

Abstract
Recent studies have revealed profound developmental consequences of mutations in genes encoding proteins of the lectin pathway of complement activation, a central component of the innate immune system. Apart from impairment of immunity against microorganisms, it is known that hereditary deficiencies of this system predispose one to autoimmune conditions. Polymorphisms in complement genes are linked to, for example, atypical hemolytic uremia and age-dependent macular degeneration. The complement system comprises three convergent pathways of activation: the classical, the alternative, and the lectin pathway. The recently discovered lectin pathway is less studied, but polymorphisms in the plasma pattern-recognition molecule mannan-binding lectin (MBL) are known to impact its level, and polymorphisms in the MBL-associated serine protease-2 (MASP-2) result in defects of complement activation. Recent studies have described roles outside complement and immunity of another MBL-associated serine protease, MASP-3, in the etiology of 3MC syndrome, an autosomal-recessive disorder involving a spectrum of developmental features, including characteristic facial dysmorphism. Syndrome-causing mutations were identified in MASP1, encoding MASP-3 and two additional proteins, MASP-1 and MAp44. Furthermore, an association was discovered between 3MC syndrome and mutations in COLEC11, encoding CL-K1, another molecule of the lectin pathway. The findings were confirmed in zebrafish, indicating that MASP-3 and CL-K1 underlie an evolutionarily conserved pathway of embryonic development. Along with the discovery of a role of C1q in pruning synapses in mice, these recent advances point toward a broader role of complement in development. Here, we compare the functional immunologic consequences of “conventional” complement deficiencies with these newly described developmental roles.
Main Text
Complement System and Components
As a prelude to discussing the polymorphisms and genetics of the complement system, we wish to set the stage with a brief introduction to the complement pathways and the proteins involved.
The complement system is the humoral backbone of the innate immune defense. It is thus involved in a number of diverse processes involving antimicrobial defense, clearance of immune complexes, and tissue regeneration. The system comprises more than 35 humoral and cell-associated proteins forming three converging enzymatic cascades: the classical pathway, the alternative pathway, and the lectin pathway.1 An overview is shown in Figure 1, where some of the proteins mentioned in the following are placed in their context.
Figure 1.
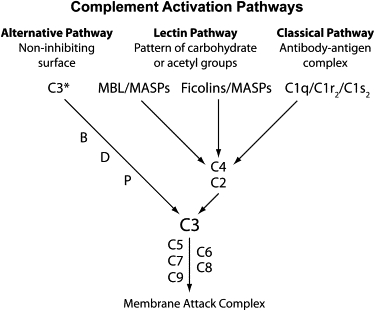
Overview of the Complement Pathways
The three activating pathways and the terminal pathway are shown. The acronyms of participating proteins are given. The lectin pathway is activated when MBL or ficolins in complex with MASPs recognize foreign patterns of carbohydrate or acetyl groups. The classical pathway is activated by C1q in complex with C1s and C1r upon binding of antibody-antigen complexes. The alternative pathway is constitutively activated and inhibited on self-surfaces, but is allowed to proceed on foreign surfaces. The three pathways converge on C3 and C5 and the common terminal pathway leading to formation of the membrane attack complex.
The classical pathway is especially important for control of infections with pyogenic encapsulated bacteria, such as Haemophilus influenzae and Streptococcus pneumoniae,2 although it has also been implicated in control of viral infections, such as influenza.3 The pathway is initiated when the recognition protein C1q binds to antibodies bound to microbes or in immune complexes.2,4 Preexisting “natural IgM” produced by naive B cells5 and IgM or IgG generated through an adaptive response (primary or secondary) will bind to, for example, determinants on the capsule of pathogenic bacteria, leading to agglutination. Binding of IgM exposes cryptic binding sites on IgM, allowing C1q to bind, and binding of arrays of IgG similarly allows high-avidity binding of C1q. C1q is in complex with two serine proteases, C1r and C1s. Upon binding of C1q, conformational changes lead to autoactivation of C1r, which in turn activates C1s. Activated C1s is capable of cleaving C4 and C2. A fragment of C4, C4b, is deposited on the activating surface, where it is covalently attached through a reactive thioester, while a small fragment, C4a, is released. The proenzyme C2 is likewise cleaved by C1s into two fragments, and C2a joins C4b on the surface. C2a is a serine protease, capable of cleaving C3. Cleaved C3 in the form of C3b is covalently bound to the surface-attached C4bC2a complex, while the concomitantly generated small fragment C3a is released to mediate inflammation. C2a in the newly formed complex, C4bC2aC3b, is now able to cleave C5, and two fragments are again generated: C5a is a potent inflammatory mediator, while C5b initiates the formation of a membrane attack complex (MAC), which is expanded through deposition of C6, C7, C8 and C9 (Figure 1), leading to the formation of a hole in the target cell membrane. More importantly, the deposited components C4b and C3b function as molecular tags (opsonins) interacting with complement receptors, hereby facilitating engulfment by phagocytes and activation of B cells.
The lectin pathway is quite similar to the classical pathway.6 An overview of the components of this pathway and the structure of some of the proteins is presented in Figure 2. The lectin pathway is also involved mainly in the control of bacterial infections, as has been observed in the case of children suffering from recurrent pyogenic infections.2,7 It would appear that the lectin pathway is especially important during the interval between the loss of passively acquired maternal antibody and the acquisition of a mature immunologic repertoire, as well as in individuals with immunosuppression. The pathway is initiated when one or more recognition molecules bind to patterns of carbohydrates or patterns of acetyl groups on the surface of, for example, bacteria or viruses. Four such recognition molecules are capable of activating the lectin pathway: mannan-binding lectin (MBL), H-ficolin, L-ficolin, and M-ficolin.8 These pattern-recognition molecules (PRMs) are found in complexes with three serine proteases (termed MBL-associated serine proteases [MASPs]), MASP-1, MASP-2, and MASP-3, as well as two nonenzymatic fragments hereof (termed MBL-associated proteins), MAp19 and MAp44 (Figure 2A). When one of the recognition molecules binds to an adequate pattern, the MASPs are activated. MASP-1 and MASP-2 are responsible for complement activation through cleavage of C2 and C4 (Figure 2B).9 The cascade then proceeds as described above for the classical pathway.
Figure 2.
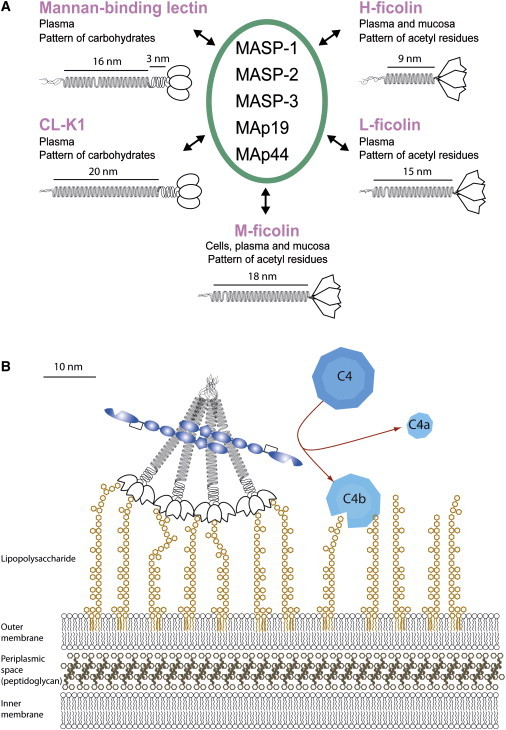
The Lectin Pathway
(A) The lectin pathway components. Overview of the five different recognition proteins of the lectin pathway, their tissue and humoral localization, and their ligand patterns. They all associate with MASPs and MAps, except for CL-K1, which has so far only been reported to associate with MASP-1/MASP-3 and is not yet known to activate complement. A schematic drawing of the structural subunit of each protein is shown, composed of three identical polypeptide chains forming a collagen-like helix with the recognition domains extending in the C-terminal end.
(B) Figure showing the binding of MBL in complex with MASP-2 to a molecular pattern on the surface of a Gram negative bacterium. MBL (and similarly the ficolins and CL-K1) are multimers (here a tetramer is shown) of the structural subunit drawn in part A of this figure. These molecules thus wield many recognition sites as indicated on the figure. MASP-2 is a homodimer and the domains are indicated with the disulphide bridge connecting the A- and B-chains after activating cleavage in the activation peptide. MASP-2 then activates C4 resulting in deposition of C4b on the surface, which initiates the tagging of the bacterium for phagocytosis. The complement cascade may then progress as indicated in Figure 1.
Recently, a fifth PRM, CL-K1,10 was reported to associate with MASPs.11 Although it reportedly binds to various pathogen-associated molecular patterns (PAMPs), it remains to be determined whether CL-K1 can activate complement.
Whereas MASP-1 and MASP-2 have been described to be true enzymes with defined substrates, MASP-3, MAp19, and MAp44 have been suggested to act as regulators of complement activation,12–14 but the relevance of these in vitro observations remains unknown. The surprising new findings of potential functions of MASP-3 and CL-K1 are described in detail below.
The third way of activating complement, the alternative pathway, is also presented in Figure 1. In many regards, this pathway can be described as an amplification mechanism, because it is always activated after activation by the classical and the lectin pathways. Thus, the alternative pathway is important for the activity of the classical and lectin pathways. When the C3b fragment is bound to a surface (see above) it forms a complex with factor B. This leads to the cleavage of factor B by factor D, giving rise to an active enzyme complex with the fragment Bb as the enzyme. The substrate for Bb is C3, and efficient deposition of more C3b fragments occurs onto nearby surfaces, again giving rise to even more complex formations. In analogy with C2a, the active Bb enzyme may now also cleave C5 and thus generate all of the functions described above.
In addition to its function as an amplification mechanism of the classical and the lectin pathway, the alternative pathway may be initiated when C3 in plasma is spontaneously activated through hydrolysis of an internal thioester (so-called “tick-over”). This spontaneous event occurs at a constant low rate wherever in the body C3 is present (blood and interstitial fluids) and is an effect of small perturbations of a somewhat unstable structure of C3, exposing the internal thioester to the solvent.15,16 Thus, generated C3(H2O) can form a complex with factor B, leading to cleavage of factor B by factor D and thus the formation of a C3-activating complex, C3(H2O)Bb, which upon further cleavage and addition of C3 forms a C5-activating complex, C3(H2O)BbC3b. The alternative pathway and the terminal lytic pathway are especially important for control of Neisserial infections, particularly Neisseria meningitidis.2
Plasma and cell surfaces harbor regulators of complement activation (e.g., factor H and CD59, respectively), immediately causing a local shutdown of the cascade, thus leaving host cells unharmed. On a foreign surface, which does not contain such regulators, the cascade continues undisturbed, ending with opsonization of the target and the formation of membrane-spanning MAC complexes. The regulators of complement activation thus add an extra layer of control to the already specific activation events of the classical and lectin pathways. Notably, in the case of the spontaneous activation of the alternative pathway, the regulators entirely determine the specificity on the basis of the recognition of so-called host-associated molecular patterns (HAMPs), a phenomenon termed “reverse recognition.”17 Importantly, activation of complement is also tightly regulated in time and space by a very short half-life of activated C3 and C4; i.e., these activated components have a limited diffusion range because of quenching by water, ensuring that only surfaces nearby the site of activation are targeted.18
Conventional Genetic Deficiencies of Complement Components
Experiments with genetically engineered complement-deficient mice have been useful in research on the complement system. However, in general, inbred mouse strains display poor complement activity,19 and numerous susceptibility differences exist between mouse and man. The study of individuals with increased susceptibility to a variety of clinical symptoms, including suspect immunodeficiency, has proven to be a most important source of information about defects of (and thus function of) the human complement system. In the following, we focus on recessive complement deficiencies based on complete absence of components or functionally defective components. The significance of partial deficiencies is less clear but may be of relevance in some situations. With regards to genetic association studies based on polymorphisms, we refer to recent reviews. As a primer to such studies, strong associations between SNPs in the gene encoding factor H (CFH [MIM 134370, 609814]) and age-related macular degeneration (AMD [MIM 610698]) have been identified.20–22 Polymorphisms in genes encoding other members of the complement system, e.g., factor B (CFB [MIM 138470]) and C3 (C3 [MIM 120700, 613779]), have also been associated with AMD.23,24 Studies are underway with regards to treatment regimens involving restoration of control mechanisms in this condition. Another example is the finding in atypical hemolytic uremic syndrome (aHUS [MIM 235400]) of a number of “loss-of function” polymorphisms in CFH and CD46 (encoding membrane cofactor protein, MCP or CD46 [MIM 120920]) and “gain-of-function” polymorphisms in CFB.25
The prevalence of inherited complement deficiencies is difficult to estimate because most deficiencies may be caused by numerous different mutations in the relevant gene. Furthermore, deficiency is only revealed when by chance the physician or clinician decides that the symptoms of the patient warrant examination of the possible involvement of the complement system. As an example, the prevalence of C2 deficiency is fairly well known in southern Sweden because researchers at Lund University have an interest in the complement system. The clinical conditions most frequently associated with complement defects are cases of hereditary angioedema (HAE [MIM 106100]), systemic lupus erythematosus (SLE [MIM 152700]), and recurrent meningococcal infection.1,26–28 But because it is a multifunctional system and due to redundancies in immune defense mechanisms, the frequency of complement deficiencies in other patient groups is likely underestimated.
Many inherited complement deficiencies have been revealed through association with invasive bacterial infections. This susceptibility appears to be restricted to a limited spectrum of bacteria, mainly encapsulated bacteria. The reason is not clear, but many bacteria (e.g., Streptococcus pyogenes, Staphylococcus aureus, Helicobacter pylori, and Pseudomonas aeruginosa) and several other microorganisms29 have evolved mechanisms to counteract the activities of the complement system and are thus less likely to be associated with complement deficiency.
It is interesting that the strongest disease-susceptibility gene candidates for the development of SLE are found within members of the classical pathway of complement activation. This is speculated to be due to impaired immune complex handling and inefficient clearance of apoptotic cells in such patients.30 An alternative model has been presented in which complement—together with other components of the innate immune system—participates in the “presentation” of SLE-inducing self-antigens to developing B cells. In this model, the complement system and innate immunity protect against responses to SLE (self) antigens by enhancing the elimination of self-reactive lymphocytes.31 Homozygous deficiency of C1q is very strongly associated with a lupus-like disease with rash and glomerulonephritis. More than 90% of C1q-deficient individuals were reported to have such symptoms.30 However, C1q deficiency appears to be very rare (Table 1), and out of all cases of SLE only a tiny fraction are associated with complement deficiency, indicating that SLE is a highly multifactorial disease.
Table 1.
Conventional Complement Deficiencies
| Protein | Gene symbol | MIM ID | Cases / Homozygosity | Disease Associations | References |
|---|---|---|---|---|---|
| Recognition Proteins | |||||
| MBL | MBL2 | 154545 | 5% | infections in immunocompromised individuals | 28,106–108 |
| H-ficolin | FCN3 |
604973, 613860 |
3 | immune deficiency, necrotizing enterocolitis | 40,41 |
| C1q |
C1QA, C1QB, C1QC |
120550, 120570, 120575, 613652 |
43 | SLE-like syndrome, recurrent bacterial infections | 26,33,51 |
| Enzymes | |||||
| MASP-2 | MASP2 | 605102 | 9 | immune deficiency | 37,38 |
| C1r/s |
C1R, C1S |
613785, 120580, 613783, 216950 |
19 | SLE-like syndrome, recurrent bacterial infections | 26,51 |
| C2 | C2 |
613927, 217000 |
1:20,000 | autoimmune disease | 28,32 |
| Factor D | CFD |
134350, 613912 |
<10 | meningococcal and encapsulated bacterial infections | 26 |
| Factor I | CFI |
217030, 610984 |
31 | encapsulated bacterial infections | 109,110 |
| Structural Proteins | |||||
| C3 | C3 |
120700, 613779 |
27 | bacterial infections, SLE-like syndrome | 111,112 |
| C4 |
C4A, C4B |
120810, 120820 |
26 | SLE-like syndrome, encapsulated bacterial infections | 33,51 |
| C5 | C5 |
120900, 609536 |
40 | meningococcal infection | 113,114 |
| C6 | C6 |
217050, 612446 |
>50 | meningococcal infection | 115–117 |
| C7 | C7 |
217070, 610102 |
>50 | meningococcal infection | 116,118,119 |
| C8 |
C8A, C8B, C8G |
120950, 120960, 120930, 613790, 613789 |
>50 | meningococcal infection | 120 |
| C9 | C9 |
120940, 613825 |
1:1,000 (Japan) | meningococcal infection | 46 |
| Control Proteins | |||||
| Properdin | CFP | 300383 | >50 | meningococcal infection | 43 |
| Factor H | CFH |
134370, 609814 |
27 | hemolytic uremic syndrome (HUS), dense deposit disease | 111,121,122 |
| C1-INH | SERPING1 | 606860 | 1:50,000 | hereditary angioedema (HAE) | 33,123 |
| CD11a (LFA-1), CD11b (CR3), CD11c (CR4) /CD181 |
ITGAL, ITGAM, ITGAX, ITGB2 |
153370, 120980, 151510, 600065, 116920 |
1:1,000,000 | leukocyte adhesion deficiency type I (LAD I) | 124,125 |
| CD46 (MCP) | CD46 | 120920 | atypical hemolytic uremic syndrome (aHUS) | 25,126,127 | |
| CD55, CD59 (PIGA)2 |
CD55, CD59, PIGA |
125240, 107271, 612300, 311770, 300818 |
1:1,000,000 | paroxysmal nocturnal hemoglobinuria (PNH) | 128 |
Summary of reported cases of homozygosity for complement-system gene mutations leading to functionally deficient proteins. The proteins in question are given on the left, followed by the gene symbol, the MIM ID number (associated with the gene and/or disease), the number of cases or incidence of homozygosity, and associated clinical findings. The numbers in the last column refer to reviews in which most of the original literature is mentioned or to recent case reports where the literature is discussed. Large ethnical differences are seen for C5, C6, C7, C8, and C9 deficiencies; for example, much higher frequencies of C9 deficiency in Japan as compared to people in western Europe. As is evident, most cases of homozygosity are rare; i.e., below 50 cases. Some are marked “ >50,” which indicates more than 50 but less than 200 cases.1 Concomitant loss of LFA-1, CR3, and CR4 is due to loss of the common CD18 chain.2 Please note that PNH may be caused either directly by a mutation in CD59 or by loss of PIGA function leading to a secondary loss of CD55 and CD59.
C4 deficiency is also extremely rare, with only 26 cases having been reported. This could be due to variation in C4 gene copy number. Because two to seven copies are found in the diploid genome, multiple mutations have to occur for deficiency to result.
Deficiency of C2, which is nearly always due to a 28 bp deletion in C2 (MIM 613927, 217000), occurs at higher frequency; for example, there are probably more than 450 in a population of 9 million in Sweden.32 But whereas the majority of patients with deficiencies of C1 or C4 develop SLE, the association of SLE with C2 deficiency is much weaker.30 Deficiency of C2 is, on the other hand, generally associated with a slight increase in the prevalence of autoimmune disease. Furthermore, about half of the patients identified as having C2 deficiency had a history of invasive infection with encapsulated bacteria. However, family members lacking C2 but not showing clinical symptoms were often encountered. A recent review33 provides a detailed picture of the C1q, C2, and C4 deficiencies described above.
With regards to the lectin pathway, MBL deficiency has a surprisingly high prevalence. About 5% of people of European descent show MBL deficiency when it is defined as having a protein level less than 100 ng/ml. The frequency is about twice this in Sub-Saharan Africans. MBL-deficient adults appear healthy, but low levels of MBL are associated with increased risk of infection in toddlers,7 in cancer patients undergoing chemotherapy, and in organ-transplant patients receiving immunosuppressive drugs, especially recipients of liver transplants.34,35 The deficiency is due to homozygosity for one of three quite common nonsynonymous SNPs in the coding region of MBL2 (MIM 154545) (gene frequency in people of European descent: 0.14), as well as a promoter polymorphism (gene frequency: 0.20).36 A case of MASP-2 deficiency was originally described in an immunodeficient patient with autoimmune manifestations.37 The deficiency was due to homozygosity of a missense mutation giving rise to a nonfunctional protein and resulting in a very low MASP-2 level. Additional studies revealed that the allele occurs in the general population at a frequency suggesting homozygosity in approximately 6 in 10,000 individuals, but only a low clinical penetrance of MASP-2 deficiency is seen.38,39 Three cases of H-ficolin deficiency have been described, two in neonates with necrotizing enterocolitis40 and one in an adult with mixed symptoms of immunodeficiency.41 All were due to a frameshift mutation occurring at a gene frequency of 0.011 among suspected immunodeficient patients as well as among controls.
With regards to deficiency in proteins of the alternative pathway, factor D deficiency, which is very rare (fewer than 10 cases reported42), and properdin deficiency both result in selective impairment of the alternative pathway.32 In both cases meningococcal disease is the cause for investigating the complement system, and indeed this bacterium is the most prevalent infectious agent in factor-D-deficient individuals. Properdin deficiency is inherited in an X-linked manner and is found as absence of the protein or as a dysfunctional protein.43 In some families with properdin deficiency, fulminant meningococcal infections, particularly meningococcal meningitis, were the most frequent problem encountered. Considering the complement system as a general antimicrobial defense system, it is surprising that susceptibility to other bacteria is not evident. Associations of factor D or P deficiency with autoimmune diseases have not been reported.
Inherited deficiency of the central component, complement factor C3, is very rare (27 cases reported), but when identified it has been found in patients with life-threatening infections with encapsulated bacteria.28 C3 deficiency is also seen secondary to genetic deficiency of regulatory proteins of the alternative pathway. Lack of factor I and of factor H thus leads to uncontrolled complement activation and consumption of C3. aHUS has been associated with mutations in the genes encoding factor H and factor I leading to failure of regulation of the alternative pathway.44 This disorder is characterized by hemolytic anemia, thrombocytopenia, and acute renal failure, which is probably caused by uncontrolled activation of the complement system at the renal endothelium.
Deficiencies in the terminal MAC components are typically identified in patients with recurrent invasive infections caused by N. meningitidis and N. gonorrhoeae.45 However, deficiencies of proteins in the MAC may remain undetected until old age, if at all detected. Deficiencies have been observed in all of the MAC components, C5, C6, C7, and C8, at a very low frequency in European populations (Table 1). There are large differences among ethnicities, and the highest frequency is found in Japan, where 1 in 1000 individuals are C9 deficient (due to a nonsense mutation).46 This high frequency may even indicate an advantage of MAC deficiency under certain conditions. There seems to be no association between MAC deficiency and autoimmune diseases.
Studies on the genes encoding regulatory complement components have revealed a number of interesting findings. HAE, resulting from C1 inhibitor (C1-INH) deficiency, is an autosomal-dominant disorder characterized by recurrent episodes of angioedema with abdominal attacks. The incidence of HAE is approximately 1:50,000, with no ethnicity-based differences. C1-INH is a serine protease inhibitor of the serpin family, named after its inhibitory activity toward the C1 enzymes, C1r and C1s, but it also controls the MASPs of the lectin pathway. However, the angioedema is due to loss of control of kallikrein of the contact system and is not a consequence of failure of control of the classical or the lectin pathway. The attacks frequently lead to hospitalization and are associated with a substantial mortality. Two forms of hereditary angioedema are found: type I (85% of cases), characterized by low levels and low functional activity of C1-INH, and type II, characterized by normal levels of C1-INH but low functional activity. Both types are due to mutations in SERPING1 (MIM 606860), encoding C1-INH. The genetics and therapy of C1-INH deficiency (using C1-INH purified from donor plasma) was recently reviewed by Cugno et al.47
Two of the important membrane-bound inhibitors of the complement system, decay-accelerating factor (DAF, CD55) and membrane inhibitor of reactive lysis (MIRL, CD59), are tethered to the cell surface through a glycosyl phosphatidylinositol (GPI) anchor. Rare mutations (1 in 1,000,000) in PIGA (MIM 311770), encoding phosphatidylinositol glycan class A, which is involved in GPI production, thus lead to loss of these regulators. This results in paroxysmal nocturnal hemoglobinuria (PNH [MIM 300818]) because the complement system autoattacks the erythrocytes lacking the control proteins. Such patients can be treated with compounds inhibiting the complement system, such as anti-C5 antibody.48 Direct mutations in CD59 (MIM 107271) can similarly result in CD59 deficiency (MIM 612300), causing PNH.
Deficiency of complement receptors may also cause pathology, as seen in the case of leukocyte adhesion deficiency (LAD) type I (MIM 116920), an autosomal-recessive disorder of leukocyte function resulting from a deficiency of the beta-2 integrin subunit (CD18, encoded by ITGB2 [MIM 600065]) of the CD11/CD18 complex.49,50 The CD11/CD18 complex is important in adhesion and phagocytosis. Three CD11 alpha chains, CD11a (integrin alpha-L, encoded by ITGAL [MIM 153370]), CD11b (integrin alpha-M, encoded by ITGAM [MIM 120980]), and CD11c (integrin alpha-X, encoded by ITGAX [MIM 151510]), form heterodimer transmembrane complexes with the CD18 beta chain: CD11a/CD18 (lymphocyte function-associated antigen 1, LFA-1), CD11b/CD18 (complement receptor 3, CR3), and CD11c/CD18 (CR4). Thus, CD18 deficiency results in a combined loss of expression of LFA-1, CR3, and CR4, causing defects in neutrophil, macrophage, T cell, and B cell adhesion to the vascular endothelium and inefficient migration into infectious sites, as well as impairment of normal T cell and B cell functions.51 Clinically, LAD is characterized by recurrent bacterial infections and impaired pus formation and wound healing.52
We have summarized these conventional complement deficiencies in Table 1. Investigations of more and larger groups of patients are expanding our understanding of the role of complement in disease. Young patients with invasive infections caused by encapsulated bacteria should be screened for complement deficiency. As evident from recent findings in studies on gene associations with AMD, surprising angles on the clinical importance of the complement system may be found through the analysis of other patient groups. Although the association of, for example, factor H deficiency with AMD fits its role in complement-mediated reactions, genetic analyses have now revealed completely unexpected roles of complement components beyond the hitherto known functions of the complement system.
Unexpected Roles of Proteins of the Lectin Pathway of Complement
In a recent study in this journal, Sirmaci and colleagues harnessed the power of homozygosity mapping and next-generation exome sequencing to identify a novel missense mutation in the exon encoding the serine protease domain of the protein MASP-3 (mannan-binding lectin-associated serine protease 3) in a family afflicted by the autosomal-recessive 3MC syndrome.53 The 3MC syndrome is a term encompassing the overlapping Mingarelli (MIM 265050),54 Malpuech (MIM 248340),55 Michels (MIM 257920),56 and Carnevale (also MIM 265050)57 syndromes, which were independently described but appear clinically to be unified58 and, as will be clear, have underlying mutations and etiology that now appear to be unified as well. Patients with 3MC syndrome (see Figure 1 in 53) may exhibit a spectrum of developmental features, including developmental delay, growth and mental retardation, and characteristic facial dysmorphism, such as hypertelorism, telecanthus, blepharophimosis, blepharoptosis, and epicanthus inversus. They may also suffer from anterior-chamber eye anomalies, mixed hearing loss, cleft lip and/or palate, periumbilical defects, and skeletal anomalies with radioulnar synostosis and caudal appendage.
MASP-3, MASP-1, and MAp44 are all encoded by MASP1. Sirmaci and colleagues proceeded to identify a nonsense mutation in a part of MASP1 (MIM 600521) common to all three splice variants in an unrelated family with 3MC syndrome. Thus, a novel and unexpected role was discovered of what was so far regarded “only” as a complement component. A more recent paper by Rooryck and colleagues presented an independent confirmation of this finding and also demonstrated 3MC-causing mutations in another gene, COLEC11 (MIM 612502), encoding a protein, collectin kidney 1 (CL-K1 or collectin-11), of the same innate immune defense system, the lectin pathway of complement activation.59 Their studies on zebrafish further support the developmental influence of mutations in these two genes, as morphant fish showed severe craniofacial abnormalities. Importantly, this finding also indicates the presence of an evolutionarily conserved pathway of embryogenesis. In the following, we will give a more detailed description of the proteins encoded by COLEC11 and MASP1 and their suggested function.
CL-K1
As mentioned above, CL-K1 is a recently described member of the collectin family of humoral pattern-recognition molecules.10,11 So far, only two papers have been directed at describing this protein. The evidence for its function in the innate immune defense and in the complement system is only fragmentary and is largely deduced from structural similarities with MBL and ficolins (Figure 2A). These molecules are composed of subunits consisting of three identical polypeptides with a collagen region and a C-terminal carbohydrate-recognition domain (CRD). Three or more subunits are joined into oligomers forming collagen-like coiled regions. Dimers of MASPs associate with the collagen region and mediate complement activation when MBL or ficolins bind to their targets (Figure 2B). It appears that CL-K1, composed of 34 kDa polypeptides with a collagen region and a C-type CRD, must present a similar overall structure (Figure 3), and it seems also to associate with MASPs11 and to bind to a number of microbial polysaccharides.10 The synthesis of CL-K1 is widely distributed in the body, and, like MBL and ficolins, it is secreted and circulates in the blood.
Figure 3.
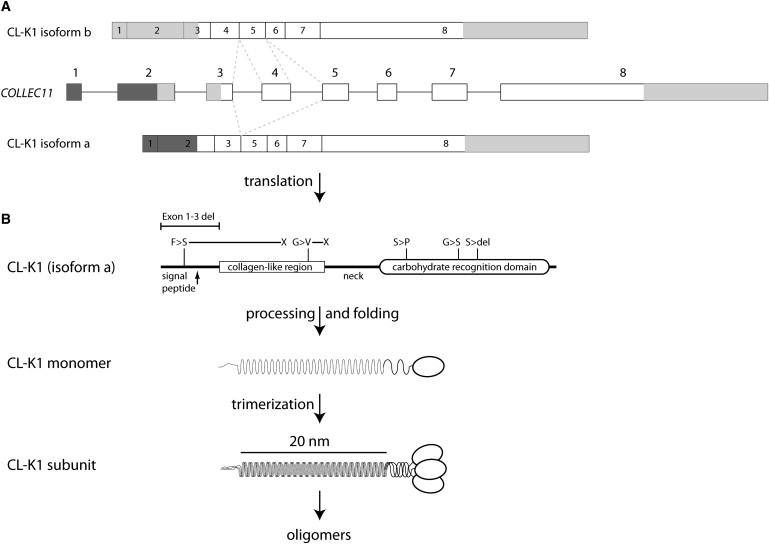
Gene Structure, Alternative Splicing, and Protein Products of COLEC11
(A) Overview of the gene/primary transcript and the two suggested alternative splice variants. Mutually exclusive splice events are indicated by the dotted lines. Greyed areas indicate the 5′ and 3′ UTRs. Notice the predicted use of alternative translation initiation sites in isoforms a and b.
(B) The protein structure of the secreted isoform is illustrated and polymorphisms are indicated, along with the mutations reported by Rooryck et al. The exact locations of the mutations are: c.496T>C (p.Ser169Pro), c.610G>A (p.Gly204Ser), c.45delC (p.Phe16SerfsX85), c.648_650delCTC (p.Ser217del), and c.300delT (p.Gly101ValfsX113), as well as the 27 kb exon 1–3 deletion. The formation of the structural subunit composed by three identical polypeptide chains and the ensuing oligomerization is illustrated.
The CL-K1-Encoding Gene, COLEC11
COLEC11, encoding CL-K1, is located on chromosome 2, at 2p25.3. It spans about 50 kb and encodes two splice forms (Figure 3). Transcript variant 1 comprises 7 exons (1–3, 5–8) and 1320 nt. Transcript variant 2 is 8 exons (1–8) and 1399 nt long. The resulting isoforms a and b are 271 aa and 268 aa long, respectively. Isoform a is predicted by SignalP60 to contain a signal peptide (aa 1–25), whereas isoform b does not harbor a signal peptide and cleavage motif. The existence of both isoforms at the mRNA level is well supported by expressed sequence tags (ESTs). At the protein level, the secreted isoform a of CL-K1 is the form described in the literature. It has 24 Gly-Xaa-Yaa repeats, starting at aa position 41, constituting a collagen-like stalk, and a C-terminal CRD (aa 151–266), thus identifying it as a member of the collectins (Figure 3).
The MASPs
The MASPs are modular proteases composed of well-described domains in the order CUB-EGF-CUB-CCP-CCP-SP (Figure 2B and Figure 4). The CUB (C1r/C1s, embryonic sea Urchin protein [Uefg], and Bone-morphogenetic protein 1 [Bmp1]) domain is around 110 aa and is found in a diverse set of proteins involved mainly in developmental processes.61 The EGF (epidermal growth factor) domain of approximately 50 aa is also found in many proteins and is known to mediate protein-protein interactions. The CUB and EGF domains of the MASPs are responsible for dimerization and for the calcium-dependent binding to MBL and ficolins.62–64 The CCP (complement control protein) domains of around 60 aa are found in a number of complement factors and other proteins.65 The contiguous CCPs of MASP-1 and MASP-2, especially the second CCP domain, have been implicated in the binding of macromolecular substrates.66–71 Finally, the SP (serine protease) domain is the catalytically active unit of the proteases and defines them as part of the S1A family of chymotrypsin-like proteases. The second CCP domain and the SP domain are connected by a somewhat flexible activation peptide, which lies just upstream of a site, which is cleaved upon activation of the MASP (Figure 4).
Figure 4.
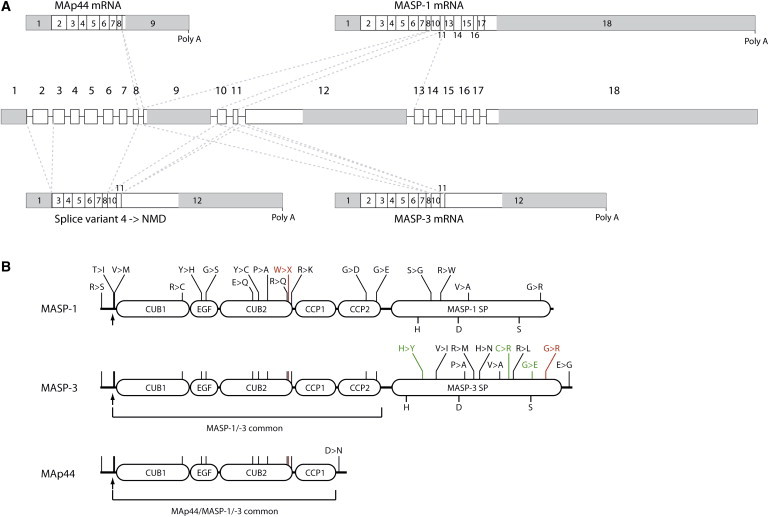
Gene Structure, Alternative Splicing, and Protein Products of MASP1
(A) Overview of the gene or primary transcript and the four resulting alternative splice variants. MAp44, MASP-1, and MASP-3 arise by mutually exclusive differential splicing (indicated by the dotted lines), each product having its own 3′ UTR and poly A signal. A fourth splice product is very similar to MASP-3 mRNA, but with the exclusion of exon 2. This leads to a frameshift, and the product is predicted to be degraded by nonsense-mediated decay.
(B) Overview of the resulting protein structures of the three mRNAs produced from MASP1. The arrow indicates the cleavage site for the signal peptide. Nonsilent polymorphisms are indicated (in black), along with the mutations reported by Sirmaci et al. (in red) and by Rooryck et al. (in green). The exact locations of the indicated nonsilent polymorphisms and mutations are given in Table 2. The part of the polypeptide chain common to all three proteins, as well as the part common to MASP-1 and MASP-3, is indicated.
MASP1
MASP1 is located on chromosome 3q27-28 and gives rise to three alternative splice products: MASP-1, MASP-3, and MAp44 (Figure 4).12,13,72,73 The gene encompasses 18 exons, of which the first 8 exons plus exons 10 and 11 encode the five N-terminal domains shared by MASP-1 and MASP-3, CUB-EGF-CUB-CCP-CCP. The serine protease domain of MASP-1 is encoded by six separate exons (13–18), whereas for MASP-3 it is encoded by a single exon (12)12,74,75 (Figure 4). Thus MASP-1 and MASP-3 share their first five domains but have distinct serine protease domains (Figure 4). The third splice product, encoding MAp44, is composed of nine exons: the first eight exons are shared with MASP-1 and MASP-3, and they encode the first four domains, CUB-EGF-CUB-CCP, whereas the ninth exon is unique to MAp44 (Figure 4). An additional adenosine nucleotide from exon 8 combined with the first 50 nt of exon 9 encodes the unique 17 C-terminal aa of MAp44.
In addition to the three expressed splice products, a fourth splice variant has been recorded in the NCBI database. However, the RNA arising from this splice pattern does not constitute mRNA, but is predicted to be degraded by the nonsense-mediated mRNA decay (NMD) pathway.76 The fourth splice variant is identical to MASP-3 mRNA, save for the exclusion of exon 2, giving rise to a premature termination (nonsense) codon. Whether this RNA splice product is truly a “waste product” or whether it is a “regulatory product” of controlled splice patterns is unknown. In this context it is exciting to note that in the recent report of the “splicing code,” a class of exons were detected whose inclusion silenced expression in adult tissues by activating nonsense-mediated messenger RNA decay. Exclusion of such exons, on the other hand, promoted expression during embryogenesis.77 Of course, in the case of MASP1, it seems that it is rather the exclusion of an exon that drives generation of a transcript, which is subject to NMD. Nonetheless, these findings point to the importance of regulated alternative splicing events on a genome-wide scale, and one may speculate that MASP1 is also subject to such regulation.
The CL-K1 Gene and 3MC
Rooryck and colleagues aimed to identify causative mutations in one or more genes in 3MC syndrome by homozygosity mapping in a total of 11 families and discovered two regions of homozygosity shared by affected individuals.59 Four families were initially identified, and they shared a homozygous region of 2.2 MB on 2p25.3. Sequencing revealed two homozygous nonsynonymous mutations (c.496T>C, c.610G>A), one single-base deletion (c.45delC), and one in-frame deletion (c.648_650delCTC) in COLEC11, encoding CL-K1 (resulting in, respectively: p.Ser169Pro, p.Gly204Ser, p.Phe16SerfsX85, and p.Ser217del) (Figure 3). Subsequent sequencing of COLEC11 in other 3MC patients revealed two additional mutations in two previously described probands. The first was a dramatic 27 kb homozygous deletion encompassing exons 1–3 of COLEC11 (exon 1–3 deletion), the second a homozygous single-base deletion causing a frameshift and premature stop codon (c.300 delT, resulting in p.Gly101ValfsX113) (Figure 3).
The two single-nucleotide deletions dictating frameshifts and premature termination, as well as the 27 kb deletion, are obviously detrimental. The in-frame deletion resulting in loss of a single amino acid and the two amino acid substitutions all occur in the CRD of CL-K1. These are otherwise well-conserved residues, and none of the mutations were found in 368 ethnically matched controls or in the dbSNP or 1000 Genomes databases. Indeed, no CL-K1 was found in the serum of two patients with the p.Gly204Ser mutation.59
Analysis of the localization of the protein in the mouse revealed broad tissue distribution during embryogenesis. This was also found by in situ expression analyses in zebrafish. The effect of loss of function of this protein during zebrafish embryogenesis was examined by the use of morpholinos directed against the initiation site and the exon 2-intron 2 splice site. Notably, these morpholinos target both isoforms. There were evident differences in the craniofacial skeleton compared with uninjected or control morpholino-injected embryos. Importantly, it was found that the phenotype of COLEC11 morphant fish could be rescued by coinjecting embryos with the full-length human CL-K1 mRNA.
MASP1 Mutations and 3MC Syndrome
A large number of SNPs have been reported for MASP1, but until now, no mutations with impact on the function of either of the three protein products have been reported. An overview of the nonsilent polymorphisms from dbSNP as well as those reported by Sirmaci and colleagues and Rooryck and colleagues is given in Table 2. The fact that two completely different mutations were found by Sirmaci and colleagues in two independent families, and that three additional mutations were found independently by Rooryck and colleagues in another four families, lends credibility to the conclusion that MASP1 dysfunctionality is causative of the phenotype. One of the mutations identified by Sirmaci and colleagues is a nonsense mutation in the common part of the three alternative splice products (Figure 4). However, it would appear that MASP-3 is the central product, as this is the only component affected in the other family as well as the four families reported by Rooryck and colleagues. The affected residue in the MASP-3-specific mutation of Sirmaci et al., G687, is located within the active site close to the junction of two β-barrels and in close proximity to D658 (Figure 4). On the basis of structural modeling, Sirmaci and colleagues present the elegant hypothesis that given the close proximity of D658 to G687, the G687R variant would likely result in the formation of an intermolecular salt bridge, thereby inhibiting catalytic activity.53 Indeed, Asp658 in MASP-3 corresponds to Asp189 in chymotrypsin (c189, chymotrypsin numbering used in the following for reference), and this residue is responsible for primary substrate specificity in the chymotrypsin family. In MASP-1, the side chain of Arg677 (c224) is rotated toward the Asp640 (c189) and forms a salt bridge, which competes with the S1-P1 interaction upon substrate binding.78 This is envisioned to modulate the activity of MASP-1. Thus, it seems plausible that the reported mutation in the MASP-3-specific exon could similarly disrupt or suppress the protease activity.
Table 2.
Overview of the Nonsilent Polymorphisms in the Coding Parts of MASP1 and the Resulting Amino Acid Substitutions
| Chr. Position | Coding Pos. | dbSNP rs #/Ref. | Heterozygosity | Contig Ref. | Missense | Amino Acid | Contig Ref. | Substitution |
|---|---|---|---|---|---|---|---|---|
| Common | ||||||||
| 187003844 | 6 | rs116384821 | N.D. | G | T | 2 | Arg [R] | Ser [S] |
| 187003788 | 62 | rs1062049 | 0.005 | C | T | 21 | Thr [T] | Ile [I] |
| 187003786 | 64 | rs77189011 | 0.022 | G | A | 22 | Val [V] | Met [M] |
| 186980367 | 379 | rs115647447 | N.D. | C | T | 127 | Arg [R] | Cys [C] |
| 186978610 | 466 | rs78137557 | 0.236 | T | C | 156 | Tyr [Y] | His [H] |
| 186978589 | 487 | rs115241263 | N.D. | G | A | 163 | Gly [G] | Ser [S] |
| 186974493 | 703 | rs3203210 | N.D. | G | C | 235 | Glu [E] | Gln [Q] |
| 186974465 | 731 | rs28945071 | N.D. | A | G | 244 | Tyr [Y] | Cys [C] |
| 186971076 | 772 | rs866085 | N.D. | C | G | 258 | Pro [P] | Ala [A] |
| 186970985 | 863 | rs116001173 | N.D. | G | A | 288 | Arg [R] | Gln [Q] |
| 186970978 | 870 | Sirmaci et al., 20101 | N.D. | G | A | 290 | Trp[W] | Stop[X] |
| 186970964 | 884 | rs73886128 | N.D. | G | A | 295 | Arg [R] | Lys [K] |
| MAp44 | ||||||||
| 186965162 | 1102 | rs113938200 | N.D. | G | A | 368 | Asp [D] | Asn [N] |
| MASP-1/3 | ||||||||
| 186959343 | 1229 | rs114365879 | N.D. | G | A | 410 | Gly [G] | Asp [D] |
| 186959295 | 1277 | rs28945068 | 0.034 | G | A | 426 | Gly [G] | Glu [E] |
| MASP-3 | ||||||||
| 186954170 | 1489 | Rooryck et al., 20117 | N.D. | C | T | 497 | His [H] | Tyr [Y] |
| 186954107 | 1552 | rs73068950 | N.D. | G | A | 518 | Val [V] | Ile [I] |
| 186953975 | 1684 | rs72549155 | 0.010 | C | G | 562 | Pro [P] | Ala [A] |
| 186953932 | 1727 | rs72549154 | 0.058 | G | T | 576 | Arg [R] | Met [M] |
| 186953906 | 1753 | rs111908734 | N.D. | C | A | 585 | His [H] | Asn [N] |
| 186953812 | 1847 | rs2461280 | N.D. | T | C | 616 | Val [V] | Ala [A] |
| 186953771 | 1888 | Rooryck et al., 20117 | N.D. | T | C | 630 | Cys [C] | Arg [R] |
| 186953749 | 1910 | rs115022399 | N.D. | G | T | 637 | Arg [R] | Leu [L] |
| 186953662 | 1997 | Rooryck et al., 20117 | N.D. | G | A | 666 | Gly [G] | Glu [E] |
| 186953600 | 2059 | Sirmaci et al., 20101 | N.D. | G | A | 687 | Gly [G] | Arg [R] |
| 186953491 | 2168 | rs115897757 | N.D. | A | G | 723 | Glu [E] | Gly [G] |
| MASP-1 | ||||||||
| 186944222 | 1528 | rs28945070 | 0.010 | A | G | 510 | Ser [S] | Gly [G] |
| 186943280 | 1573 | rs28945073 | N.D. | C | T | 525 | Arg [R] | Trp [W] |
| 186943150 | 1703 | rs13322090 | 0.041 | T | C | 568 | Val [V] | Ala [A] |
| 186937924 | 2035 | rs3774266 | 0.006 | G | A | 679 | Gly [G] | Arg [R] |
The three mutations identified by Rooryck and colleagues are all located in the MASP-3-specific exon (Figure 4). They find two homozygous missense mutations in two new 3MC families (c.1489C>T and c.1888T>C, at the protein level resulting in p.His497Tyr and p.Cys630Arg, respectively) and a third homozygous missense mutation in two previously described 3MC families (c.1997G>A, encoding p.Gly666Glu). All of these mutations are found in well-conserved positions, are predicted to be damaging (Polyphen), and are not found in 286 ethnically matched controls.59
Rooryck et al. targeted MASP1 expression in developing zebrafish embryos by use of morpholinos specific for the initiation site and the exon 3-intron 3 splice site.59 Such targeting should disrupt expression of the two splice forms (MAp44 and MASP-3), which are predicted to be found in zebrafish.13 Pigment and craniofacial cartilage defects similar to those seen in CL-K1 morphants resulted.
MASP-3 and CL-K1 Likely Act in the Same Pathway, Possibly as Chemoattractants
Rooryck and colleagues stab at the mechanistic link by examining the effects of COLEC11 and MASP1 mutations on the migration of cranial neural crest cells during zebrafish development. It appears that they may be early guidance cues directing the migration of neural crest cells, and this is followed up by the demonstration that recombinant CL-K1 attracts neural crest cells. The effect of CL-K1 was confirmed in a quail neural tube explant assay.59 Other proteins with collagen-like regions linked to a CRD domain (surfactant protein D79–81 and surfactant protein A82) have previously been noted to act as chemoattractants. Whereas the possibility remains that several members of this family have dual roles in immunity and development, the observed directional cell movement may be caused by a “matrix-like” effect of the collagen helix in the proteins, or, indeed, these molecules may activate cells to secrete other chemoattractants.
Notwithstanding the zeal of the investigators, the mechanism behind the link between MASP1 and COLEC11 mutations and 3MC syndrome remains obscure. Sirmaci and colleagues speculate that the effect may be mediated through an activity of MASP-3 toward insulin-like growth factor-binding protein 5 (IGFBP-5).53 This is based on one study demonstrating cleavage of IGFBP-5 by MASP-3.83 However, such an activity has not been firmly established, but is based on the observation of a weak activity of recombinant serine protease domain of MASP-3 toward IGFBP-5. IGFBP-5-cleaving activity has also been reported for a number of other enzymes; for example, thrombin,84 C1s,85 and the inflammation-related neutrophil proteases cathepsin G and elastase,86 all of which are serine proteases involved in or related to host defense. It is tempting to suggest a function in the release of sequestered IGF for promotion of wound healing and repair. A more cynical explanation, based on the large number of reported IGFBP-5 proteases,87 is that IGFBP-5 is a quite promiscuous substrate. The weak activity of MASP-3 could also be explained as an evolutionary remnant on the basis of homology with the proteases known to cleave IGFBP-5. It would be worthwhile to examine whether mutated MASP-3 is produced and whether it is indeed catalytically active or not. Unfortunately, the latter is difficult because a convincing substrate for MASP-3 remains to be identified.
Notably, there exists a mouse that was targeted in exon two of the MASP1 ortholog (masp1) and is hence (presumably) deficient in MASP-1, MASP-3, and MAp44.88 It has been reported to be more susceptible to influenza virus infection, and more recently it was found that the mouse is deficient in alternative pathway function, with MASP-1 reportedly being the enzyme responsible for activation of murine factor D by cleavage of pro-factor D.89 Apart from small size and low weight, no phenotypic characterization of the mouse has been presented. One would have expected a murine “3MC” phenotype to be immediately apparent.
Evolutionary Aspects
MASPs are found all the way back to cephalochordates and urochordates. MASP-2 and MASP-3 are believed to have evolved from an ancestral MASP-1 by gene duplication and divergence.72,90 In this connection, it is peculiar that birds91 and fish seem to have no MASP-1, whereas they have MASP-2 and MASP-3, but this could represent a loss or inactivation of the MASP-1 exon(s) in each of their ancestors subsequent to the duplication of MASP-1.92 Interestingly, the chicken also lacks the related haptoglobin, which is believed to have evolved from an ancestral MASP.93
Krem and colleagues created a large phylogenetic tree of the protease domains of all the then-known members of the S1 family.94 The tree was rooted with the use of marine arthropod enzymes, chosen for the primordiality of the species and certain molecular markers. This phylogenetic tree suggested two origins of proteases that participate in the innate immune system. Proteases of mast cells, neutrophils, and other immune cells are most closely related to kallikrein enzymes, which probably evolved from an ancestral gene distinct from degradative, clotting, and complement lineages.95,96 Complement proteases, with the exceptions of factors D and I, share ancestry with clotting proteases,97 most likely having evolved from clotting-immune systems of the type found in the horseshoe crab.98,99 Interestingly, complement and coagulation proteases diverge in close relation to embryonic developmental proteases. There are some functional overlaps between arthropod developmental and hemolymph proteases, and these evolutionary connections between development, clotting, and immunity extend beyond the proteases to their substrates.95 In the complement system there is a clear distinction of origin between enzymes of the classical/lectin and alternative pathways. Factor D diverges early with enzymes of cell-mediated immunity and shares common ancestry with arthropod degradative enzymes. Factor I also groups with degradative enzymes, but of later origin, whereas factors B and C2 diverge together and share common ancestry with fibrinolytic proteases. Finally, the MASPs, C1r, and C1s emerge in close connection with enzymes of blood coagulation.95
Extending the Scope
Notably, the 3MC patients suffer moderate mental retardation. In recent years, evidence has been mounting of unrelated or alternative roles of complement outside immunity. In the mouse, Stevens and colleagues reported that the classical complement cascade mediates CNS synapse elimination100 and that consequently C1q knockout mice display enhanced synaptic connectivity and suffer from epilepsy.101 No neural defects have been reported in the identified human C1q-deficiency cases. In contrast, the finding of elevated levels of MBL/MASP-2 complexes within the schizophrenic brain102 may implicate the lectin pathway in neural functions in humans. A model describing a possible mechanism for activity-dependent synapse elimination was suggested nearly two decades ago.103 According to this model, strong synapses facilitate elimination of weaker synapses by sending both a protective signal for the stronger synapse, resulting in stabilization and strengthening, and a punishment signal for weaker synapses, resulting in their elimination. It has recently been suggested that immune molecules may mediate the punishment signals.104 Synapse elimination is a prominent process both during development and during neurodegenerative diseases and aging, in the former as a requirement for correct formation of synaptic networks and in the latter as a detrimental process destroying such synaptic networks. An intriguing hypothesis has been proposed: that a common mechanism may underlie this elimination of synapses during development and disease.104 Indeed, a recent review has highlighted the currently known roles, some detrimental and some protective, of complement in neurodegenerative disorders.105 The new results on MASP-3 and CL-K1 may spur renewed interest in studies on intellect and the immune system.
Conclusion
A number of conventional complement deficiencies with immunological consequences have been characterized. Here, we have focused on recessive complement deficiencies based on complete absence of components or functionally defective components. Partial deficiencies and polymorphisms also account for numerous immunological defects. Commonly, these conventional complement deficiencies have not been very severe since the advent of antibiotics and modern treatment modalities, and hence such genetic deficiencies are quite widespread in various populations.
In contrast, newly discovered defects of components of the lectin pathway have implications outside innate immunity, namely in development. These appear to be more severe and are caused by de novo mutations, which have arisen in a handful of families and have been fixed by consanguinity. A role for MASP-3 in developmental processes is supported by several lines of circumstantial evidence: (1) a general link between immunity and development, (2) a specific evolutionary relationship between complement and coagulation proteases and embryonic developmental proteases, (3) broad expression of MASP-3 in developing chick and rat embryos,91 (4) enzymatic activity toward IGFBP-5 and similar broad developmental expression of this purported substrate, itself suggested to be involved in developmental processes, (5) homology with classical pathway proteases involved in developmental processes in the mouse, (6) CUB-domain-containing proteins often being involved in developmental processes,61 and (7) a very high degree of phylogenetic conservation of MASP-3. The report by Sirmaci and colleagues has provided the first solid evidence for an important function of MASP-3. This finding is confirmed and substantiated by the report of Rooryck and colleagues, and it is extended with the discovery of a similar role for CL-K1, as well as by the evidence that the two proteins collaborate in the same pathway. Future studies should elucidate the mechanism behind these fascinating observations.
Acknowledgments
Financial support was provided by The Danish Council for Independent Research, Medical Sciences.
Contributor Information
Søren E. Degn, Email: sdegn@microbiology.au.dk.
Steffen Thiel, Email: st@microbiology.au.dk.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
References
- 1.Ricklin D., Hajishengallis G., Yang K., Lambris J.D. Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walport M.J. Complement. First of two parts. N. Engl. J. Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 3.Jayasekera J.P., Moseman E.A., Carroll M.C. Natural antibody and complement mediate neutralization of influenza virus in the absence of prior immunity. J. Virol. 2007;81:3487–3494. doi: 10.1128/JVI.02128-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walport M.J. Complement. Second of two parts. N. Engl. J. Med. 2001;344:1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 5.Alugupalli K.R., Gerstein R.M. Divide and conquer: division of labor by B-1 B cells. Immunity. 2005;23:1–2. doi: 10.1016/j.immuni.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Degn S.E., Jensenius J.C., Bjerre M. The lectin pathway and its implications in coagulation, infections and auto-immunity. Current Opin. Organ Transplant. 2010 doi: 10.1097/MOT.0b013e32834253df. Published online December 9, 2010. [DOI] [PubMed] [Google Scholar]
- 7.Super M., Thiel S., Lu J., Levinsky R.J., Turner M.W. Association of low levels of mannan-binding protein with a common defect of opsonisation. Lancet. 1989;2:1236–1239. doi: 10.1016/s0140-6736(89)91849-7. [DOI] [PubMed] [Google Scholar]
- 8.Thiel S. Complement activating soluble pattern recognition molecules with collagen-like regions, mannan-binding lectin, ficolins and associated proteins. Mol. Immunol. 2007;44:3875–3888. doi: 10.1016/j.molimm.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 9.Møller-Kristensen M., Thiel S., Sjöholm A., Matsushita M., Jensenius J.C. Cooperation between MASP-1 and MASP-2 in the generation of C3 convertase through the MBL pathway. Int. Immunol. 2007;19:141–149. doi: 10.1093/intimm/dxl131. [DOI] [PubMed] [Google Scholar]
- 10.Keshi H., Sakamoto T., Kawai T., Ohtani K., Katoh T., Jang S.J., Motomura W., Yoshizaki T., Fukuda M., Koyama S. Identification and characterization of a novel human collectin CL-K1. Microbiol. Immunol. 2006;50:1001–1013. doi: 10.1111/j.1348-0421.2006.tb03868.x. [DOI] [PubMed] [Google Scholar]
- 11.Hansen S., Selman L., Palaniyar N., Ziegler K., Brandt J., Kliem A., Jonasson M., Skjoedt M.-O., Nielsen O., Hartshorn K. Collectin 11 (CL-11, CL-K1) is a MASP-1/3-associated plasma collectin with microbial-binding activity. J. Immunol. 2010;185:6096–6104. doi: 10.4049/jimmunol.1002185. [DOI] [PubMed] [Google Scholar]
- 12.Dahl M.R., Thiel S., Matsushita M., Fujita T., Willis A.C., Christensen T., Vorup-Jensen T., Jensenius J.C. MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity. 2001;15:127–135. doi: 10.1016/s1074-7613(01)00161-3. [DOI] [PubMed] [Google Scholar]
- 13.Degn S.E., Hansen A.G., Steffensen R., Jacobsen C., Jensenius J.C., Thiel S. MAp44, a human protein associated with pattern recognition molecules of the complement system and regulating the lectin pathway of complement activation. J. Immunol. 2009;183:7371–7378. doi: 10.4049/jimmunol.0902388. [DOI] [PubMed] [Google Scholar]
- 14.Iwaki D., Kanno K., Takahashi M., Endo Y., Lynch N.J., Schwaeble W.J., Matsushita M., Okabe M., Fujita T. Small mannose-binding lectin-associated protein plays a regulatory role in the lectin complement pathway. J. Immunol. 2006;177:8626–8632. doi: 10.4049/jimmunol.177.12.8626. [DOI] [PubMed] [Google Scholar]
- 15.Carroll M. Immunology: exposure of an executioner. Nature. 2006;444:159–160. doi: 10.1038/nature05307. [DOI] [PubMed] [Google Scholar]
- 16.Janssen B.J.C., Christodoulidou A., McCarthy A., Lambris J.D., Gros P. Structure of C3b reveals conformational changes that underlie complement activity. Nature. 2006;444:213–216. doi: 10.1038/nature05172. [DOI] [PubMed] [Google Scholar]
- 17.Pangburn M.K., Ferreira V.P., Cortes C. Discrimination between host and pathogens by the complement system. Vaccine. 2008;26(Suppl 8):I15–I21. doi: 10.1016/j.vaccine.2008.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dodds A.W., Ren X.D., Willis A.C., Law S.K. The reaction mechanism of the internal thioester in the human complement component C4. Nature. 1996;379:177–179. doi: 10.1038/379177a0. [DOI] [PubMed] [Google Scholar]
- 19.Lachmann P.J. Preparing serum for functional complement assays. J. Immunol. Methods. 2010;352:195–197. doi: 10.1016/j.jim.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 20.de Córdoba S.R., de Jorge E.G. Translational mini-review series on complement factor H: genetics and disease associations of human complement factor H. Clin. Exp. Immunol. 2008;151:1–13. doi: 10.1111/j.1365-2249.2007.03552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zipfel P.F., Lauer N., Skerka C. The role of complement in AMD. Adv. Exp. Med. Biol. 2010;703:9–24. doi: 10.1007/978-1-4419-5635-4_2. [DOI] [PubMed] [Google Scholar]
- 22.Ryu E., Fridley B.L., Tosakulwong N., Bailey K.R., Edwards A.O. Genome-wide association analyses of genetic, phenotypic, and environmental risks in the age-related eye disease study. Mol. Vis. 2010;16:2811–2821. [PMC free article] [PubMed] [Google Scholar]
- 23.Gold B., Merriam J.E., Zernant J., Hancox L.S., Taiber A.J., Gehrs K., Cramer K., Neel J., Bergeron J., Barile G.R., AMD Genetics Clinical Study Group Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat. Genet. 2006;38:458–462. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yates J.R., Sepp T., Matharu B.K., Khan J.C., Thurlby D.A., Shahid H., Clayton D.G., Hayward C., Morgan J., Wright A.F., Genetic Factors in AMD Study Group Complement C3 variant and the risk of age-related macular degeneration. N. Engl. J. Med. 2007;357:553–561. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- 25.Hirt-Minkowski P., Dickenmann M., Schifferli J.A. Atypical hemolytic uremic syndrome: update on the complement system and what is new. Nephron Clin. Pract. 2010;114:c219–c235. doi: 10.1159/000276545. [DOI] [PubMed] [Google Scholar]
- 26.Botto M., Kirschfink M., Macor P., Pickering M.C., Würzner R., Tedesco F. Complement in human diseases: Lessons from complement deficiencies. Mol. Immunol. 2009;46:2774–2783. doi: 10.1016/j.molimm.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 27.Frank M.M. Complement disorders and hereditary angioedema. J. Allergy Clin. Immunol. 2010;125(2, Suppl 2):S262–S271. doi: 10.1016/j.jaci.2009.10.063. [DOI] [PubMed] [Google Scholar]
- 28.Ram S., Lewis L.A., Rice P.A. Infections of people with complement deficiencies and patients who have undergone splenectomy. Clin. Microbiol. Rev. 2010;23:740–780. doi: 10.1128/CMR.00048-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambris J.D., Ricklin D., Geisbrecht B.V. Complement evasion by human pathogens. Nat. Rev. Microbiol. 2008;6:132–142. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pickering M.C., Botto M., Taylor P.R., Lachmann P.J., Walport M.J. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv. Immunol. 2000;76:227–324. doi: 10.1016/s0065-2776(01)76021-x. [DOI] [PubMed] [Google Scholar]
- 31.Carroll M.C. A protective role for innate immunity in systemic lupus erythematosus. Nat. Rev. Immunol. 2004;4:825–831. doi: 10.1038/nri1456. [DOI] [PubMed] [Google Scholar]
- 32.Sjöholm A.G., Jönsson G., Braconier J.H., Sturfelt G., Truedsson L. Complement deficiency and disease: an update. Mol. Immunol. 2006;43:78–85. doi: 10.1016/j.molimm.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 33.Lipsker D., Hauptmann G. Cutaneous manifestations of complement deficiencies. Lupus. 2010;19:1096–1106. doi: 10.1177/0961203310373370. [DOI] [PubMed] [Google Scholar]
- 34.de Rooij B.J., van Hoek B., ten Hove W.R., Roos A., Bouwman L.H., Schaapherder A.F., Porte R.J., Daha M.R., van der Reijden J.J., Coenraad M.J. Lectin complement pathway gene profile of donor and recipient determine the risk of bacterial infections after orthotopic liver transplantation. Hepatology. 2010;52:1100–1110. doi: 10.1002/hep.23782. [DOI] [PubMed] [Google Scholar]
- 35.Vekemans M., Robinson J., Georgala A., Heymans C., Muanza F., Paesmans M., Klastersky J., Barette M., Meuleman N., Huet F. Low mannose-binding lectin concentration is associated with severe infection in patients with hematological cancer who are undergoing chemotherapy. Clin. Infect. Dis. 2007;44:1593–1601. doi: 10.1086/518171. [DOI] [PubMed] [Google Scholar]
- 36.Steffensen R., Thiel S., Varming K., Jersild C., Jensenius J.C. Detection of structural gene mutations and promoter polymorphisms in the mannan-binding lectin (MBL) gene by polymerase chain reaction with sequence-specific primers. J. Immunol. Methods. 2000;241:33–42. doi: 10.1016/s0022-1759(00)00198-8. [DOI] [PubMed] [Google Scholar]
- 37.Stengaard-Pedersen K., Thiel S., Gadjeva M., Møller-Kristensen M., Sørensen R., Jensen L.T., Sjøholm A.G., Fugger L., Jensenius J.C. Inherited deficiency of mannan-binding lectin-associated serine protease 2. N. Engl. J. Med. 2003;349:554–560. doi: 10.1056/NEJMoa022836. [DOI] [PubMed] [Google Scholar]
- 38.Garcia-Laorden M.I., Sole-Violan J., Rodriguez de Castro F., Aspa J., Briones M.L., Garcia-Saavedra A., Rajas O., Blanquer J., Caballero-Hidalgo A., Marcos-Ramos J.A. Mannose-binding lectin and mannose-binding lectin-associated serine protease 2 in susceptibility, severity, and outcome of pneumonia in adults. J. Allergy Clin. Immunol. 2008;122:368–374. doi: 10.1016/j.jaci.2008.05.037. 374, e1–e2. [DOI] [PubMed] [Google Scholar]
- 39.Olesen H.V., Jensenius J.C., Steffensen R., Thiel S., Schiøtz P.O. The mannan-binding lectin pathway and lung disease in cystic fibrosis—disfunction of mannan-binding lectin-associated serine protease 2 (MASP-2) may be a major modifier. Clin. Immunol. 2006;121:324–331. doi: 10.1016/j.clim.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 40.Schlapbach L.J., Thiel S., Kessler U., Ammann R.A., Aebi C., Jensenius J.C. Congenital H-ficolin deficiency in premature infants with severe necrotising enterocolitis. Gut. 2010 doi: 10.1136/gut.2010.226027. Published online October 22, 2010. [DOI] [PubMed] [Google Scholar]
- 41.Munthe-Fog L., Hummelshøj T., Honoré C., Madsen H.O., Permin H., Garred P. Immunodeficiency associated with FCN3 mutation and ficolin-3 deficiency. N. Engl. J. Med. 2009;360:2637–2644. doi: 10.1056/NEJMoa0900381. [DOI] [PubMed] [Google Scholar]
- 42.Sprong T., Roos D., Weemaes C., Neeleman C., Geesing C.L., Mollnes T.E., van Deuren M. Deficient alternative complement pathway activation due to factor D deficiency by 2 novel mutations in the complement factor D gene in a family with meningococcal infections. Blood. 2006;107:4865–4870. doi: 10.1182/blood-2005-07-2820. [DOI] [PubMed] [Google Scholar]
- 43.van den Bogaard R., Fijen C.A., Schipper M.G., de Galan L., Kuijper E.J., Mannens M.M. Molecular characterisation of 10 Dutch properdin type I deficient families: mutation analysis and X-inactivation studies. Eur. J. Hum. Genet. 2000;8:513–518. doi: 10.1038/sj.ejhg.5200496. [DOI] [PubMed] [Google Scholar]
- 44.Roumenina L.T., Loirat C., Dragon-Durey M.A., Halbwachs-Mecarelli L., Sautes-Fridman C., Fremeaux-Bacchi V. Alternative complement pathway assessment in patients with atypical HUS. J. Immunol. Methods. 2011;365:8–26. doi: 10.1016/j.jim.2010.12.020. [DOI] [PubMed] [Google Scholar]
- 45.Figueroa J.E., Densen P. Infectious diseases associated with complement deficiencies. Clin. Microbiol. Rev. 1991;4:359–395. doi: 10.1128/cmr.4.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khajoee V., Ihara K., Kira R., Takemoto M., Torisu H., Sakai Y., Guanjun J., Hee P.M., Tokunaga K., Hara T. Founder effect of the C9 R95X mutation in Orientals. Hum. Genet. 2003;112:244–248. doi: 10.1007/s00439-002-0870-8. [DOI] [PubMed] [Google Scholar]
- 47.Cugno M., Zanichelli A., Foieni F., Caccia S., Cicardi M. C1-inhibitor deficiency and angioedema: molecular mechanisms and clinical progress. Trends Mol. Med. 2009;15:69–78. doi: 10.1016/j.molmed.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 48.Rachidi S., Musallam K.M., Taher A.T. A closer look at paroxysmal nocturnal hemoglobinuria. Eur. J. Intern. Med. 2010;21:260–267. doi: 10.1016/j.ejim.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 49.Springer T.A., Thompson W.S., Miller L.J., Schmalstieg F.C., Anderson D.C. Inherited deficiency of the Mac-1, LFA-1, p150,95 glycoprotein family and its molecular basis. J. Exp. Med. 1984;160:1901–1918. doi: 10.1084/jem.160.6.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Springer T.A., Miller L.J., Anderson D.C. p150,95, the third member of the Mac-1, LFA-1 human leukocyte adhesion glycoprotein family. J. Immunol. 1986;136:240–245. [PubMed] [Google Scholar]
- 51.Pettigrew H.D., Teuber S.S., Gershwin M.E. Clinical significance of complement deficiencies. Ann. N Y Acad. Sci. 2009;1173:108–123. doi: 10.1111/j.1749-6632.2009.04633.x. [DOI] [PubMed] [Google Scholar]
- 52.Etzioni A. Defects in the leukocyte adhesion cascade. Clin. Rev. Allergy Immunol. 2010;38:54–60. doi: 10.1007/s12016-009-8132-3. [DOI] [PubMed] [Google Scholar]
- 53.Sirmaci A., Walsh T., Akay H., Spiliopoulos M., Sakalar Y.B., Hasanefendioğlu-Bayrak A., Duman D., Farooq A., King M.-C., Tekin M. MASP1 mutations in patients with facial, umbilical, coccygeal, and auditory findings of Carnevale, Malpuech, OSA, and Michels syndromes. Am. J. Hum. Genet. 2010;87:679–686. doi: 10.1016/j.ajhg.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mingarelli R., Castriota Scanderbeg A., Dallapiccola B. Two sisters with a syndrome of ocular, skeletal, and abdominal abnormalities (OSA syndrome) J. Med. Genet. 1996;33:884–886. doi: 10.1136/jmg.33.10.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Malpuech G., Demeocq F., Palcoux J.B., Vanlieferinghen P. A previously undescribed autosomal recessive multiple congenital anomalies/mental retardation (MCA/MR) syndrome with growth failure, lip/palate cleft(s), and urogenital anomalies. Am. J. Med. Genet. 1983;16:475–480. doi: 10.1002/ajmg.1320160405. [DOI] [PubMed] [Google Scholar]
- 56.Michels V.V., Hittner H.M., Beaudet A.L. A clefting syndrome with ocular anterior chamber defect and lid anomalies. J. Pediatr. 1978;93:444–446. doi: 10.1016/s0022-3476(78)81154-8. [DOI] [PubMed] [Google Scholar]
- 57.Carnevale F., Krajewska G., Fischetto R., Greco M.G., Bonvino A. Ptosis of eyelids, strabismus, diastasis recti, hip defect, cryptorchidism, and developmental delay in two sibs. Am. J. Med. Genet. 1989;33:186–189. doi: 10.1002/ajmg.1320330210. [DOI] [PubMed] [Google Scholar]
- 58.Titomanlio L., Bennaceur S., Bremond-Gignac D., Baumann C., Dupuy O., Verloes A. Michels syndrome, Carnevale syndrome, OSA syndrome, and Malpuech syndrome: variable expression of a single disorder (3MC syndrome)? Am. J. Med. Genet. A. 2005;137A:332–335. doi: 10.1002/ajmg.a.30878. [DOI] [PubMed] [Google Scholar]
- 59.Rooryck C., Diaz-Font A., Osborn D.P.S., Chabchoub E., Hernandez-Hernandez V., Shamseldin H., Kenny J., Waters A., Jenkins D., Kaissi A.A. Mutations in lectin complement pathway genes COLEC11 and MASP1 cause 3MC syndrome. Nat. Genet. 2011;43:197–203. doi: 10.1038/ng.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bendtsen J.D., Nielsen H., von Heijne G., Brunak S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 61.Bork P., Beckmann G. The CUB domain. A widespread module in developmentally regulated proteins. J. Mol. Biol. 1993;231:539–545. doi: 10.1006/jmbi.1993.1305. [DOI] [PubMed] [Google Scholar]
- 62.Gregory L.A., Thielens N.M., Arlaud G.J., Fontecilla-Camps J.C., Gaboriaud C. X-ray structure of the Ca2+-binding interaction domain of C1s. Insights into the assembly of the C1 complex of complement. J. Biol. Chem. 2003;278:32157–32164. doi: 10.1074/jbc.M305175200. [DOI] [PubMed] [Google Scholar]
- 63.Gregory L.A., Thielens N.M., Matsushita M., Sorensen R., Arlaud G.J., Fontecilla-Camps J.C., Gaboriaud C. The X-ray structure of human mannan-binding lectin-associated protein 19 (MAp19) and its interaction site with mannan-binding lectin and L-ficolin. J. Biol. Chem. 2004;279:29391–29397. doi: 10.1074/jbc.M402687200. [DOI] [PubMed] [Google Scholar]
- 64.Thielens N.M., Enrie K., Lacroix M., Jaquinod M., Hernandez J.F., Esser A.F., Arlaud G.J. The N-terminal CUB-epidermal growth factor module pair of human complement protease C1r binds Ca2+ with high affinity and mediates Ca2+-dependent interaction with C1s. J. Biol. Chem. 1999;274:9149–9159. doi: 10.1074/jbc.274.14.9149. [DOI] [PubMed] [Google Scholar]
- 65.Kirkitadze M.D., Barlow P.N. Structure and flexibility of the multiple domain proteins that regulate complement activation. Immunol. Rev. 2001;180:146–161. doi: 10.1034/j.1600-065x.2001.1800113.x. [DOI] [PubMed] [Google Scholar]
- 66.Bally I., Rossi V., Thielens N.M., Gaboriaud C., Arlaud G.J. Functional role of the linker between the complement control protein modules of complement protease C1s. J. Immunol. 2005;175:4536–4542. doi: 10.4049/jimmunol.175.7.4536. [DOI] [PubMed] [Google Scholar]
- 67.Gál P., Harmat V., Kocsis A., Bián T., Barna L., Ambrus G., Végh B., Balczer J., Sim R.B., Náray-Szabó G., Závodszky P. A true autoactivating enzyme. Structural insight into mannose-binding lectin-associated serine protease-2 activations. J. Biol. Chem. 2005;280:33435–33444. doi: 10.1074/jbc.M506051200. [DOI] [PubMed] [Google Scholar]
- 68.Harmat V., Gál P., Kardos J., Szilágyi K., Ambrus G., Végh B., Náray-Szabó G., Závodszky P. The structure of MBL-associated serine protease-2 reveals that identical substrate specificities of C1s and MASP-2 are realized through different sets of enzyme-substrate interactions. J. Mol. Biol. 2004;342:1533–1546. doi: 10.1016/j.jmb.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 69.Kardos J., Gál P., Szilágyi L., Thielens N.M., Szilágyi K., Lõrincz Z., Kulcsár P., Gráf L., Arlaud G.J., Závodszky P. The role of the individual domains in the structure and function of the catalytic region of a modular serine protease, C1r. J. Immunol. 2001;167:5202–5208. doi: 10.4049/jimmunol.167.9.5202. [DOI] [PubMed] [Google Scholar]
- 70.Lacroix M., Ebel C., Kardos J., Dobó J., Gál P., Závodszky P., Arlaud G.J., Thielens N.M. Assembly and enzymatic properties of the catalytic domain of human complement protease C1r. J. Biol. Chem. 2001;276:36233–36240. doi: 10.1074/jbc.M105688200. [DOI] [PubMed] [Google Scholar]
- 71.Rossi V., Teillet F., Thielens N.M., Bally I., Arlaud G.J. Functional characterization of complement proteases C1s/mannan-binding lectin-associated serine protease-2 (MASP-2) chimeras reveals the higher C4 recognition efficacy of the MASP-2 complement control protein modules. J. Biol. Chem. 2005;280:41811–41818. doi: 10.1074/jbc.M503813200. [DOI] [PubMed] [Google Scholar]
- 72.Nonaka M., Miyazawa S. Evolution of the initiating enzymes of the complement system. Genome Biol. 2002;3:S1001. doi: 10.1186/gb-2001-3-1-reviews1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takada F., Seki N., Matsuda Y., Takayama Y., Kawakami M. Localization of the genes for the 100-kDa complement-activating components of Ra-reactive factor (CRARF and Crarf) to human 3q27-q28 and mouse 16B2-B3. Genomics. 1995;25:757–759. doi: 10.1016/0888-7543(95)80027-j. [DOI] [PubMed] [Google Scholar]
- 74.Endo Y., Sato T., Matsushita M., Fujita T. Exon structure of the gene encoding the human mannose-binding protein-associated serine protease light chain: comparison with complement C1r and C1s genes. Int. Immunol. 1996;8:1355–1358. doi: 10.1093/intimm/8.9.1355. [DOI] [PubMed] [Google Scholar]
- 75.Endo Y., Takahashi M., Nakao M., Saiga H., Sekine H., Matsushita M., Nonaka M., Fujita T. Two lineages of mannose-binding lectin-associated serine protease (MASP) in vertebrates. J. Immunol. 1998;161:4924–4930. [PubMed] [Google Scholar]
- 76.Chang Y.F., Imam J.S., Wilkinson M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 77.Barash Y., Calarco J.A., Gao W., Pan Q., Wang X., Shai O., Blencowe B.J., Frey B.J. Deciphering the splicing code. Nature. 2010;465:53–59. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- 78.Dobó J., Harmat V., Beinrohr L., Sebestyén E., Závodszky P., Gál P. MASP-1, a promiscuous complement protease: structure of its catalytic region reveals the basis of its broad specificity. J. Immunol. 2009;183:1207–1214. doi: 10.4049/jimmunol.0901141. [DOI] [PubMed] [Google Scholar]
- 79.Cai G.Z., Griffin G.L., Senior R.M., Longmore W.J., Moxley M.A. Recombinant SP-D carbohydrate recognition domain is a chemoattractant for human neutrophils. Am. J. Physiol. 1999;276:L131–L136. doi: 10.1152/ajplung.1999.276.1.L131. [DOI] [PubMed] [Google Scholar]
- 80.Crouch E.C., Persson A., Griffin G.L., Chang D., Senior R.M. Interactions of pulmonary surfactant protein D (SP-D) with human blood leukocytes. Am. J. Respir. Cell Mol. Biol. 1995;12:410–415. doi: 10.1165/ajrcmb.12.4.7695920. [DOI] [PubMed] [Google Scholar]
- 81.Tino M.J., Wright J.R. Surfactant proteins A and D specifically stimulate directed actin-based responses in alveolar macrophages. Am. J. Physiol. 1999;276:L164–L174. doi: 10.1152/ajplung.1999.276.1.L164. [DOI] [PubMed] [Google Scholar]
- 82.Schagat T.L., Wofford J.A., Greene K.E., Wright J.R. Surfactant protein A differentially regulates peripheral and inflammatory neutrophil chemotaxis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003;284:L140–L147. doi: 10.1152/ajplung.00125.2002. [DOI] [PubMed] [Google Scholar]
- 83.Cortesio C.L., Jiang W. Mannan-binding lectin-associated serine protease 3 cleaves synthetic peptides and insulin-like growth factor-binding protein 5. Arch. Biochem. Biophys. 2006;449:164–170. doi: 10.1016/j.abb.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 84.Zheng B., Clarke J.B., Busby W.H., Duan C., Clemmons D.R. Insulin-like growth factor-binding protein-5 is cleaved by physiological concentrations of thrombin. Endocrinology. 1998;139:1708–1714. doi: 10.1210/endo.139.4.5945. [DOI] [PubMed] [Google Scholar]
- 85.Busby W.H., Jr., Nam T.J., Moralez A., Smith C., Jennings M., Clemmons D.R. The complement component C1s is the protease that accounts for cleavage of insulin-like growth factor-binding protein-5 in fibroblast medium. J. Biol. Chem. 2000;275:37638–37644. doi: 10.1074/jbc.M006107200. [DOI] [PubMed] [Google Scholar]
- 86.Gibson T.L., Cohen P. Inflammation-related neutrophil proteases, cathepsin G and elastase, function as insulin-like growth factor binding protein proteases. Growth Horm. IGF Res. 1999;9:241–253. doi: 10.1054/ghir.1999.0115. [DOI] [PubMed] [Google Scholar]
- 87.Beattie J., Allan G.J., Lochrie J.D., Flint D.J. Insulin-like growth factor-binding protein-5 (IGFBP-5): a critical member of the IGF axis. Biochem. J. 2006;395:1–19. doi: 10.1042/BJ20060086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takahashi M., Mori S., Shigeta S., Fujita T. Role of MBL-associated serine protease (MASP) on activation of the lectin complement pathway. Adv. Exp. Med. Biol. 2007;598:93–104. doi: 10.1007/978-0-387-71767-8_8. [DOI] [PubMed] [Google Scholar]
- 89.Takahashi M., Ishida Y., Iwaki D., Kanno K., Suzuki T., Endo Y., Homma Y., Fujita T. Essential role of mannose-binding lectin-associated serine protease-1 in activation of the complement factor D. J. Exp. Med. 2010;207:29–37. doi: 10.1084/jem.20090633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sato T., Endo Y., Matsushita M., Fujita T. Molecular characterization of a novel serine protease involved in activation of the complement system by mannose-binding protein. Int. Immunol. 1994;6:665–669. doi: 10.1093/intimm/6.4.665. [DOI] [PubMed] [Google Scholar]
- 91.Lynch N.J., Khan S.U., Stover C.M., Sandrini S.M., Marston D., Presanis J.S., Schwaeble W.J. Composition of the lectin pathway of complement in Gallus gallus: absence of mannan-binding lectin-associated serine protease-1 in birds. J. Immunol. 2005;174:4998–5006. doi: 10.4049/jimmunol.174.8.4998. [DOI] [PubMed] [Google Scholar]
- 92.Nonaka M., Kimura A. Genomic view of the evolution of the complement system. Immunogenetics. 2006;58:701–713. doi: 10.1007/s00251-006-0142-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wicher K.B., Fries E. Haptoglobin, a hemoglobin-binding plasma protein, is present in bony fish and mammals but not in frog and chicken. Proc. Natl. Acad. Sci. USA. 2006;103:4168–4173. doi: 10.1073/pnas.0508723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Krem M.M., Rose T., Di Cera E. The C-terminal sequence encodes function in serine proteases. J. Biol. Chem. 1999;274:28063–28066. doi: 10.1074/jbc.274.40.28063. [DOI] [PubMed] [Google Scholar]
- 95.Krem M.M., Rose T., Di Cera E. Sequence determinants of function and evolution in serine proteases. Trends Cardiovasc. Med. 2000;10:171–176. doi: 10.1016/s1050-1738(00)00068-2. [DOI] [PubMed] [Google Scholar]
- 96.Salvesen G., Enghild J.J. Zymogen activation specificity and genomic structures of human neutrophil elastase and cathepsin G reveal a new branch of the chymotrypsinogen superfamily of serine proteinases. Biomed. Biochim. Acta. 1991;50:665–671. [PubMed] [Google Scholar]
- 97.Patthy L. Evolution of blood coagulation and fibrinolysis. Blood Coagul. Fibrinolysis. 1990;1:153–166. [PubMed] [Google Scholar]
- 98.Iwanaga S., Kawabata S. Evolution and phylogeny of defense molecules associated with innate immunity in horseshoe crab. Front. Biosci. 1998;3:D973–D984. doi: 10.2741/a337. [DOI] [PubMed] [Google Scholar]
- 99.Muta T., Iwanaga S. The role of hemolymph coagulation in innate immunity. Curr. Opin. Immunol. 1996;8:41–47. doi: 10.1016/s0952-7915(96)80103-8. [DOI] [PubMed] [Google Scholar]
- 100.Stevens B., Allen N.J., Vazquez L.E., Howell G.R., Christopherson K.S., Nouri N., Micheva K.D., Mehalow A.K., Huberman A.D., Stafford B. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 101.Chu Y., Jin X., Parada I., Pesic A., Stevens B., Barres B., Prince D.A. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc. Natl. Acad. Sci. USA. 2010;107:7975–7980. doi: 10.1073/pnas.0913449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mayilyan K.R., Arnold J.N., Presanis J.S., Soghoyan A.F., Sim R.B. Increased complement classical and mannan-binding lectin pathway activities in schizophrenia. Neurosci. Lett. 2006;404:336–341. doi: 10.1016/j.neulet.2006.06.051. [DOI] [PubMed] [Google Scholar]
- 103.Jennings C. Developmental neurobiology. Death of a synapse. Nature. 1994;372:498–499. doi: 10.1038/372498a0. [DOI] [PubMed] [Google Scholar]
- 104.Schafer D.P., Stevens B. Synapse elimination during development and disease: immune molecules take centre stage. Biochem. Soc. Trans. 2010;38:476–481. doi: 10.1042/BST0380476. [DOI] [PubMed] [Google Scholar]
- 105.Yanamadala V., Friedlander R.M. Complement in neuroprotection and neurodegeneration. Trends Mol. Med. 2010;16:69–76. doi: 10.1016/j.molmed.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Thiel S., Frederiksen P.D., Jensenius J.C. Clinical manifestations of mannan-binding lectin deficiency. Mol. Immunol. 2006;43:86–96. doi: 10.1016/j.molimm.2005.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Israëls J., Frakking F.N., Kremer L.C., Offringa M., Kuijpers T.W., van de Wetering M.D. Mannose-binding lectin and infection risk in newborns: a systematic review. Arch. Dis. Child. Fetal Neonatal Ed. 2010;95:F452–F461. doi: 10.1136/adc.2009.172122. [DOI] [PubMed] [Google Scholar]
- 108.Møller-Kristensen M., Thiel S., Jensenius J.C. Mannan-Binding Lectin Polymorphisms and Infectious Diseases. In: Vasta G.R., Ahmed H., editors. Animal Lectins: A Functional View. C R C Press LLC; Boca Raton, FL: 2009. pp. 303–332. [Google Scholar]
- 109.Nilsson S.C., Trouw L.A., Renault N., Miteva M.A., Genel F., Zelazko M., Marquart H., Muller K., Sjöholm A.G., Truedsson L. Genetic, molecular and functional analyses of complement factor I deficiency. Eur. J. Immunol. 2009;39:310–323. doi: 10.1002/eji.200838702. [DOI] [PubMed] [Google Scholar]
- 110.Nita I.M., Genel F., Nilsson S.C., Smart J., Truedsson L., Choo S., Blom A.M. Molecular characterization of two novel cases of complete complement inhibitor Factor I deficiency. Mol. Immunol. 2011;48:1068–1072. doi: 10.1016/j.molimm.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 111.S Reis E., Falcão D.A., Isaac L. Clinical aspects and molecular basis of primary deficiencies of complement component C3 and its regulatory proteins factor I and factor H. Scand. J. Immunol. 2006;63:155–168. doi: 10.1111/j.1365-3083.2006.01729.x. [DOI] [PubMed] [Google Scholar]
- 112.Ghannam A., Pernollet M., Fauquert J.L., Monnier N., Ponard D., Villiers M.B., Péguet-Navarro J., Tridon A., Lunardi J., Gerlier D., Drouet C. Human C3 deficiency associated with impairments in dendritic cell differentiation, memory B cells, and regulatory T cells. J. Immunol. 2008;181:5158–5166. doi: 10.4049/jimmunol.181.7.5158. [DOI] [PubMed] [Google Scholar]
- 113.Aguilar-Ramirez P., Reis E.S., Florido M.P., Barbosa A.S., Farah C.S., Costa-Carvalho B.T., Isaac L. Skipping of exon 30 in C5 gene results in complete human C5 deficiency and demonstrates the importance of C5d and CUB domains for stability. Mol. Immunol. 2009;46:2116–2123. doi: 10.1016/j.molimm.2008.10.035. [DOI] [PubMed] [Google Scholar]
- 114.López-Lera A., Garrido S., de la Cruz R.M., Fontán G., López-Trascasa M. Molecular characterization of three new mutations causing C5 deficiency in two non-related families. Mol. Immunol. 2009;46:2340–2347. doi: 10.1016/j.molimm.2009.03.026. [DOI] [PubMed] [Google Scholar]
- 115.Zhu Z., Atkinson T.P., Hovanky K.T., Boppana S.B., Dai Y.L., Densen P., Go R.C., Jablecki J.S., Volanakis J.E. High prevalence of complement component C6 deficiency among African-Americans in the south-eastern USA. Clin. Exp. Immunol. 2000;119:305–310. doi: 10.1046/j.1365-2249.2000.01113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Würzner R. Deficiencies of the complement MAC II gene cluster (C6, C7, C9): is subtotal C6 deficiency of particular evolutionary benefit? Clin. Exp. Immunol. 2003;133:156–159. doi: 10.1046/j.1365-2249.2003.02230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Parham K.L., Roberts A., Thomas A., Würzner R., Henderson H.E., Potter P.C., Morgan B.P., Orren A. Prevalence of mutations leading to complete C6 deficiency (C6Q0) in the Western Cape, South Africa and detection of novel mutations leading to C6Q0 in an Irish family. Mol. Immunol. 2007;44:2756–2760. doi: 10.1016/j.molimm.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 118.Rameix-Welti M.A., Régnier C.H., Bienaimé F., Blouin J., Schifferli J., Fridman W.H., Sautès-Fridman C., Frémeaux-Bacchi V. Hereditary complement C7 deficiency in nine families: subtotal C7 deficiency revisited. Eur. J. Immunol. 2007;37:1377–1385. doi: 10.1002/eji.200636812. [DOI] [PubMed] [Google Scholar]
- 119.Barroso S., López-Trascasa M., Merino D., Alvarez A.J., Núñez-Roldán A., Sánchez B. C7 deficiency and meningococcal infection susceptibility in two spanish families. Scand. J. Immunol. 2010;72:38–43. doi: 10.1111/j.1365-3083.2010.02403.x. [DOI] [PubMed] [Google Scholar]
- 120.Arnold D.F., Roberts A.G., Thomas A., Ferry B., Morgan B.P., Chapel H. A novel mutation in a patient with a deficiency of the eighth component of complement associated with recurrent meningococcal meningitis. J. Clin. Immunol. 2009;29:691–695. doi: 10.1007/s10875-009-9295-7. [DOI] [PubMed] [Google Scholar]
- 121.Pickering M.C., Cook H.T. Translational mini-review series on complement factor H: renal diseases associated with complement factor H: novel insights from humans and animals. Clin. Exp. Immunol. 2008;151:210–230. doi: 10.1111/j.1365-2249.2007.03574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Servais A., Noël L.H., Dragon-Durey M.A., Gübler M.C., Rémy P., Buob D., Cordonnier C., Makdassi R., Jaber W., Boulanger E. Heterogeneous pattern of renal disease associated with homozygous Factor H deficiency. Hum. Pathol. 2011 doi: 10.1016/j.humpath.2010.11.023. Published online March 9, 2011. [DOI] [PubMed] [Google Scholar]
- 123.Firszt R., Frank M.M. An overview of novel therapies for acute hereditary angioedema. Am. J. Clin. Dermatol. 2010;11:383–388. doi: 10.2165/11537030-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 124.Roos D., Meischl C., de Boer M., Simsek S., Weening R.S., Sanal O., Tezcan I., Güngör T., Law S.K. Genetic analysis of patients with leukocyte adhesion deficiency: genomic sequencing reveals otherwise undetectable mutations. Exp. Hematol. 2002;30:252–261. doi: 10.1016/s0301-472x(01)00782-2. [DOI] [PubMed] [Google Scholar]
- 125.Bernard Cher T.H., Chan H.S., Klein G.F., Jabkowski J., Schadenböck-Kranzl G., Zach O., Roca X., Law S.K. A novel 3′ splice-site mutation and a novel gross deletion in leukocyte adhesion deficiency (LAD)-1. Biochem. Biophys. Res. Commun. 2011;404:1099–1104. doi: 10.1016/j.bbrc.2010.12.124. [DOI] [PubMed] [Google Scholar]
- 126.Kavanagh D., Richards A., Atkinson J. Complement regulatory genes and hemolytic uremic syndromes. Annu. Rev. Med. 2008;59:293–309. doi: 10.1146/annurev.med.59.060106.185110. [DOI] [PubMed] [Google Scholar]
- 127.Fremeaux-Bacchi V., Kemp E.J., Goodship J.A., Dragon-Durey M.A., Strain L., Loirat C., Deng H.W., Goodship T.H. The development of atypical haemolytic-uraemic syndrome is influenced by susceptibility factors in factor H and membrane cofactor protein: evidence from two independent cohorts. J. Med. Genet. 2005;42:852–856. doi: 10.1136/jmg.2005.030783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kelly R.J., Hill A., Arnold L.M., Brooksbank G.L., Richards S.J., Cullen M., Mitchell L.D., Cohen D.R., Gregory W.M., Hillmen P. Long term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood. 2011 doi: 10.1182/blood-2011-02-333997. Published online April 1, 2011. [DOI] [PubMed] [Google Scholar]