

Abstract
X-linked congenital generalized hypertrichosis (CGH), an extremely rare condition characterized by universal overgrowth of terminal hair, was first mapped to chromosome Xq24-q27.1 in a Mexican family. However, the underlying genetic defect remains unknown. We ascertained a large Chinese family with an X-linked congenital hypertrichosis syndrome combining CGH, scoliosis, and spina bifida and mapped the disease locus to a 5.6 Mb critical region within the interval defined by the previously reported Mexican family. Through the combination of a high-resolution copy-number variation (CNV) scan and targeted genomic sequencing, we identified an interchromosomal insertion at Xq27.1 of a 125,577 bp intragenic fragment of COL23A1 on 5q35.3, with one X breakpoint within and the other very close to a human-specific short palindromic sequence located 82 kb downstream of SOX3. In the Mexican family, we found an interchromosomal insertion at the same Xq27.1 site of a 300,036 bp genomic fragment on 4q31.2, encompassing PRMT10 and TMEM184C and involving parts of ARHGAP10 and EDNRA. Notably, both of the two X breakpoints were within the short palindrome. The two palindrome-mediated insertions fully segregate with the CGH phenotype in each of the families, and the CNV gains of the respective autosomal genomic segments are not present in the public database and were not found in 1274 control individuals. Analysis of control individuals revealed deletions ranging from 173 bp to 9104 bp at the site of the insertions with no phenotypic consequence. Taken together, our results strongly support the pathogenicity of the identified insertions and establish X-linked congenital hypertrichosis syndrome as a genomic disorder.
Main Text
Congenital generalized hypertrichosis (CGH) is a genetically and phenotypically heterogeneous group of rare conditions characterized by universal hair overgrowth. It is the major phenotypic feature of many distinct genetic syndromes and can be inherited as an autosomal or X-linked dominant trait.1–5 The CGH phenotype has generated much scientific interest and media attention, largely because of the striking hairy phenotype and its apparently atavistic nature.6,7 To date, genetic defects have been found in two forms of autosomal-dominant CGH. Autosomal-dominant congenital generalized hypertrichosis terminalis with or without gingival hyperplasia (MIM 135400) is associated with copy-number variations (CNVs), either microdeletions or microduplication, on chromosome 17q24 in both familial and sporadic cases.5 Rearrangements of chromosome 8 and a possible position effect have been detected in hypertrichosis universalis congenita, Ambras type (MIM 145701).8 Figuera et al. mapped the first CGH locus to chromosome Xq24-q27.1 in 1995 in a large Mexican family segregating X-linked hypertrichosis (MIM 307150).3 Subsequently, the genetic mapping was confirmed in a Mexican kindred with an X-linked congenital hypertrichosis syndrome consisting of CGH, deafness, and dental anomalies.4 However, the underlying mutation remains unidentified.
Genomic disorders are a growing class of human disorders that are caused by a genomic rearrangement that might lead to the complete loss or gain of a gene(s) sensitive to a dosage effect or, alternatively, might disrupt the structural integrity of a gene.9 Microdeletions and microduplications are most frequently associated with human genomic disorders.9–11 Another type of genomic rearrangement, seen less frequently, is an interchromosomal insertion where there is an intercalation of a part of one chromosome into another, nonhomologous chromosome and is also referred to as an interchromosomal insertional translocation.12
Recent application of massively parallel sequencing (MPS) and high-resolution genome-wide CNV analysis has rapidly unraveled the genetic basis of many rare Mendelian and genomic disorders.10–14 Here, we add X-linked congenital hypertrichosis syndrome, among the rarest of rare conditions, to the list of these disorders by reporting on the identification of independent interchromosomal insertions involving different autosomal segments mediated by the same small palindrome at Xq27.1 in each of two X-linked CGH families of different ethnic backgrounds.
We first ascertained a five-generation Chinese family with a distinct syndrome of CGH, scoliosis, and spina bifida (Figures 1A and 1B). The family had 11 affected individuals (Figure 1A). All of the four affected males available for phenotypic evaluation had severe hypertrichosis associated with scoliosis, whereas all affected females had only mild hypertrichosis, which was consistent with X-linked inheritance (Figures 1B–1D). The proband, a 41-year-old male, also had spina bifida presenting as cervical and sacral meningoceles (Figure 1C). The other ten affected individuals did not exhibit spina bifida. We collected blood samples from 19 participating family members (eight affected, seven unaffected, and four spouses) after obtaining informed consent from participants and approval from the Peking Union Medical College institutional review board. Chorionic villus sampling was performed for participant V1 (Figure 1A). Genomic DNA was extracted via standard methods. To determine whether the X-linked congenital hypertrichosis syndrome mapped to the same locus as reported previously in the Mexican CGH family,3 we determined genotypes in 17 family members at 14 polymorphic microsatellite marker loci from the Xq26.3-q27.2 region (Table S1 available online), 11 of which were designed with the use of the UCSC Human Genome Browser. Two-point linkage analysis and LOD score calculation were carried out with the MLINK program of the LINKAGE package software (version 5.2). The parameters used in linkage analysis were X-linked dominant inheritance, complete penetrance, a mutation rate of zero, equal microsatellite-allele frequency, and a disease-allele frequency of 1 in 10,000. Our two-point linkage analysis produced a maximum LOD score of 3.91 at θ = 0 for five markers (Table S2), confirming genetic linkage to the same locus in the Chinese family. On the basis of the haplotypes, we observed two recombination events, one in III5 and the other in IV2 (Figure S1). Further haplotype analysis in these two recombinants defined the critical region to an interval between ZLS3 and ZLS10, representing a genomic interval of 5.6 Mb containing 40 RefSeq genes (Figure 1E and Figure S1). We performed PCR amplification and sequencing of all exons and their flanking intronic sequences for these genes in the proband (Figure 1E; primer information is available upon request) but did not identify any pathogenic mutation.
Figure 1.
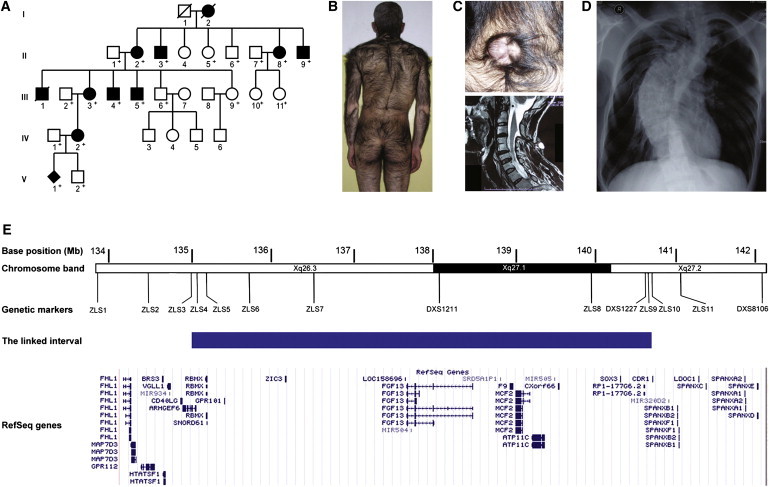
Phenotypes and Genetic Locus of a Distinct X-Linked Congenital Hypertrichosis Syndrome
(A) Pedigree of the five-generation Chinese family with eleven affected individuals. For V1, the diagnosis was made from a chorionic villus sample. Individuals whose DNA was available are indicated by “+.”
(B) Photograph of the proband showing severe CGH.
(C) Photograph and MRI image showing a cervical meningocele in the proband.
(D) X-ray image of the proband showing scoliosis.
(E) Schematic diagram of Xq26.3-q27.2 showing the RefSeq genes in the critical region for the X-linked congenital hypertrichosis syndrome. A solid blue bar represents the critical region. The positions of all genetic markers used in linkage analysis are shown.
To determine whether the X-linked congenital hypertrichosis syndrome was caused by an unknown microdeletion or microduplication, we performed a genome-wide high-resolution CNV scan in four affected individuals (two males and two females), using the Affymetrix Genome-Wide Human SNP Array 6.0 containing over 906,600 SNPs and over 946,000 copy-number probes. Genomic DNA samples from four affected individuals (two males, II9 and III5; and two females, III3 and IV2) were genotyped at the CapitalBio Corporation (Beijing, China) with the SNP Array 6.0 in accordance with the manufacturer's protocols. Genotype calling, genotyping quality control, and CNV identification were performed with the Affymetrix Genotyping Console 3.0 software. Copy-number-state calls were determined with the Canary algorithm embedded in the Affymetrix Genotyping Console 3.0 package. We did not detect potential pathogenic CNVs in the critical region but found a >121 kb microduplication of the COL23A1 locus on 5q35.3 (CN_143030 to CN_1143071) in all four individuals (Figure 2A).
Figure 2.
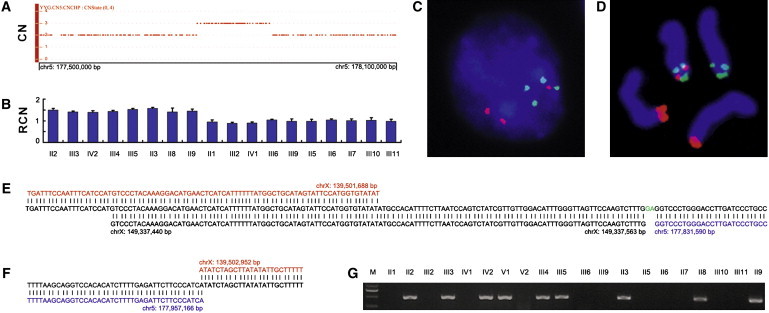
Identification of an Inherited Interchromosomal Insertion at Xq27.1 in the Chinese Family with a Distinct X-Linked Congenital Hypertrichosis Syndrome
(A) Copy-number state of a 600 kb genomic region on chromosome 5q35.3 showing the presence of a microduplication in the proband. CN, copy number.
(B) Validation of the microduplication and its segregation with the disease phenotype by qPCR. RCN, relative copy number. Error bars represent SD.
(C and D) Two-color FISH signals on a representative interphase nucleus (C) and typical metaphase chromosomes (D) demonstrating the insertion event. BAC probes are CTD-2507I18 (red), RP11-55E17 (green), and RP11-671F22 (green). See Figure 4A for the positions of BAC clones.
(E and F) Sequence analysis of the proximal (E) and distal (F) insertion junctions. Reference sequences on Xq27.1 and 5q35.3 are indicated in red and blue, respectively. The proximal junction contains a microinsertion from the X chromosome (black) and a 2 bp microinsertion (green) of unknown origin.
(G) PCR amplification of the distal insertion junction showing segregation of the insertion with the phenotype.
All genomic positions correspond to the February 2009 human reference sequence (GRCh37).
To confirm the genomic duplication of the 5q35.3 region, we designed primers for a real-time quantitative PCR (qPCR) assay by using the Primer Express v2.0 software (Applied Biosystems). We performed qPCR as previously described.15 The relative copy number (RCN) of the target sequences was determined with the comparative ΔΔCT method. A ∼1.5-fold RCN was used for duplication. The qPCR experiments were repeated three times. Primers used for the qPCR assays are given in Table S1. Our qPCR assay confirmed the microduplication and also showed full segregation of the microduplication with the disease phenotype in the family (Figure 2B), suggesting a possible interchromosomal insertion event within the critical region.
We performed two-color interphase and metaphase fluorescence in situ hybridization (FISH) to confirm the insertion in the Chinese family. The bacterial artificial chromosome (BAC) clones CTD-2507I18, RP11-55E17, and RP11-671F22 were selected to cover the duplicated region of COL23A1 (MIM 610043) on 5q35.3, FHL1 (MIM 300163) on Xq26.3, and F8 (MIM 300841) on Xq28, respectively (Figure 4A). The RP11-55E17 and RP11-671F22 BAC DNA samples were individually labeled with SpectrumGreen-dUTP (Abbott Molecular), and the CTD-2507I18 DNA was labeled with Cy3-dUTP (GE Healthcare Life Sciences). The interphase nuclei and metaphase chromosomes were cohybridized with a pair of SpectrumGreen-dUTP-labeled reference probes (green signal) and Cy3-dUTP-labeled test probe (red signal), counterstained with DAPI, and visualized by fluorescence microscopy. The two-color FISH signal patterns observed on both interphase nuclei (Figure 2C) and metaphase chromosomes (Figure 2D and Figure S2) were consistent with an interchromosomal insertion event, as indicated by the presence of red signals for COL23A1 on chromosome 5 homologs and on the X chromosome between the green signals corresponding to FHL1 on X26.3 and F8 on Xq28 (Figure 2D and Figure S2).
Figure 4.
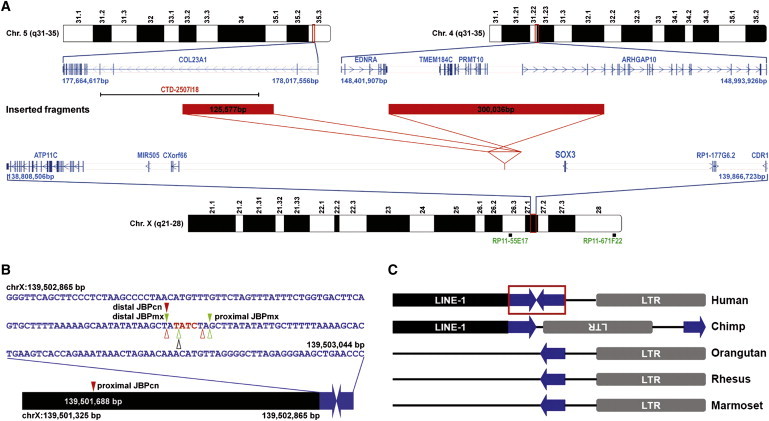
Interchromosomal Insertions Mediated by the Same Human-Specific Palindrome
(A) Schematic diagram depicting the two independent insertions found in the present study. Red solid bars represent the inserted fragments, and indicated sizes correspond to base pairs (bp). Red lines display the orientation of insertions. Positions of the BAC probes used in two-color FISH and of the RefSeq genes on the corresponding chromosomal regions are shown.
(B) Schematic diagram of the 180 bp human-specific palindrome with a summary of the breakpoints identified in the present study. Solid triangles indicate insertion breakpoints in the two study families (Chinese in red and Mexican in green). JBPcn, junction breakpoint in the Chinese family; JBPmx, junction breakpoint in the Mexican family. Open triangles represent the deletion breakpoints in normal individuals (Chinese in red, Mexican in green, and Yoruban in black). A black solid bar represents the LINE-1 element that contains the proximal JBPcn. The precise position of the JBPcn is given.
(C) Schematic diagram showing the small human-specific palindrome at Xq27.1 and its flanking sequences. The 180 bp palindromic sequence is boxed. A chimp has two halves of the palindrome but in direct orientation. All other three nonhuman primates only have one half of the palindrome.
All genomic positions correspond to the February 2009 human reference sequence (GRCh37).
In parallel with the above-described CNV and FISH analyses, we conducted targeted capture and MPS in the proband by using the Roche NimbleGen SeqCap and 454 Sequencing technologies. Two custom Sequence Capture 385K human arrays were first designed and manufactured at Roche NimbleGen, each having 385,000 unique SeqCap probes, as determined by the SSAHA algorithm, covering 88.1% of the targeted critical region between ZLS3 and ZLS10 (chrX: 135,204,482–140,853,096; GRCh37, hg19) (Figure 1E). The captured DNA was sequenced on a Genome Sequencer FLX System with long-read GS FLX Titanium series chemistry at Roche 454 Life Sciences. The resulting sequence reads were mapped to the hg18 human reference with the GS Reference Mapper software and amounted to 555 Mb of sequence data with about 98% mapped back to the targeted region. Further analysis with the 454 GS Reference Mapper software revealed the distal insertion junction sequences (Figure S3), pinpointing the breakpoint within the X chromosome (chrX: 139,502,951) to the center of a 180 bp short palindromic sequence at Xq27.1, which is located 82 kb downstream of SOX3 (MIM 313430) (Figure S2, Figures 4A and 4B). We used long-range PCR to amplify the insertion junctions. Primers were designed from the sequences flanking the palindrome at Xq27.1 and the breakpoints on the basis of the SNP Array 6.0 genomic coordinates. Sequence analysis of the resultant amplicons by Sanger sequencing verified the distal junction found in the targeted genomic sequencing (Figure 2F), placed the other X chromosome breakpoint forming the proximal insertion junction in the middle of the LINE-1 element beside the small palindrome (Figure 2E and Figure 4B), and indicated that the mutant X chromosome had a direct insertion of a 125,577 bp fragment from within COL23A1 (Figures 2E and 2F, Figure 4A); i.e., der(X)dir ins(X;5)(q27.1;q35.3). This insertion was shown to occur concomitantly with a deletion of 1263 bp between the two X chromosome breakpoints at Xq27.1 (Figures 2E and 2F). Consistent with the qPCR assay, our junction PCR assay in the family showed specific fragments of the expected sizes in all affected individuals, but in none of the unaffected family members (Figure 2G).
The discovery of the insertion mediated by a short palindromic sequence in the Chinese family prompted us to examine the originally reported X-linked Mexican CGH family (Figure 3A). Use of the SNP Array 6.0 for the examination of four family members (two affected and two unaffected) for CNVs revealed a microduplication of a >278 kb fragment on 4q31 (SNP_A-2053483 to SNP_A-1883747), encompassing PRMT10 and TMEM184C and involving parts of ARHGAP10 (MIM 609746) and EDNRA (MIM 131243), in affected members (Figure 3B and Figure 4A). We validated this microduplication by a qPCR assay (Figure 3C) and obtained breakpoint information by using a similar junction PCR approach (Figures 3D and 3E). Our results suggested that the affected members in the Mexican family had inherited a mutant X chromosome with an inverted insertion of a 300,036 bp fragment from the 4q31.22-q31.23 region (Figures 3D and 3E, Figure 4A); i.e., der(X)inv ins(X;4)(q27.1;q31.23q31.22). Examination of all available family members for the insertion with the use of a junction PCR test confirmed segregation of the insertional event with the CGH phenotype (Figure 3F). Very interestingly, both of the two X breakpoints identified in the Mexican family were at the center of the palindrome (chrX: 139,502,951 and 139,502,958) (Figures 3D and 3E, Figure 4B), thus reinforcing the role of this small palindrome as a mediator in the initiation of the two insertions identified in this study.
Figure 3.
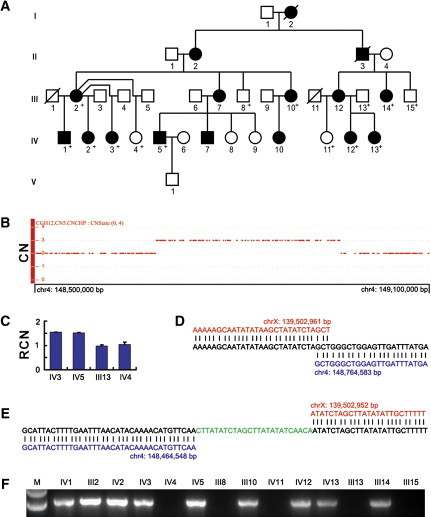
Identification of an Inherited Interchromosomal Insertion at Xq27.1 in the Mexican Family with X-Linked CGH
(A) Pedigree of the five-generation Mexican family with X-linked CGH. Individuals whose DNA was available are indicated by “+.”
(B) Copy-number state of a 600 kb genomic region on chromosome 4q31 showing the presence of a microduplication in an affected individual. CN, copy number.
(C) Validation of the microduplication by qPCR. RCN, relative copy number. Error bars represent SD.
(D and E) Sequence analysis of the proximal (D) and distal (E) insertion junctions. Reference sequences on Xq27.1 and 4q31 are indicated in red and blue, respectively. The distal junction contains a 25 bp microinsertion.
(F) PCR amplification of the proximal insertion junction showing segregation of the insertion with the phenotype.
All genomic positions correspond to the February 2009 human reference sequence (GRCh37).
We have identified independent insertions mediated by the same small palindrome at Xq27.1 in two different families affected with X-linked hypertrichosis. Furthermore, complete segregation of these insertions with the phenotypes has provided additional supportive evidence toward their causal role. To determine whether these insertions were unique to these families and to rule out the possibility of their representing normal genomic variation in the population, we screened 740 male control individuals (215 Chinese, 118 Mexican, and 407 Asian Indian) by PCR using primers derived from the sequences flanking the palindrome (Table S1), and we found no detectable insertion. Furthermore, CNVs involving the two identified inserted segments are not reported in the public Database of Genomic Variants and are not present in 1274 Chinese control individuals (1074 from Shanghai and 200 from Hong Kong). Taken together, our results suggest that the palindrome-mediated insertions are the underlying cause for isolated and syndromic X-linked CGH.
Palindromic sequences are not stable and can induce genomic rearrangements, including deletions and recurrent translocations.16–18 The 180 bp palindromic sequence is present only in humans. It is flanked by a LINE-1 repeat and an LTR sequence (Figure 4C). Examination of the orthologous region in the chimp genome reveals the two halves of the palindrome, but they are in direct orientation and are separated by the LTR sequence. Other nonhuman primates, including the orangutan, rhesus, and marmoset, have only one half of the palindrome (Figure 4C). These results suggest that the palindrome is evolutionally very young. The aforementioned PCR analysis in control individuals did, however, detect deletions ranging in size from 173 bp to 9104 bp in nine individuals (two Chinese, two Mexican, and five Asian Indian) (Table S3). In addition, a deletion of 209 bp was evident in a Yoruban individual subjected to whole-genome sequencing.19 Noticeably, all of these ten deletions had one breakpoint at the center of the palindrome (Table S3), thereby deleting one half of the palindrome. Thus, it appears that the human-specific palindromic sequence at Xq27.1 is prone to breakage and, hence, represents a hotspot for genomic rearrangements, including insertion.
An interchromosomal insertion event requires at least three breakage events and therefore is not only rarer but also more complex in comparison to the common types of genomic rearrangements (microdeletion, microduplication, and terminal translocation). Earlier studies of microscopically visible interchromosomal insertions estimated the incidence to be 1:80,000 live births.20 However, recently, high-resolution array-based comparative genomic hybridization (aCGH) analysis and confirmatory FISH of 18,000 clinical samples identified 40 interchromosomal insertions (1:500), thus indicating that they may not be as rare as previously thought.12 Interchromosomal insertions can produce a disease phenotype by altering the activity of a gene. If a gene(s) within the insertion is dosage-sensitive, its expression may be increased. The insertion event may disrupt a gene and cause either a loss or a gain of function. Aberrant expression of a gene may result from insertion of a novel sequence within or near the gene because of either a position effect or the introduction of novel regulatory sequences. In the spontaneous Dancer (Dc) mouse mutant, an interchromosomal insertion in the first intron of Tbx10 of a genomic fragment containing the p23 promoter can cause ectopic Tbx10 expression, thereby producing cleft lip and plate in homozygotes.21 Through a combination of a high-resolution microarray-based CNV scan and targeted genomic sequencing, we were able to find independent pathogenic insertions in X-linked CGH. The two insertion events were shown to be mediated by the same small palindrome that is situated in a 485 kb gene-desert region flanked telomerically by SOX3 encoding the SRY (sex determining region Y)-box 3 transcription factor (Figure 4A). SOX3 is the most intriguing candidate gene because its 3′ end lies 82 kb telomeric to the palindrome and the SOX family of transcription factors is among the most important groups of developmental regulators. We postulate that the palindrome-mediated insertions identified in our present study might have introduced tissue-specific regulatory elements and hence induced ectopic expression of SOX3 in hair follicles (HFs) or precursor cells, which might cause aberrant patterning of hair and result in the abnormal phenotype.
Mutations in SOX3 have been associated with X-linked mental retardation with isolated growth hormone deficiency (MIM 300123),22 and also with X-linked hypopituitarism (MIM 312000).23 Microduplications encompassing SOX3 have been found in X-linked hypopituitarism.23 In X-linked recessive hypoparathyroidism (MIM 307700), an insertion of an approximately 340 kb intragenic fragment of SNTG2 (MIM 608715) on 2p25.3 into an Xq27.1 site 67 kb downstream of SOX3 has been identified.24 This insertion event accompanies a 23–25 kb deletion that includes the human-specific palindrome. A position effect on SOX3 expression has been suggested as the underlying pathogenic mechanism.24 Recently, Sutton et al. have reported genomic rearrangements, including microduplications and a deletion in the SOX3 region, in three patients with XX male sex reversal (MIM 300833) with breakpoints in the SOX3 regulatory region.25 Although the precise breakpoints of these rearrangements are not available from their study, it does appear that the genomic region involved in the insertion events reported in our study is duplicated in two of three patients reported. The above-described patients with X-linked hypopituitarism, X-linked recessive hypoparathyroidism, or XX male sex reversal do not have the hypertrichosis phenotype.23–25 Additionally, females with Xq26-q28 deletions, which can result in a loss of SOX3, develop premature ovary failure 1 (MIM 311360) but not hypertrichosis.26–29 Together, these provide support for our hypothesis that X-linked CGH with or without scoliosis and spina bifida results from the insertion events introducing novel DNA regulatory elements within each of the inserted sequences, rather than from the creation of a “position effect” by physical separation of the gene from its intrinsic regulatory elements.
SOX3 is closely related to SOX2 (MIM 184429), the gene encoding a key regulator for stem cells, but to date, it is not known to be expressed in HFs. It has been shown that the Sox2-expressing dermal papilla cells specify mouse HF types and induce HF morphogenesis.30,31 With use of RT-PCR (Table S1), we could not detect SOX3 mRNA expression in HFs isolated from scalp skin tissue donated by healthy individuals after cosmetic surgery or from a skin specimen taken from the upper arm of the proband of the Chinese family (Figure S4). Thus, it seems reasonable to speculate that the palindrome-mediated insertions might have induced ectopic SOX3 expression at an early stage of HF development. The inserted fragment in the Chinese family might contain additional regulatory elements and, thus, lead to additional malformations, including scoliosis. Coincidentally, CNVs on 17q24 near SOX9 (MIM 608160), the gene encoding an essential regulator of HF stem cells,32,33 cause autosomal-dominant CGH.5 Further studies are required to determine whether aberrant expression of SOX3 or SOX9 alters the patterning of hair as implicated by these studies.
Acknowledgments
We thank the family members for their participation in the study. This work was supported mainly by the National Natural Science Foundation of China (fund 30730097 to X.Z. and T.J.) and the State Key Laboratory Program of the Ministry of Science and Technology of China (to X.Z.). X.Z. is a Chang Jiang Scholar of Genetic Medicine supported by the Ministry of Education of China. X.Z. is also supported by the 111 Project (B08007).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Structural Variation (dbVar), http://www.ncbi.nlm.nih.gov/dbvar
Database of Genomic Variants, http://projects.tcag.ca/variation
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
Accession Numbers
The dbVar accession number for the study reported in this paper is nstd55.
References
- 1.Macías-Flores M.A., García-Cruz D., Rivera H., Escobar-Luján M., Melendrez-Vega A., Rivas-Campos D., Rodríguez-Collazo F., Moreno-Arellano I., Cantú J.M. A new form of hypertrichosis inherited as an X-linked dominant trait. Hum. Genet. 1984;66:66–70. doi: 10.1007/BF00275189. [DOI] [PubMed] [Google Scholar]
- 2.Garcia-Cruz D., Figuera L.E., Cantu J.M. Inherited hypertrichoses. Clin. Genet. 2002;61:321–329. doi: 10.1034/j.1399-0004.2002.610501.x. [DOI] [PubMed] [Google Scholar]
- 3.Figuera L.E., Pandolfo M., Dunne P.W., Cantú J.M., Patel P.I. Mapping of the congenital generalized hypertrichosis locus to chromosome Xq24-q27.1. Nat. Genet. 1995;10:202–207. doi: 10.1038/ng0695-202. [DOI] [PubMed] [Google Scholar]
- 4.Tadin-Strapps M., Salas-Alanis J.C., Moreno L., Warburton D., Martinez-Mir A., Christiano A.M. Congenital universal hypertrichosis with deafness and dental anomalies inherited as an X-linked trait. Clin. Genet. 2003;63:418–422. doi: 10.1034/j.1399-0004.2003.00069.x. [DOI] [PubMed] [Google Scholar]
- 5.Sun M., Li N., Dong W., Chen Z., Liu Q., Xu Y., He G., Shi Y., Li X., Hao J. Copy-number mutations on chromosome 17q24.2-q24.3 in congenital generalized hypertrichosis terminalis with or without gingival hyperplasia. Am. J. Hum. Genet. 2009;84:807–813. doi: 10.1016/j.ajhg.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hall B.K. Atavisms and atavistic mutations. Nat. Genet. 1995;10:126–127. doi: 10.1038/ng0695-126. [DOI] [PubMed] [Google Scholar]
- 7.Davies K. Nature, genetics and the Niven factor. Nat. Genet. 2007;39:805–806. doi: 10.1038/ng0707-805. [DOI] [PubMed] [Google Scholar]
- 8.Fantauzzo K.A., Tadin-Strapps M., You Y., Mentzer S.E., Baumeister F.A., Cianfarani S., Van Maldergem L., Warburton D., Sundberg J.P., Christiano A.M. A position effect on TRPS1 is associated with Ambras syndrome in humans and the Koala phenotype in mice. Hum. Mol. Genet. 2008;17:3539–3551. doi: 10.1093/hmg/ddn247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lupski J.R., Stankiewicz P. Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 2005;1:e49. doi: 10.1371/journal.pgen.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stankiewicz P., Lupski J.R. Structural variation in the human genome and its role in disease. Annu. Rev. Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- 11.Mefford H.C., Eichler E.E. Duplication hotspots, rare genomic disorders, and common disease. Curr. Opin. Genet. Dev. 2009;19:196–204. doi: 10.1016/j.gde.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang S.H., Shaw C., Ou Z., Eng P.A., Cooper M.L., Pursley A.N., Sahoo T., Bacino C.A., Chinault A.C., Stankiewicz P. Insertional translocation detected using FISH confirmation of array-comparative genomic hybridization (aCGH) results. Am. J. Med. Genet. A. 2010;152A:1111–1126. doi: 10.1002/ajmg.a.33278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ng S.B., Nickerson D.A., Bamshad M.J., Shendure J. Massively parallel sequencing and rare disease. Hum. Mol. Genet. 2010;19(R2):R119–R124. doi: 10.1093/hmg/ddq390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maxmen A. Exome sequencing deciphers rare diseases. Cell. 2011;144:635–637. doi: 10.1016/j.cell.2011.02.033. [DOI] [PubMed] [Google Scholar]
- 15.Sun M., Ma F., Zeng X., Liu Q., Zhao X.L., Wu F.X., Wu G.P., Zhang Z.F., Gu B., Zhao Y.F. Triphalangeal thumb-polysyndactyly syndrome and syndactyly type IV are caused by genomic duplications involving the long range, limb-specific SHH enhancer. J. Med. Genet. 2008;45:589–595. doi: 10.1136/jmg.2008.057646. [DOI] [PubMed] [Google Scholar]
- 16.Kurahashi H., Inagaki H., Ohye T., Kogo H., Kato T., Emanuel B.S. Palindrome-mediated chromosomal translocations in humans. DNA Repair (Amst.) 2006;5:1136–1145. doi: 10.1016/j.dnarep.2006.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheridan M.B., Kato T., Haldeman-Englert C., Jalali G.R., Milunsky J.M., Zou Y., Klaes R., Gimelli G., Gimelli S., Gemmill R.M. A palindrome-mediated recurrent translocation with 3:1 meiotic nondisjunction: the t(8;22)(q24.13;q11.21) Am. J. Hum. Genet. 2010;87:209–218. doi: 10.1016/j.ajhg.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henthorn P.S., Mager D.L., Huisman T.H., Smithies O. A gene deletion ending within a complex array of repeated sequences 3′ to the human beta-globin gene cluster. Proc. Natl. Acad. Sci. USA. 1986;83:5194–5198. doi: 10.1073/pnas.83.14.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bentley D.R., Balasubramanian S., Swerdlow H.P., Smith G.P., Milton J., Brown C.G., Hall K.P., Evers D.J., Barnes C.L., Bignell H.R. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Hemel J.O., Eussen H.J. Interchromosomal insertions. Identification of five cases and a review. Hum. Genet. 2000;107:415–432. doi: 10.1007/s004390000398. [DOI] [PubMed] [Google Scholar]
- 21.Bush J.O., Lan Y., Jiang R. The cleft lip and palate defects in Dancer mutant mice result from gain of function of the Tbx10 gene. Proc. Natl. Acad. Sci. USA. 2004;101:7022–7027. doi: 10.1073/pnas.0401025101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laumonnier F., Ronce N., Hamel B.C., Thomas P., Lespinasse J., Raynaud M., Paringaux C., Van Bokhoven H., Kalscheuer V., Fryns J.P. Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am. J. Hum. Genet. 2002;71:1450–1455. doi: 10.1086/344661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woods K.S., Cundall M., Turton J., Rizotti K., Mehta A., Palmer R., Wong J., Chong W.K., Al-Zyoud M., El-Ali M. Over- and underdosage of SOX3 is associated with infundibular hypoplasia and hypopituitarism. Am. J. Hum. Genet. 2005;76:833–849. doi: 10.1086/430134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowl M.R., Nesbit M.A., Harding B., Levy E., Jefferson A., Volpi E., Rizzoti K., Lovell-Badge R., Schlessinger D., Whyte M.P., Thakker R.V. An interstitial deletion-insertion involving chromosomes 2p25.3 and Xq27.1, near SOX3, causes X-linked recessive hypoparathyroidism. J. Clin. Invest. 2005;115:2822–2831. doi: 10.1172/JCI24156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sutton E., Hughes J., White S., Sekido R., Tan J., Arboleda V., Rogers N., Knower K., Rowley L., Eyre H. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J. Clin. Invest. 2011;121:328–341. doi: 10.1172/JCI42580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tharapel A.T., Anderson K.P., Simpson J.L., Martens P.R., Wilroy R.S., Jr., Llerena J.C., Jr., Schwartz C.E. Deletion (X)(q26.1—>q28) in a proband and her mother: molecular characterization and phenotypic-karyotypic deductions. Am. J. Hum. Genet. 1993;52:463–471. [PMC free article] [PubMed] [Google Scholar]
- 27.Fimiani G., Laperuta C., Falco G., Ventruto V., D'Urso M., Ursini M.V., Miano M.G. Heterozygosity mapping by quantitative fluorescent PCR reveals an interstitial deletion in Xq26.2-q28 associated with ovarian dysfunction. Hum. Reprod. 2006;21:529–535. doi: 10.1093/humrep/dei356. [DOI] [PubMed] [Google Scholar]
- 28.Rizzolio F., Bione S., Sala C., Goegan M., Gentile M., Gregato G., Rossi E., Pramparo T., Zuffardi O., Toniolo D. Chromosomal rearrangements in Xq and premature ovarian failure: mapping of 25 new cases and review of the literature. Hum. Reprod. 2006;21:1477–1483. doi: 10.1093/humrep/dei495. [DOI] [PubMed] [Google Scholar]
- 29.Ferreira S.I., Matoso E., Pinto M., Almeida J., Liehr T., Melo J.B., Carreira I.M. X-chromosome terminal deletion in a female with premature ovarian failure: Haploinsufficiency of X-linked genes as a possible explanation. Mol. Cytogenet. 2010;3:14. doi: 10.1186/1755-8166-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Driskell R.R., Giangreco A., Jensen K.B., Mulder K.W., Watt F.M. Sox2-positive dermal papilla cells specify hair follicle type in mammalian epidermis. Development. 2009;136:2815–2823. doi: 10.1242/dev.038620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Biernaskie J., Paris M., Morozova O., Fagan B.M., Marra M., Pevny L., Miller F.D. SKPs derive from hair follicle precursors and exhibit properties of adult dermal stem cells. Cell Stem Cell. 2009;5:610–623. doi: 10.1016/j.stem.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vidal V.P., Chaboissier M.C., Lützkendorf S., Cotsarelis G., Mill P., Hui C.C., Ortonne N., Ortonne J.P., Schedl A. Sox9 is essential for outer root sheath differentiation and the formation of the hair stem cell compartment. Curr. Biol. 2005;15:1340–1351. doi: 10.1016/j.cub.2005.06.064. [DOI] [PubMed] [Google Scholar]
- 33.Nowak J.A., Polak L., Pasolli H.A., Fuchs E. Hair follicle stem cells are specified and function in early skin morphogenesis. Cell Stem Cell. 2008;3:33–43. doi: 10.1016/j.stem.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.