH1 cause retina-specific photoreceptor degeneration? Invest. Ophthalmol. Vis. Sci. 2006;47:3754–3765. doi: 10.1167/iovs.06-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barabási A.L., Oltvai Z.N. Network biology: Understanding the cell's functional organization. Nat. Rev. Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 37.Suthram S., Dudley J.T., Chiang A.P., Chen R., Hastie T.J., Butte A.J. Network-based elucidation of human disease similarities reveals common functional modules enriched for pluripotent drug targets. PLoS Comput. Biol. 2010;6:e1000662. doi: 10.1371/journal.pcbi.1000662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Y., Agarwal P. A pathway-based view of human diseases and disease relationships. PLoS ONE. 2009;4:e4346. doi: 10.1371/journal.pone.0004346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hristovski D., Peterlin B., Mitchell J.A., Humphrey S.M. Using literature-based discovery to identify disease candidate genes. Int. J. Med. Inform. 2005;74:289–298. doi: 10.1016/j.ijmedinf.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 40.Lee I., Lehner B., Vavouri T., Shin J., Fraser A.G., Marcotte E.M. Predicting genetic modifier loci using functional gene networks. Genome Res. 2010;20:1143–1153. doi: 10.1101/gr.102749.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pujana M.A., Han J.D., Starita L.M., Stevens K.N., Tewari M., Ahn J.S., Rennert G., Moreno V., Kirchhoff T., Gold B. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat. Genet. 2007;39:1338–1349. doi: 10.1038/ng.2007.2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This table shows the orphan disease gene associations extracted from Uniprot. The degree corresponds to the total number of known genes associated with an orphan disease. The networks were generated using the 1772 curated orphan disease terms and 2124 orphan disease-causing mutant genes (ODMGs). The table also includes the gene IDs and gene descriptions of the ODMGs.
These interactions form 145 connected clusters (of size 2 and larger) of genes and are associated with the same disorder (see also Figure 4 in the main text).
The first four worksheets have the details of the enriched (p = 0.05; Bonferroni correction) GO biological process (BP), cellular component (CC), mammalian phenotype (MP), and pathways for 196 orphan diseases (ODs; ODs with four or more causal genes) respectively. The next four worksheets show orphan disease (OD) connectivity based on shared GO-BP, GO-CC, MP, and pathways, respectively. The last worksheet has the results of the comparisons between gene-based and functional feature-based ODNs (node and edge overlaps) and also the results from randomization.
This table has six worksheets that include OD-OD networks based on literature, gene, or both; literature-based ODN versus gene-based ODN and comparison of the top 100 ranked nodes based on eigenvector centrality (EC) measure; literature-based ODN versus gene-based ODN and comparison of the top 100 ranked nodes based on betweenness centrality (BC) measure; literature-based ODN versus gene-based ODN and comparison of top 100 ranked nodes based on closeness centrality (CC) measure; literature-based ODN amd subnetworks and communities: This network of orphan disease is based exclusively on literature connectivity; comparison of node/edge overlaps of gene-based and literature-based ODNs and the effect of incompleteness in data.