

Abstract
Congenital muscular dystrophy is a heterogeneous group of inherited muscle diseases characterized clinically by muscle weakness and hypotonia in early infancy. A number of genes harboring causative mutations have been identified, but several cases of congenital muscular dystrophy remain molecularly unresolved. We examined 15 individuals with a congenital muscular dystrophy characterized by early-onset muscle wasting, mental retardation, and peculiar enlarged mitochondria that are prevalent toward the periphery of the fibers but are sparse in the center on muscle biopsy, and we have identified homozygous or compound heterozygous mutations in the gene encoding choline kinase beta (CHKB). This is the first enzymatic step in a biosynthetic pathway for phosphatidylcholine, the most abundant phospholipid in eukaryotes. In muscle of three affected individuals with nonsense mutations, choline kinase activities were undetectable, and phosphatidylcholine levels were decreased. We identified the human disease caused by disruption of a phospholipid de novo biosynthetic pathway, demonstrating the pivotal role of phosphatidylcholine in muscle and brain.
Main Text
A spontaneous mutant mouse with a neonatal-onset autosomal-recessive rostral-to-caudal muscular dystrophy (rmd mouse) due to a loss-of-function mutation in choline kinase beta (Chkb) was identified in 2006.1 Interestingly, rmd mice exhibit a unique mitochondrial morphology in muscle fibers, which show enlarged mitochondria at the periphery of the fiber but none at the center (Figure S1). These features are similar to those seen in a congenital muscular dystrophy (CMD) that we previously reported in four Japanese individuals.2 We therefore screened 15 genetically undiagnosed cases of CMD with fairly homogenous clinical features (Table 1) for mutations in choline kinase beta (CHKB); we included the four cases from in our previous study in these 15 cases. Features included peculiar mitochondrial changes in muscle as well as motor delay followed by the appearance of severe mental retardation and microcephaly without structural brain abnormalities (Figure 1 and Table 1).
Table 1.
Summary of Clinical and Laboratory Features
|
Phenotypic Findings |
Muscle Pathology |
Mutations |
|||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Individual | Sex | Origin | Age at Last Follow-Up | Floppy at Birth | Walk Alone | Serum Creatine Kinase (IU/liter) | Head Circumference (percentile) | Mental Retardation | Seizure | Cardiomyopathy | Skin Change | Age at Muscle Biopsy | Necrotic Fiber | Regenerative Fiber | Endomysial Fibrosis | Mitochondrial Enlargement | Status | cDNA | Consequence | Exon | Literature ref. on phenotype |
| 1 | F | Japanese | died at 13 yr | + | 2 yr 6 mo | 370 | ND | + | - | + | - | 7 yr3 mo | + | + | + | + | homo | c.810T>A | p.Tyr270X | 7 | 2 |
| 2 | M | Japanese | died at 23 yr | + | 1 yr 9 mo | 190–2676 | 25–50 | + | + | + | - | 1 yr 2 mo | + | + | + | + | homo | c.810T>A | p.Tyr270X | 7 | 2 |
| 3 | F | Japanese | 28 yr | + | 1 yr 6 mo | 502 | ND | + | + | + | - | 8 yr | + | + | + | + | het | c.116C>A | p.Ser39X | 1 | 2 |
| het | c.458dup | p.Leu153PhefsX57 | 3 | 2 | |||||||||||||||||
| 4 | M | Japanese | 22 yr | + | 2 yr 6 mo | 230 | 3–10 | + | + | - | - | 4 yr 11 mo | + | + | + | + | het | c.116C>A | p.Ser39X | 1 | |
| het | c.458dup | p.Leu153PhefsX57 | 3 | ||||||||||||||||||
| 5 | M | Turkish | 7 yr | - | 2 yr 6 mo | 843 | <3 | + | - | - | + | 6 yr | ± | + | + | + | homo | c.611_612insC | p.Thr205AsnfsX5 | 5 | |
| 6a | M | Turkish | died at 2 yr 6 mo | + | no | 258 | <3 | + | - | + | - | 1 yr 3 mo | ± | ± | + | + | homo | c.922C>T | p.Gln308X | 8 | |
| 7 | F | Turkish | 2 yr | - | no | 368 | 3-10 | + | - | -b | - | 9 mo | - | ± | + | + | homo | c.847G>A | p.Glu283Lys | 8 | |
| 8 | M | Turkish | 13 yr | ND | 2 yr | 1122 | ND | + | - | - | - | 12 yr 10 mo | ± | ± | + | + | homo | c.1130 G>T | p.Arg377Leu | 11 | |
| 9 | F | Turkish | 17 yr | + | 3 yr | 2669 | <3 | + | - | ND | - | 17 yr | ± | ± | + | + | homo | c.554_562del | p.Pro185_Trp187del | 4 | |
| 10 | F | Turkish | 16 yr | + | 3 yr | 1103 | <3 | + | - | -c | + | 3 yr | - | ± | + | + | homo | c.677+1G>A | ND | 5 | |
| 11 | F | Turkish | 3 yr 3 mo | + | no | 497 | 10-25 | + | - | ND | - | 3 yr | ± | - | + | + | homo | c.677+1G>A | ND | 5 | |
| 12 | F | Turkish | 5 yr | - | 3 yr 6 mo | 467 | 25–50 | + | - | -d | + | 4 yr 6 mo | ± | + | + | + | homo | c.677+1G>A | ND | 5 | |
| 13 | M | Turkish | 3 yr 6 mo | + | no | 428 | <3 | + | - | + | + | 3 yr | + | + | + | + | homo | c.1031+1G>A | aberrant splicing | 9 | |
| 14 | F | Turkish | 6 yr 4 mo | - | 1 yr 3 mo | 1606 | 3–10 | + | - | + | - | 4 yr | + | + | + | + | homo | c.1031+1G>A | ND | 9 | |
| 15 | M | British | died at 8 yr | - | 3 yr 4 mo | 607–1715 | <3 | + | - | + | + | 2 yr 2 mo | + | - | + | + | homo | c.852_859del | p.Trp284X | 8 | |
Detailed clinical information for individual 1 to 4 was previously described (2). Eleven CHKB mutations were identified in 15 affected individuals. All exhibited generalized muscle hypotonia and weakness from early infancy. Ambulation was delayed, and gait in those who achieved walking was limited. In addition, all displayed marked mental retardation, and most never acquired meaningful language. Microcephaly with head circumferences at or below the 3rd to 10th percentile was observed in most cases. Cranial magnetic resonance imaging showed no developmental brain defects. Six individuals had dilated cardiomyopathy, and two had cardiac anomaly. Individuals 1, 2, 6, and 15 died from cardiomyopathy at ages 13 yr, 23 yr, 2 yr 6 mo, and 8 yr, respectively. No one had respiratory insufficiency. Ichthyosiform skin changes were frequent. All showed mildly to moderately elevated serum creatine kinase (CK) levels. Individuals 7 and 9 also had homozygous single-nucleotide substitutions, c.902C>T (p.Thr301Ile) and c.983A>G (p.Gln328Arg), respectively. CHK activities of recombinant CHK-β proteins with p.Thr301Ile and p.Gln328Arg were only mildly decreased (Figure S2), suggesting these are likely to be neutral polymorphisms or only mildly hypomorphic mutations. Individuals 10, 11, and 12, who have same c.677+1G>A mutation, and individuals 13 and 14, who have same c.1031+1G>A mutation, are not siblings. Abbreviations are as follows: ND, not determined; p, percentile; F, female; and M, male.
An affected sibling had ichthyosis and died at age 6 years with cardiomyopathy.
Patent ductus arteriosus.
Atrial septal defect.
Mitral valve prolapse.
Figure 1.
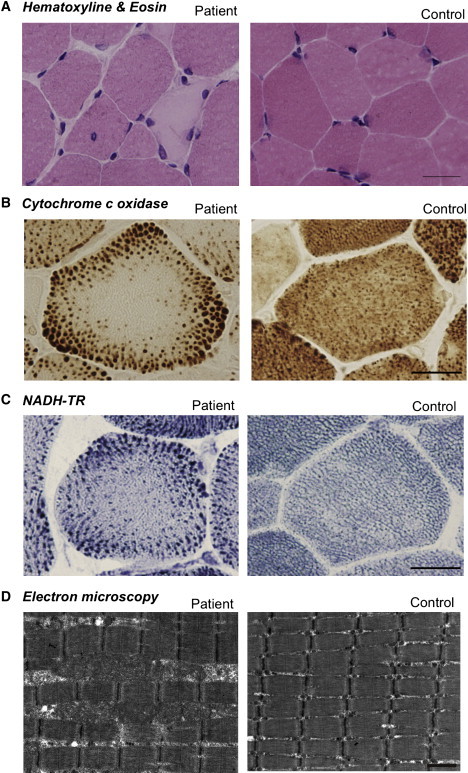
Muscle Pathology of the Affected Individuals
Cross-sections of muscle fiber from a human control and individual 4.
(A) On H&E staining, nonspecific dystrophic features with necrotic and regenerating fibers, internalized nuclei, and endomysial fibrosis are seen. The scale bar represents 25 μm.
(B) On cytochrome c oxidase staining, enlarged mitochondria at the periphery and central areas devoid of mitochondria were seen. The scale bar represents 20 μm.
(C) On NADH-TR staining, the intermyofibrillar network was preserved even in the central areas that are devoid of mitochondria, suggesting the presence of myofibrils and only absence of mitochondria. The scale bar represents 20 μm.
(D) Electron microscopy confirmed enlarged mitochondria. The scale bar represents 1 μm.
All clinical materials used in this study were obtained for diagnostic purposes with written informed consent. The study was approved by the Ethical Committee of the National Center of Neurology and Psychiatry. All mouse protocols were approved by the Ethical Review Committee on the Care and Use of Rodents in the National Institute of Neuroscience, National Center of Neurology and Psychiatry. For muscle pathology, samples of skeletal muscle were obtained from biceps brachii or quadriceps femoris in humans and from quadriceps femoris muscle in 8-week-old rmd mice. Muscles were frozen and sectioned at a thickness of 10 μm according to standard procedures, and a battery of routine histochemical stains, including hematoxylin and eosin (H&E), modified Gomori trichrome (mGT), NADH-tetrazolium reductase (NADH-TR), succinate dehydrogenase (SDH), cytochrome c oxidase (COX), and Oil Red O, were analyzed. For electron microscopic analysis, muscles were fixed as previously described,3 and ultra-thin sections were observed at 120kV or 80kV. All affected individuals exhibited nonspecific dystrophic features (Figure 1A). However, in mGT, NADH-TR, SDH, and COX staining, prominent mitochondria at the periphery as well as central areas devoid of mitochondria were seen (Figures 1B and 1C). Oil Red O staining was unremarkable (data not shown). Electron microscopy confirmed enlarged mitochondria (Figure 1D).
We directly sequenced all exons and their flanking intronic regions in CHKB (MIM 612395, NM_005198.4, GenBank Gene ID 1120) in genomic DNA extracted from individuals' peripheral lymphocytes. All 15 individuals in three different populations (Japanese, Turkish, and British) had homozygous or compound heterozygous mutations in CHKB (Table 1). Among a total of 11 mutations identified, six were nonsense, two were missense, one was a 3 amino acid deletion, and two were splice-site mutations. The six nonsense mutations, c.116C>A (p.Ser39X), c.458dup (p.Leu153PhefsX57), c.611_612insC (p.Thr205AsnfsX5), c.810T>A (p.Tyr270X), c.852_859del (p.Trp284X), and c.922C>T (p.Gln308X), were predicted to truncate the protein and eliminate highly conserved domains of CHK.4,5 Individuals 1 and 2 (unrelated, Japanese) had the same homozygous nonsense mutation of c.810T>A (p.Tyr270X). Individual 2's mother, who was healthy, had the heterozygous c.810T>A (p.Tyr270X) mutation. Unfortunately, a DNA sample from the father of individual 2 was not available. DNA samples from other family members of individual 1 and 2 were not available. Individuals 3 and 4 (siblings, Japanese) had the same compound heterozygous mutation c.116C>A (p.Ser39X) and c.458dup (p.Leu153PhefsX57). Both parents were healthy, and the father was heterozygous for mutation c.116C>A (p.Ser39X), whereas the mother was heterozygous for mutation c. 458dup (p.Leu153PhefsX57), thus confirming a recessive inheritance pattern. These mutations cosegregated with the disease phenotype in all family members tested.
We therefore measured CHK activity in biopsied muscle. For all biochemical analyses, because of the limiting amounts of remaining tissue, biopsied muscle samples were available only from individuals 2, 3, and 4. Biopsied muscle samples from these three individuals were homogenized in 3 volumes of 20 mM Tris-HCl (pH 7.5), 154 mM KCl, and 1 mM phenylmethanesulfonyl fluoride with a sonicator (MISONIX), and supernatant fractions (105,000 × g, 60 min) were prepared and analyzed for CHK activity as previously described.6 Similar to muscles of rmd mice,1 muscles from individuals 2, 3, and 4, who carried homozygous or compound heterozygous nonsense mutations, did not have any detectable CHK activity (Figure 2A). Individuals 7, 8, and 9 had homozygous missense mutations c.847G>A (p.Glu283Lys) and c.1130 G>T (p.Arg377Leu) and a homozygous 3 amino acid deletion, c.554_562 del (p.Pro185_Trp187del), respectively.We screened 210 control chromosomes for the identified missense mutations and small in-frame deletion by direct sequencing or single-strand conformation polymorphism (SSCP) analysis. SSCP was performed with Gene Gel Excel (GE Healthcare) as previously described.7 These missense mutations and this small in-frame deletion were not identified in control chromosomes. To elucidate the pathogenesis of these substitutions, we measured CHK activity in recombinant proteins with mutations. We cloned the open reading frame of CHKB into pGEM-T easy (Promega), then subcloned it into pET15b (Novagen) to make His-tagged CHK-β.8 Each mutation was induced by site-directed mutagenesis.7 Plasmids were transformed into Escherichia coli strain BL21 (DE3) and inoculated at 20°C to an OD600 of approximately 0.5, and the addition of 0.4 mM isopropyl-β-D-thiogalactopyranoside induced expression. The His-tagged CHK-β proteins were subjected to affinity purification on a nickel column (GE Healthcare) and eluted with 20 mM Tris-HCl (pH 7.4), 0.5 M NaCl, 300 mM imidazol, and 1 mM phenylmethanesulfonyl fluoride, and 25 ng protein was analyzed for CHK activity. CHK activity of recombinant proteins with these mutations decreased to less than 30% of wild-type CHK activity, suggesting that these mutations are causative in these individuals (Figure S2). For individual 13, who had a mutation at the splice site of the exon-intron border after exon 9 (c.1031+1G>A), we also analyzed cDNA sequences. Exons 4 through 10 were amplified from the first-strand cDNAs, and direct sequencing followed. cDNA analysis of CHKB in skeletal muscle from individual 13 showed four splicing variants, all of which remove consensus domains for CHKB (Figure S3). This suggests the same loss-of-function mechanism in humans and rmd mice.
Figure 2.
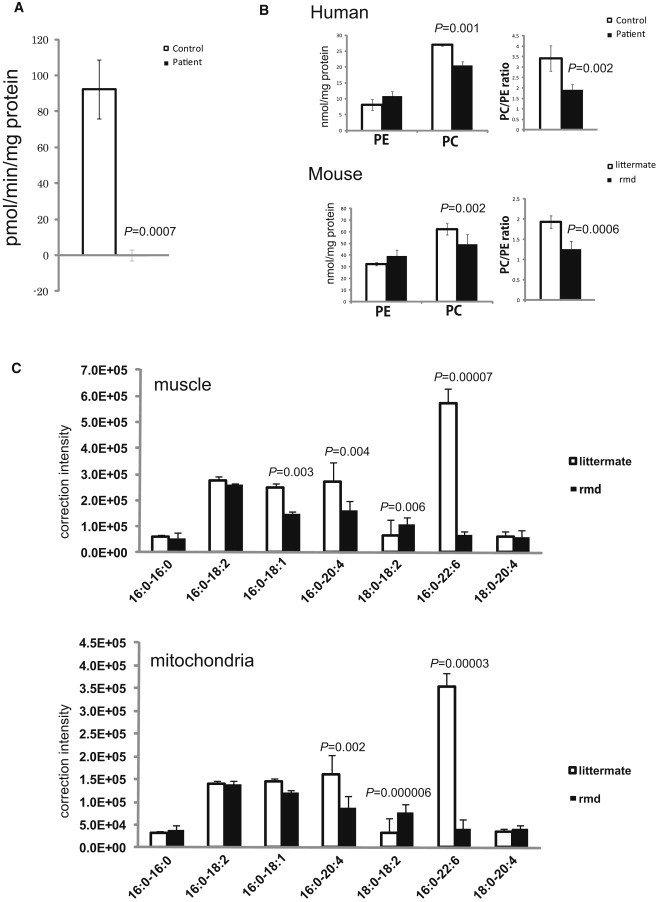
Choline Kinase Activity and Phospholipid Analyses
(A) In muscle tissue from individuals 2, 3, and 4, CHK activity cannot be detected (n = 3). Data represent the mean of three individuals.
(B) PC and PE content in frozen biopsied muscle tissues from individuals 2, 3, and 4 and hindlimb muscles from 8-week-old rmd mice (n = 4) and control littermates (n = 5) were analyzed by thin-layer chromatography followed by phosphorus analysis. PC and the PC/PE ratio are significantly decreased in affected individuals and rmd mice (n = 3 for humans, n = 4 for rmd mice, n = 5 for littermates).
(C) Fatty acid composition of PC molecular species in muscles and isolated mitochondria from hindlimb muscles of rmd mice are determined by electrospray ionization mass spectrometry (ESI-MS). We observed that 34:1-PC (16:0-18:1), 36:4-PC (16:0-20:4), and 38:6-PC (16:0-22:6) species are significantly decreased, whereas 36:2-PC (18:0-18:2) is increased in rmd muscle. Similarly, in isolated mitochondria from hindlimb muscle, 36:4-PC (16:0-20:4) and 38:6-PC (16:0-22:6) species are decreased, whereas 36:2-PC (18:0-18:2) is increased. In muscle and isolated mitochondria, the 38:6-PC molecular species is profoundly decreased (n = 6 for muscle, n = 5 for isolated mitochondria).
Mitochondria from skeletal muscles of whole hindlimbs of rmd mice were isolated by the differential centrifugation method. Fresh muscle was minced and homogenized with a motor-driven Teflon pestle homogenizer with ice-cold mitochondrial isolation buffer (10 mM Tris-HCl [pH 7.2], 320 mM sucrose, 1mM EDTA, 1mM DTT, 1 mM PMSF, 1 mg/ml BSA, and protease inhibitor cocktail [Roche]) and centrifuged at 1,500 × g for 5 min. The supernatant fraction was centrifuged at 15,000 × g for 20 min, the pellet was resuspended in mitochondrial isolation buffer, and the centrifugation/resuspension was repeated twice more.
All data are presented as means ± standard deviation (SD). Means were compared by analysis with a two-tailed t test via R software version 2.11.0.
Because phosphorylation of choline by CHK is the first enzymatic step for phosphatidylcholine (PC) biosynthesis,9 we anticipated that PC content should be altered in affected individuals' muscles. Phosphatidylcholine (PC), phosphatidylethanolamine (PE), and total phospholipid amounts were measured in biopsied muscles from individuals 2, 3, and 4 and in leg muscles from 8-week-old rmd mice by either one-dimensional or two-dimensional thin-layer chromatography (TLC) followed by phosphorus analysis.10,11 As expected, PC levels decreased in affected individuals' skeletal muscle (Figure 2B), as they did in rmd mice (Figure 2B and Sher et al.1), suggesting that the CMDs due to CHKB mutations in humans and rmd mice are not only pathologically but also pathomechanistically similar.
PC is present in all tissues and accounts for around 50% of phospholipids in biological membranes in eukaryotes. Selective tissue involvement can be explained by the different tissue distribution of CHK isoforms. There are two CHK isoforms: CHK-α and CHK-β, encoded by distinct genes, CHKA (MIM 118491) and CHKB, respectively. They are known to form both homodimers and heterodimers, with differential tissue distribution.12 In mice, disruption of Chka causes embryonic lethality,13 suggesting the importance of CHK-α in embryonic development. In skeletal muscles from rmd mice, CHK activity is absent, and PC levels are decreased.1 In other tissues, however, CHK activity is only mildly decreased, PC levels are not altered, and no obvious pathological change is seen.1 CHK activity in skeletal muscle from individuals 2, 3, and 4 is barely detectable, and PC levels are significantly decreased, suggesting that CHK-β is the major isoform in human skeletal muscle. In support of this notion, CHK-α was not detected in human muscle (Figure S4). These results suggest that muscular dystrophy in affected individuals and rmd mice is caused by a defect in muscle PC biosynthesis. In addition, in rmd mice, hindlimb muscles are more significantly affected than forelimb muscles.1 This is most likely explained by the fact that CHK activity is detected, though decreased, in forelimb muscles in rmd mice as a result of the continued post-natal expression of Chka.14 This indicates that the severity of muscle involvement is determined by the degree of deficiency of CHK activity.
Generally, phospholipids have saturated or monounsaturated fatty acids at the sn-1 position and polyunsaturated fatty acids at the sn-2 position of glycerol backbone.15 It has been shown that phospholipids have tissue-specific fatty acid composition.15 For example, heart PC and muscle PC mainly contain docosahexaenoic acid (22:6) (Nakanishi et al.15 and Figure 2C), but liver PC includes various fatty acids.15 NanoESI-MS analyses of PC molecular species in muscle and isolated mitochondria were performed with a 4000Q TRAP (AB SCIEX, Foster City, CA, USA) and a chip-based ionization source, TriVersa NanoMate (Advion BioSystems, Ithaca, NY, USA).16 Quadriceps femoris (hindlimb) and Triceps (forelimb) muscle from affected rmd mice and littermate controls were frozen with liquid nitrogen, and total lipid was extracted by the Bligh and Dyer method.10 The ion spray voltage was set at −1.25kV, gas pressure at 0.3 pound per square inch (psi), and flow rates at 200 nl/min. The scan range was set at m/z 400∼1200, declustering potential at −100V, collision energies at −35∼−45V, and resolutions at Q1 and Q3 “unit.” The mobile phase composition was chloroform:methanol (1/2) containing 5 mM ammonium formate and was normalized to the muscle weight. The total lipids were directly subjected by flow injection, and selectivity was analyzed by neutral loss scanning of the polar head group for PC in negative-ion mode.17 Interestingly, there was a 10-fold decrease (9.8%) in the 16:0-22:6-PC levels versus the control in rmd hindlimb muscle and also in muscle mitochondria (Figure 2C), indicating the importance of the PC de novo synthesis pathway for maintaining not only PC levels but also fatty acid composition of PC molecular species. Similarly, in forelimb muscle 16:0-22:6 PC levels were also decreased in comparison to the control, but to a milder extent (18.2%), suggesting an association between severity of muscle damage and fatty acid composition alteration of PC (data not shown). In rmd mice, it has been shown that muscle PC can be delivered from plasma lipoprotein,18 suggesting that non-decreased PC molecular species might be derived from the plasma, whereas 16:0-22:6 PC might be synthesized only in muscle (and possibly in brain). However, confirmation of this requires further studies.
Individuals with CHKB mutations have severe mental retardation in addition to the muscular dystrophy. Interestingly, polymorphisms near the CHKB locus and decreased CHKB expression have been associated with narcolepsy with cataplexy, suggesting a link between CHK-β activity and the maintenance of normal brain function in humans.19 Furthermore, brain damage in pneumococcal infection has been attributed to the inhibition of de novo PC synthesis, suggesting the importance of PC synthesis for the brain.20 Our data provide evidence that altered phospholipid biosynthesis is a causative agent for a human congenital muscular dystrophy, and further studies will elucidate the detailed molecular mechanisms of the disease in both muscle and brain.
Acknowledgments
We are grateful to the patients and their family for their participation, to Megumu Ogawa, Etsuko Keduka, Yuriko Kure, Mieko Ohnishi, Kaoru Tatezawa, and Kazu Iwasawa (National Center of Neurology and Psychiatry) for their technical assistance, to Naoki Kondou and Hiroyuki Taguchi (Kao Corporation) for their kind support on mass analysis, to Osamu Fujino and Kiyoshi Takahashi (Department of Pediatrics, Nippon Medical School) for providing patient information, and to Ken Inoue (National Center of Neurology and Psychiatry) for thoughtful comments on genetics. This study was supported partly by the Research on Psychiatric and Neurological Diseases and Mental Health of Health and Labour Sciences research grants; partly by Research on Intractable Diseases of Health and Labor Sciences research grants; partly by a Research Grant for Nervous and Mental Disorders (20B-12, 20B-13) from the Ministry of Health, Labour and Welfare; partly by an Intramural Research Grant (23-4, 23-5) for Neurological and Psychiatric Disorders from NCNP; partly by KAKENHI (20390250, 22791019); partly by Research on Publicly Essential Drugs and Medical Devices of Health and Labor Sciences research grants; partly by the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NIBIO); and partly by a grant from the Japan Foundation for Neuroscience and Mental Health. G.A.C. and R.B S. were supported in part by a National Institutes of Health grant (AR-49043 to G.A.C.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GenBank, http://www.ncbi.nlm.nih.gov/Genbank
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
R software version 2.11.0, http://www.r-project.org/
References
- 1.Sher R.B., Aoyama C., Huebsch K.A., Ji S., Kerner J., Yang Y., Frankel W.N., Hoppel C.L., Wood P.A., Vance D.E., Cox G.A. A rostrocaudal muscular dystrophy caused by a defect in choline kinase beta, the first enzyme in phosphatidylcholine biosynthesis. J. Biol. Chem. 2006;281:4938–4948. doi: 10.1074/jbc.M512578200. [DOI] [PubMed] [Google Scholar]
- 2.Nishino I., Kobayashi O., Goto Y., Kurihara M., Kumagai K., Fujita T., Hashimoto K., Horai S., Nonaka I. A new congenital muscular dystrophy with mitochondrial structural abnormalities. Muscle Nerve. 1998;21:40–47. doi: 10.1002/(sici)1097-4598(199801)21:1<40::aid-mus6>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 3.Hayashi Y.K., Matsuda C., Ogawa M., Goto K., Tominaga K., Mitsuhashi S., Park Y.E., Nonaka I., Hino-Fukuyo N., Haginoya K. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J. Clin. Invest. 2009;119:2623–2633. doi: 10.1172/JCI38660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liao H., Aoyama C., Ishidate K., Teraoka H. Deletion and alanine mutation analyses for the formation of active homo- or hetero-dimer complexes of mouse choline kinase-α and -β. Biochim. Biophys. Acta. 2006;1761:111–120. doi: 10.1016/j.bbalip.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Aoyama C., Yamazaki N., Terada H., Ishidate K. Structure and characterization of the genes for murine choline/ethanolamine kinase isozymes alpha and beta. J. Lipid Res. 2000;41:452–464. [PubMed] [Google Scholar]
- 6.Ishidate K., Nakazawa Y. Choline/ethanolamine kinase from rat kidney. Methods Enzymol. 1992;209:121–134. doi: 10.1016/0076-6879(92)09016-v. [DOI] [PubMed] [Google Scholar]
- 7.Matsumoto H., Hayashi Y.K., Kim D.S., Ogawa M., Murakami T., Noguchi S., Nonaka I., Nakazawa T., Matsuo T., Futagami S. Congenital muscular dystrophy with glycosylation defects of α-dystroglycan in Japan. Neuromuscul. Disord. 2005;15:342–348. doi: 10.1016/j.nmd.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 8.Mitsuhashi H., Futai E., Sasagawa N., Hayashi Y., Nishino I., Ishiura S. Csk-homologous kinase interacts with SHPS-1 and enhances neurite outgrowth of PC12 cells. J. Neurochem. 2008;105:101–112. doi: 10.1111/j.1471-4159.2007.05121.x. [DOI] [PubMed] [Google Scholar]
- 9.Aoyama C., Liao H., Ishidate K. Structure and function of choline kinase isoforms in mammalian cells. Prog. Lipid Res. 2004;43:266–281. doi: 10.1016/j.plipres.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Bligh E.G., Dyer W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 11.Rouser G., Fkeischer S., Yamamoto A. Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids. 1970;5:494–496. doi: 10.1007/BF02531316. [DOI] [PubMed] [Google Scholar]
- 12.Aoyama C., Ohtani A., Ishidate K. Expression and characterization of the active molecular forms of choline/ethanolamine kinase-α and -β in mouse tissues, including carbon tetrachloride-induced liver. Biochem. J. 2002;363:777–784. doi: 10.1042/0264-6021:3630777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu G., Aoyama C., Young S.G., Vance D.E. Early embryonic lethality caused by disruption of the gene for choline kinase alpha, the first enzyme in phosphatidylcholine biosynthesis. J. Biol. Chem. 2008;283:1456–1462. doi: 10.1074/jbc.M708766200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu G., Sher R.B., Cox G.A., Vance D.E. Differential expression of choline kinase isoforms in skeletal muscle explains the phenotypic variability in the rostrocaudal muscular dystrophy mouse. Biochim. Biophys. Acta. 2010;1801:446–454. doi: 10.1016/j.bbalip.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 15.Nakanishi H., Iida Y., Shimizu T., Taguchi R. Separation and quantification of sn-1 and sn-2 fatty acid positional isomers in phosphatidylcholine by RPLC-ESIMS/MS. J. Biochem. 2010;147:245–256. doi: 10.1093/jb/mvp171. [DOI] [PubMed] [Google Scholar]
- 16.Ikeda K., Mutoh M., Teraoka N., Nakanishi H., Wakabayashi K., Taguchi R. Increase of oxidant-related triglycerides and phosphatidylcholines in serum and small intestinal mucosa during development of intestinal polyp formation in Min mice. Cancer Sci. 2011;102:79–87. doi: 10.1111/j.1349-7006.2010.01754.x. [DOI] [PubMed] [Google Scholar]
- 17.Taguchi R., Houjou T., Nakanishi H., Yamazaki T., Ishida M., Imagawa M., Shimizu T. Focused lipidomics by tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2005;823:26–36. doi: 10.1016/j.jchromb.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 18.Wu G., Sher R.B., Cox G.A., Vance D.E. Understanding the muscular dystrophy caused by deletion of choline kinase beta in mice. Biochim. Biophys. Acta. 2009;1791:347–356. doi: 10.1016/j.bbalip.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 19.Miyagawa T., Kawashima M., Nishida N., Ohashi J., Kimura R., Fujimoto A., Shimada M., Morishita S., Shigeta T., Lin L. Variant between CPT1B and CHKB associated with susceptibility to narcolepsy. Nat. Genet. 2008;40:1324–1328. doi: 10.1038/ng.231. [DOI] [PubMed] [Google Scholar]
- 20.Zweigner J., Jackowski S., Smith S.H., Van Der Merwe M., Weber J.R., Tuomanen E.I. Bacterial inhibition of phosphatidylcholine synthesis triggers apoptosis in the brain. J. Exp. Med. 2004;200:99–106. doi: 10.1084/jem.20032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.