

Abstract
Autosomal-recessive immunodeficiency, centromeric instability, and facial anomalies (ICF) syndrome is mainly characterized by recurrent, often fatal, respiratory and gastrointestinal infections. About 50% of patients carry mutations in the DNA methyltransferase 3B gene (DNMT3B) (ICF1). The remaining patients carry unknown genetic defects (ICF2) but share with ICF1 patients the same immunological and epigenetic features, including hypomethylation of juxtacentromeric repeat sequences. We performed homozygosity mapping in five unrelated ICF2 patients with consanguineous parents and then performed whole-exome sequencing in one of these patients and Sanger sequencing in all to identify mutations in the zinc-finger- and BTB (bric-a-bric, tramtrack, broad complex)-domain-containing 24 (ZBTB24) gene in four consanguineously descended ICF2 patients. Additionally, we found ZBTB24 mutations in an affected sibling pair and in one patient for whom it was not known whether his parents were consanguineous. ZBTB24 belongs to a large family of transcriptional repressors that include members, such as BCL6 and PATZ1, with prominent regulatory roles in hematopoietic development and malignancy. These data thus indicate that ZBTB24 is involved in DNA methylation of juxtacentromeric DNA and in B cell development and/or B and T cell interactions. Because ZBTB24 is a putative DNA-binding protein highly expressed in the lymphoid lineage, we predict that by studying the molecular function of ZBTB24, we will improve our understanding of the molecular pathophysiology of ICF syndrome and of lymphocyte biology in general.
Main Text
Patients with autosomal-recessive immunodeficiency, centromeric instability, and facial anomalies (ICF [MIM 242860]) syndrome suffer from recurrent and often fatal respiratory and gastrointestinal infections because of hypogammaglobulinemia, even in the presence of normal B cell counts. ICF syndrome also presents with facial anomalies, most notably a broad and flat nasal bridge, hypertelorism, and epicanthal folds. The molecular hallmark of this condition is the presence of chromosomal abnormalities in phytohemagglutinin (PHA)-stimulated peripheral blood lymphocytes (PBLs) of patients. These abnormalities are not restricted to hematopoietic cell lineages and include the formation of radial chromosomes involving the juxtacentromeric heterochromatin regions of chromosomes 1, 9, and 16.1,2
Approximately half of ICF cases have mutations in the DNA methyltransferase 3B gene (DNMT3B [MIM 602900)] at chromosomal locus 20q11.2 (these cases are said to have ICF1).3,4 DNMT3B is one of the three main mammalian DNA methyltransferases that play an important but not exclusive role in de novo DNA methylation.5 In ICF1 patients, the enzymatic activity of DNMT3B is strongly, but not completely, reduced,6 and as a consequence, specific genomic regions of ICF patients show significant loss of DNA methylation. This hypomethylation is most strongly observed in satellite 2 (Sat2) repeats on the long arm of chromosomes 1 and 16, satellite 3 (Sat3) repeats on the long arm of chromosome 9, and in the nonsatellite repeats NBL2 on acrocentric chromosomes and D4Z4 in the subtelomeres of the long arms of chromosomes 4 and 10.7,8
In the remainder of ICF patients, no DNMT3B mutations can be found (these cases are said to have ICF2).9 These patients are clinically identical to ICF1 patients,2 but they have additional DNA hypomethylation of α-satellite repeats.10 In two ICF2 patients, the DNMT3B locus was excluded by haplotype analysis,9 and the most common splice variant of DNMT3B mRNA was detected in several ICF2 patients.10 Furthermore, in one ICF2 patient no mutations were identified in two putative DNMT3B promoter regions,11 and no evidence for changes in DNMT3B splicing was obtained in another ICF2 patient.12 We therefore hypothesized that the ICF syndrome is genetically heterogeneous.
For our studies genomic DNA was available from 11 ICF2 patients (Figure 1A and Table 1).
Figure 1.
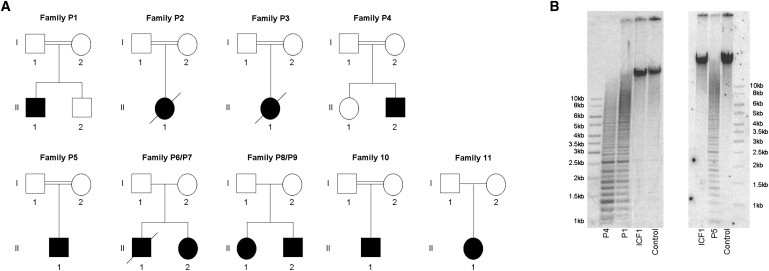
Genetics and Epigenetics of ICF2 Patients
(A) Pedigrees of 11 ICF2 families studied. The parents of P1, P2, P3, and P4 were first-degree relatives. The great-grandmothers of patient P5 were sisters. The history of consanguinity for P10 was unknown before this study. However, our SNP array data show large regions of homozygosity, suggesting that the parents of P10 are related (data not shown).
(B) α-satellite hypomethylation in ICF2 patients. Southern-blot analysis of the α-satellite repeat on chromosome 9 in a control individual, an ICF1 patient with a DNMT3B mutation, two ICF2 patients (P1 and P5) with a mutation in ZBTB24, and one ICF2 patient (P4) in whom no mutations in DNMT3B or ZBTB24 were identified and in whom the DNMT3B and ZBTB24 loci were excluded by homozygosity mapping. DNA samples (2 μg) were digested overnight with the restriction enzyme HhaI, separated by linear gel electrophoresis on a 0.8% agarose gel. Subsequently, Southern blotting of the DNA and hybridization of the resulting membrane with a probe targeted to the α-satellite repeat on chromosome 9 were performed.48
Table 1.
Clinical and Genetic Data of ICF2 Patients
| Case, Gender | Clinical Phenotype | Status | Origin | ZBTB24 Mutation | Number in Hagleitner et al.2 |
|---|---|---|---|---|---|
| P1, male | agammaglobulinemia, facial anomalies, motor development delay, mental retardation | 4 years old | Turkish | p.Asn306IlefsX4 (c.917delA) | – |
| P2, female | agammaglobulinemia, facial anomalies, motor development delay, mental retardation | died at age 13 | Scottish | p.Ser16X (c.47C>G) | 11 |
| P3, female | agammaglobulinemia, facial anomalies, motor development delay, mental retardation | died at age 11 | Turkish | p.Arg320X (c.958C>T) | 17 |
| P4, male | agammaglobulinemia, facial anomalies, mental retardation | 6 years old | Turkish | No mutation identified | – |
| P5, male | agammaglobulinemia, facial anomalies, mental retardation | 13 years old | Turkish | p.Val168SerfsX28 (c.501dup) | – |
| P6, male | agammaglobulinemia, facial anomalies, mental retardation | died at age 4 | German | p.Ser278X (c.833C>G) and p.Cys408Gly (c.1222T>G) | 37 |
| P7, female | hypogammaglobulinemia, facial anomalies, motor development delay, mental retardation | 9 years old | German | p.Ser278X (c.833C>G) and p.Cys408Gly (c.1222T>G) | 38 |
| P8, female | agammaglobulinemia, facial anomalies, normal intelligence | 11 years old | British | No mutation identified | 34 |
| P9, male | agammaglobulinemia, facial anomalies, normal intelligence | 4 years old | British | No mutation identified | – |
| P10, male | hypogammaglobulinemia, facial anomalies, mental retardation | adult | Italian | p.Arg457X (c.1369C>T) | 6 |
| P11, female | IgM deficiency, facial anomalies, mental retardation | adult | Italian | No mutation identified | 13 |
All studies were carried out with informed consent of the probands or their legal guardians and were approved by the institutional ethic review board of the Radboud University Nijmegen Medical Centre. P1 (II.1 in family P1 in Figure 1A) is a consanguineously descended Turkish male patient with agammaglobulinemia, facial anomalies, motor development delay, and mental retardation. We found no DNMT3B mutation, but we did observe α-satellite hypomethylation indicative of ICF2 (Figure 1B).10 P2 (II.1 in family P2 in Figure 1A) was a consanguineously descended Scottish female patient who presented with agammaglobulinemia, facial anomalies, motor development delay, and mental retardation and who was previously included in the homozygosity mapping study aimed at identifying the gene in which variation is associated with ICF1.13 However, she did not show autozygosity for the DNMT3B locus, and no DNMT3B mutation was identified.14 P3 (II.1 in family P3 in Figure 1A) was a consanguineously descended Dutch female patient of Turkish descent. This patient was also part of the patient cohort used for identification of the gene in which variation is associated with ICF1, and she did not show autozygosity for the DNMT3B locus either.14 In addition, no DNMT3B mutation was identified.9 She presented with agammaglobulinemia, facial anomalies, motor development delay, and mental retardation.14 Because of limited material, we could not perform α-satellite DNA methylation analysis in patients P2 and P3. P4 (II.2 in family P4 in Figure 1A) is a consanguineously descended Turkish male patient with agammaglobulinemia, facial anomalies, and mental retardation. We observed α-satellite DNA hypomethylation in this patient (Figure 1B); we could not detect a mutation in DNMT3B. P5 (II.1 in family P5 in Figure 1A) is a consanguineously descended Belgian male patient of Turkish descent who presented with agammaglobulinemia, facial anomalies, and mental retardation. We did not identify a DNMT3B mutation, but we did observe α-satellite hypomethylation (Figure 1B). P6 (II.1 in family P6/P7 in Figure 1A) and P7 (II.2 in family P6/P7 in Figure 1A) were a German brother and sister with no known history of consanguinity. Both presented with facial anomalies, mental retardation, and agammaglobulinemia or hypogammaglobulinemia. In addition, P7 presented with a motor development delay. No DNMT3B mutation was observed in either patient, but both patients showed α-satellite hypomethylation.15 P8 (II.1 in family P8/P9 in Figure 1A) and P9 (II.2 in family P8/P9 in Figure 1A) are a British sister and brother who have no history of consanguinity. Initially, both presented with agammaglobulinemia and facial anomalies. The agammaglobulinemia in P8 and P9 was corrected by hematopoietic stem cell transplantation.2,16 We did not find DNMT3B mutations, but we observed α-satellite hypomethylation (data not shown). P10 (II.1 in family P10 in Figure 1A) is an Italian male patient who has no known history of consanguinity and who presented with hypogammaglobulinemia, facial anomalies, and mental retardation.17 We did not detect a DNMT3B mutation. Finally, P11 (II.1 in family P11 in Figure 1A) is an Italian female patient with a IgM deficiency, facial anomalies, mental retardation, and no known history of consanguinity.18,19 No mutation was found in DNMT3B. Because of scarcity of material, no α-satellite DNA methylation analysis was performed in P10 and P11.
To identify genes in which variation is associated with ICF, we performed homozygosity mapping20 in the five unrelated ICF2 patients born to consanguineous parents (P1–P5). We used the Sentrix HumanHap-300 Genotyping BeadChips containing 317,503 loci of HapMap I and II TagSNP (Illumina). Seven hundred and fifty nanograms of DNA was processed with the Infinium II Whole-Genome Genotyping Assay (Illumina). In short, DNA was amplified, fragmented, precipitated with ethanol, and resuspended. Next, the DNA was applied to the BeadChip and incubated overnight; enzymatic base extension, fluorescent staining of the beads, and detection of fluorescent intensities by the BeadArray Reader (Illumina) followed. Finally, to detect regions of homozygosity, we assessed B allele frequencies for all SNPs by using BeadStudio version 3.2 (Illumina). Large regions of homozygosity were identified in all patients and confirmed that the patients were each of consanguineous descent. However, no region of homozygosity was shared by all patients, suggesting that ICF2 itself is also heterogeneous.
Next, we performed exome sequencing21 to study all exons in the genome of one consanguineously descended ICF2 patient (P3).14 Five micrograms of DNA was fragmented to yield 200–300 bp fragments with the Bioruptor (Diagenode). End repair of DNA fragments was subsequently carried out per the manufacturer's instructions (New England Biolabs). Next, paired-end adaptor oligonucleotides (Illumina) were added to the ends of the DNA fragments, and the resulting DNA-adaptor-ligated fragments were hybridized for 72 hr against the Sequence Capture Human Exome 2.1M Array (Roche NimbleGen); washing of unbound fragments and elution of the exome-enriched pool of DNA followed. The eluted DNA fragments were amplified with adaptor-specific primers (Illumina) and Phusion DNA polymerase (Finnzymes). Finally, the exome-enriched DNA fragments were sequenced with the Genome Analyzer IIx system (Illumina). We prepared paired-end flow cells on the supplied cluster station according to the manufacturer's instructions. The resulting reads were aligned to the reference human genome (hg 19, NCBI build 37) by Bowtie software,22 and sequence variants were identified with SAMtools software.23 In a homozygous region that was shared between four of the five patients of consanguineous descent, we identified a homozygous sequence variant (c.958C>T; NM_014797.2) in exon 3 of the zinc-finger- and BTB (bric-a-bric, tramtrack, broad complex)-domain-containing (ZBTB24) gene. This sequence variant creating a premature stop codon at amino acid position 320 (p.Arg320X; NP_055612.2) was confirmed by PCR amplification of exon 3 of ZBTB24 and subsequent Sanger sequencing (Figure 2A).
Figure 2.
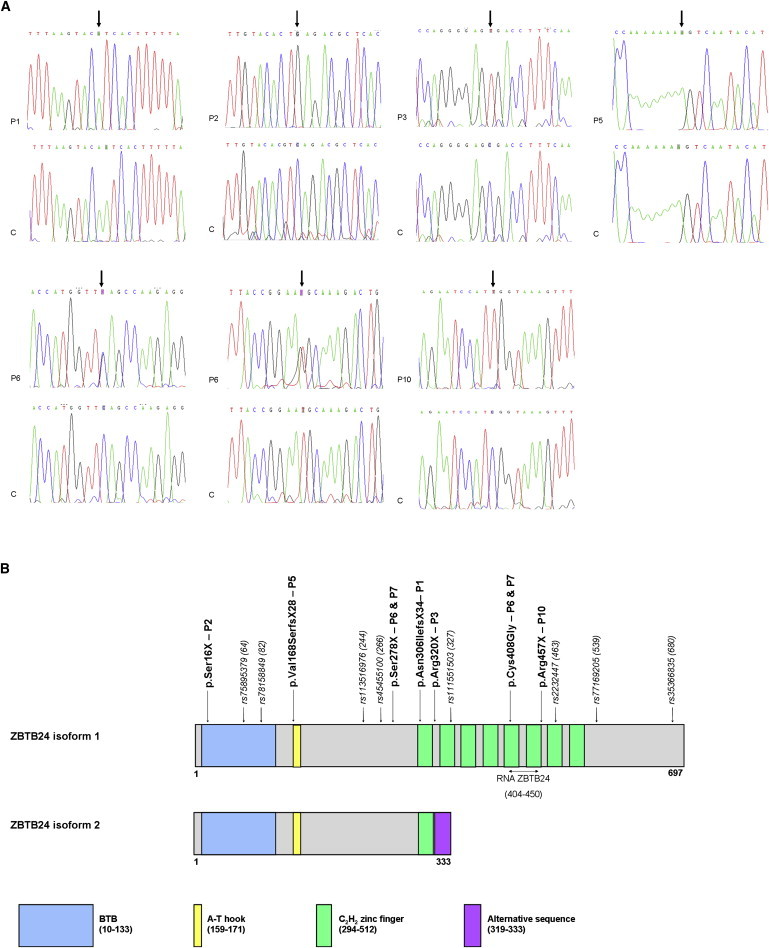
ZBTB24 Mutations at the DNA and Protein Level
(A) Sanger sequencing results of seven ICF2 patients with ZBTB24 mutations and an unrelated control individual. P1 carries a homozygous 1 bp deletion resulting in a frameshift and a premature stop codon three amino acids downstream (p.Asn306IlefsX4). Both parents are heterozygous carriers for the mutation, and the healthy brother is heterozygous as well (data not shown). P2 is homozygous for a nonsense mutation resulting in a serine-to-stop conversion (p.Ser16X). P3 carries a homozygous sequence variant creating a premature stop codon (p.Arg320X). Both parents are heterozygous carriers for the mutation (data not shown). P5 carries a homozygous 1 bp insertion resulting in a frameshift and a premature stop codon 27 amino acids downstream (p.Val168SerfsX28). P6 and P7 are compound heterozygous for a serine-to-stop nonsense mutation (p.Ser278X) and a cysteine-to-glycine missense mutation (p.Cys408Gly). P10 is homozygous for a nonsense mutation resulting in an arginine-to-stop conversion (p.Arg457X). The mother is a heterozygous carrier for the mutation (data not shown).
(B) A schematic of the two isoforms of the ZBTB24 protein. The full-length ZBTB24 protein (isoform 1) contains a BTB domain (amino acids 10–133), a DNA-binding A-T hook domain (amino acids 159–171), and eight C2H2 zinc finger domains of 23 amino acids each (amino acids 294–512). The shorter isoform 2 lacks seven C2H2 zinc finger domains, and the last 15 amino acids are an alternative sequence (an alternative stop codon is used in the intron between exon 2 and exon 3). RNA ZBTB24 denotes the location of the primers used for RT-PCR. At least eight SNPs have been found in the coding sequence of ZBTB24. The numbers and locations of these SNPs are indicated as well.
Finally, we performed mutation analysis in all ZBTB24 coding exons by PCR amplification and Sanger sequencing in the remaining four consanguineously descended ICF2 patients (P1, P2, P4, and P5) and in the six additional ICF2 patients for whom genomic DNA was available (P6–P11). The primers used for the ZBTB24 mutation analysis are shown in Table S1, available online. We identified homozygous ZBTB24 sequence variants in three additional consanguineously descended patients (P1, P2, and P5) and in one patient for whom consanguineous descent had not been previously established (P10). An affected sibling pair of nonconsanguineous descent was compound heterozygote for ZBTB24 mutations (P6 and P7). We did not find any ZBTB24 sequence variants in an affected sibling pair (P8 and P9), one patient of nonconsanguineous descent (P11), or one consanguineously descended patient (P4), confirming additional genetic heterogeneity. P4 was also excluded for the ZBTB24 locus based on our homozygosity mapping data.
P1 is homozygous for a 1 bp deletion in exon 2 (c.917delA; NM_014797.2), resulting in a frameshift and a premature stop codon three amino acids downstream (p.Asn306IlefsX4; NP_055612.2). P2 carries a homozygous nonsense mutation in exon 2 (c.47C>G; NM_014797.2), resulting in a serine-to-stop conversion (p.Ser16X; NP_055612.2). P5 is homozygous for a 1 bp duplication in exon 2 (c.501dup; NM_014797.2), resulting in a frameshift and a premature stop codon 27 amino acids downstream (p.Val168SerfsX28; NP_055612.2). P10 carries a homozygous nonsense mutation in exon 6 (c.1369C>T; NM_014797.2), resulting in an arginine-to-stop conversion (p.Arg457X; NP_055612.2). Finally, P6 and P7 carried a serine-to-stop nonsense mutation in exon 2 (c.833C>G, p.Ser278X; NM_014797.2 and NP_055612.2) and a cysteine-to-glycine missense mutation in exon 5 (c.1222T>G, p.Cys408Gly; NM_014797.2 and NP_055612.2); the latter mutation affects a conserved cysteine in one of the eight C2H2 zinc fingers of the ZBTB24 protein. In conclusion, all but one of the identified mutations created premature stop codons in ZBTB24. This suggests that ICF2 is caused by loss-of-function mutations in ZBTB24. The identified ZBTB24 mutations are summarized in Table 1; Sanger sequencing results are shown in Figure 2A, and the locations of the mutations in ZBTB24 are shown in Figure 2B. Finally, in Table S2 the clinical and immunological characteristics that the ICF2 patients with ZBTB24 mutations had at diagnosis are summarized and compared with those that ICF1 patients with DNMT3B mutations had at diagnosis, and no difference in phenotype between ICF1 and ICF2 patients is revealed.
Using quantitative reverse transcriptase PCR (qRT-PCR), we studied full-length ZBTB24 mRNA expression in human primary cell cultures, tissues, and cell lines. In addition, we combined fluorescence-activated cell sorting (FACS) analysis and qRT-PCR to study ZBTB24 mRNA expression in subpopulations of normal human B cells, CD4+ T cells, and natural killer (NK) cells more specifically. We obtained human peripheral blood from two healthy blood-bank donors after receiving informed consent. Peripheral blood mononuclear cells (PBMCs) were isolated by centrifugation over Ficoll-Hypaque density gradients. CD19+ B cells were purified by positive selection with the Dynabeads CD19 Pan B (Invitrogen) and then detached by DETACHaBEAD CD19 (Invitrogen). Purified B cells were then stained with a combination of CD19percp-cy5.5 (SJ25C1), CD27phycoerythrin (L128), IgMallophycocyanin (G20-127), and IgDfluorescein isothiocyanate (IA6-2) antibodies. CD4+ T cells were purified by positive selection with the Dynabeads FlowComp Human CD4 T cell isolation kit (Invitrogen). Purified T cells were then incubated with CD4pacific blue (RPA-T4), CD45RAfluorescein isothiocyanate (L48), and CCR7alexa fluor 647 (3D12) antibodies. All antibodies were obtained from BD Biosciences. The following cell fractions were sorted with a FACS Aria cell sorter (Becton Dickinson) with greater than 98% purity: total CD19+, naive CD19+CD27-IgD+, unswitched memory CD19+CD27+IgD+IgM+ and switched memory CD19+CD27+IgD−IgM− B cells, total CD4+, naive CD4+CD45RA+CCR7+, and memory CD4+CD45RA− T cells. The analysis and sort gates were restricted to the small lymphocyte gate as determined by their characteristic forward and side-scatter properties. NK cells were isolated with the MACS NK Cell Isolation Kit (Miltenyi Biotech). NK cell purity was at least 95% of isolated cells. Next, total RNA was isolated from a minimum of 100,000 cells with the NucleoSpin RNA II kit (Macherey-Nagel). The RNA sample was then used for cDNA synthesis, which was performed with random hexamers (MBI-Fermentas) and RevertAid H Minus Reverse Transcriptase (MBI-Fermentas). The final cDNA sample was used in a RT-PCR reaction containing iQ SYBR Green Supermix (Bio-Rad) and 1 μM of both forward and reverse primers. All RT-PCR reactions were performed in duplicate with the MyiQ2 Two-Color Real-Time PCR Detection System (Bio-Rad). Primer sequences are shown in Table S3. These experiments showed that ZBTB24 is ubiquitously expressed, and the highest expression levels are in B cell subpopulations, most notably in naive B cells (Figure 3). This corroborates the role of ZBTB24 in B cell differentiation and is consistent with the observed B cell defects in ICF patients.24
Figure 3.

ZBTB24 Expression
Full-length ZBTB24 expression levels were measured in human primary cell cultures (ES cells and fibroblasts; n = 2), in human tissues (alveoli, kidney, liver, and thymus; n = 1), in human cell lines (HeLa, HCT116, JVM-2, Jurkat, LCL, MOLT-4, Raji, Ramos, Tera-2, and THP-1; n = 1), in human B cells (naive B cells [CD19+CD27−IgD+)], unswitched memory B cells [CD19+CD27+IgD+IgM+], switched memory B cells [CD19+CD27+IgD−IgM−], and total B cells [CD19+]; n = 2), in human CD4+ T cells (naive CD4+ T cells [CD4+CD45RA+CCR7+], memory CD4+ T cells [CD4+CD45RA−], and total CD4+ T cells [CD4+]; n = 2), and in human NK cells (CD3−CD56+; n = 2). Expression levels were measured in duplicate and are presented relative to the expression levels of the three housekeeping genes GAPDH, GUSB, and ACTB, which were also measured in duplicate. Data are presented as mean ± standard deviation.
ZBTB24 (also known as ZNF450, BIF1, or PATZ2) contains a BTB domain, a DNA-binding A-T hook domain, and eight C2H2 zinc finger domains (Figure 2B). BTB domains are found near the N terminus of several zinc finger proteins. These domains mediate homomeric or heteromeric dimerization between proteins25 and transcriptional repression26 and have been shown to interact with histone deacteylase corepressor complexes such as NCoR (nuclear receptor corepressor), SMRT (silencing mediator of retinoic acid and thyroid hormone receptor),27 and BCoR (BCL6 corepressor).28 A-T hook domains are DNA-binding motifs present in many proteins, including the chromosomal high-mobility group A (HMGA) proteins that are involved in gene transcription, retroviral integration, neoplastic transformation, and cancer cell metastasis.29
ZBTB24 is a member of a family of more than 40 human ZBTB proteins, several of which play prominent roles in hematologic differentiation.30 A well-studied ZBTB protein is BCL6 (ZBTB27 [MIM 109565]), which was initially identified as deregulated in certain B cell lymphomas by chromosomal translocations.31 BCL6 seems to be required for germinal center (GC) formation,32 represses gene functioning during the terminal differentiation of B cells into plasma cells,33 and binds to the p53 promoter, thereby downregulating p53 expression in GC B lymphocytes and lymphomas.34 Other ZBTB proteins, including BCL6B (ZBTB28 [MIM 608992]),35,36 PATZ1 (ZBTB19 [MIM 605165]),37 ZBTB7B (MIM 607646),38,39 and ZBTB32 (MIM 605859),40,41 have been implicated in hematologic development and malignancies.42
Explaining the molecular mechanisms underlying ICF1 syndrome has been a challenge. DNMT3B is a ubiquitously expressed enzyme central to mammalian biology. Dnmt3b-knockout mice are embryonic lethal, and ICF1 syndrome alleles should be considered hypomorphs.5 Two mechanisms have been proposed to link DNMT3B defects to compromised B cell immune responses. First, DNA methylation of promoter regions of genes involved in B cell differentiation might be disturbed. Indeed, a number of transcriptionally deregulated genes have been identified in EBV-transformed B cell lines of ICF patients, but only a small proportion of them have significantly altered DNA methylation promoter levels.43,44 Nevertheless, it is conceivable that altered chromatin structures of some master genes of B cell development contribute to ICF pathology. A second mechanism postulates that the altered chromatin structure of juxtacentromeric heterochromatin and changes in the nuclear architecture lead to defects in gene expression through effects in trans. Indeed, several reports support this hypothesis by demonstrating altered nuclear architecture and changes in chromosome territories with consequences for gene expression.45,46
Intriguingly, mice carrying ICF-like missense mutations in Dnmt3b recapitulate some, but not all, molecular and clinical features of ICF syndrome. These mice have hypomethylated repetitive DNA, low body weight, and cranial facial abnormalities. However, these mice do not present with a B cell defect; they have a normal spectrum of B cell precursors and mature B cell populations. Instead, these mice have reduced amounts of thymocytes and increased levels of fragmented nuclei in the thymus.47 Although T cell defects have been rarely reported in ICF syndrome, the presence of opportunistic infections in ICF patients strongly argues for a (subtle) T cell defect in ICF syndrome.2 In ICF patients, the early stages of B cell development are reported to be normal, and the defect mainly seems to arise in the peripheral terminal steps of B cell differentiation. ICF patients have immature naive B cells and impaired negative B cell selection. Although patients have strongly reduced or absent immunoglobulin levels, ICF B cells produce normal amounts of immunoglobulins upon in vitro stimulation.24 These findings strongly argue that the defect in ICF syndrome is restricted to the latest stages of B cell development, possibly in combination with an impaired T and B cell interaction.
The expression of ZBTB24 seems to be coregulated with DNMT3B during B cell differentiation (Figure S1). Therefore, to investigate direct effects of DNTM3B mutations on ZBTB24 expression or vice versa, we studied the major known splice variants of DNTMT3B (DNMT3B1, DNMT3B2, DNMT3B3, DNMT3B4, and DNMT3B5) and the presence of ZBTB24 in ICF1, ICF2, and control RNA samples by nonquantitative RT-PCR. RNA and cDNA samples were prepared in the same way as before, but the final cDNA sample was used in a standard PCR reaction containing DreamTaq DNA polymerase (Fermentas) and 1 μM of both forward and reverse primers (Table S3). All DNMT3B splice variants and the ZBTB24 transcript could be detected in ICF1, ICF2, and control samples (Figure S2). Because our RNA sources came from different origins, both primary cells and immortalized cell lines, we refrained from quantitative analysis of DNMT3B and ZBTB24 expression.
With the identification of mutations in ZBTB24, encoding a member of a family of transcription factors important for B and T cell differentiation, we expect that molecular studies of ZBTB24 will soon shed more light on the pathophysiology of ICF syndrome and lymphocyte biology in general.
Acknowledgments
We thank the ICF patients and their families for their participation. We thank J. van Reeuwijk, (Radboud University Nijmegen Medical Centre, Nijmegen, the Netherlands) for his help with the homozygosity mapping data analysis, J.J. Houwing-Duistermaat, (Leiden University Medical Center, Leiden, the Netherlands) for statistical advice, and S. Klingeman Plati for technical assistance. This research was supported by funding from the National Institutes of Health/National Institute of Allergy and Infectious Diseases R21 AI090135.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Maraschio P., Zuffardi O., Dalla Fior T., Tiepolo L. Immunodeficiency, centromeric heterochromatin instability of chromosomes 1, 9, and 16, and facial anomalies: The ICF syndrome. J. Med. Genet. 1988;25:173–180. doi: 10.1136/jmg.25.3.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hagleitner M.M., Lankester A., Maraschio P., Hultén M., Fryns J.P., Schuetz C., Gimelli G., Davies E.G., Gennery A., Belohradsky B.H. Clinical spectrum of immunodeficiency, centromeric instability and facial dysmorphism (ICF syndrome) J. Med. Genet. 2008;45:93–99. doi: 10.1136/jmg.2007.053397. [DOI] [PubMed] [Google Scholar]
- 3.Hansen R.S. X inactivation-specific methylation of LINE-1 elements by DNMT3B: Implications for the Lyon repeat hypothesis. Hum. Mol. Genet. 2003;12:2559–2567. doi: 10.1093/hmg/ddg268. [DOI] [PubMed] [Google Scholar]
- 4.Xu G.L., Bestor T.H., Bourc'his D., Hsieh C.L., Tommerup N., Bugge M., Hulten M., Qu X., Russo J.J., Viegas-Péquignot E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402:187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- 5.Okano M., Bell D.W., Haber D.A., Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 6.Gowher H., Jeltsch A. Molecular enzymology of the catalytic domains of the Dnmt3a and Dnmt3b DNA methyltransferases. J. Biol. Chem. 2002;277:20409–20414. doi: 10.1074/jbc.M202148200. [DOI] [PubMed] [Google Scholar]
- 7.Jeanpierre M., Turleau C., Aurias A., Prieur M., Ledeist F., Fischer A., Viegas-Pequignot E. An embryonic-like methylation pattern of classical satellite DNA is observed in ICF syndrome. Hum. Mol. Genet. 1993;2:731–735. doi: 10.1093/hmg/2.6.731. [DOI] [PubMed] [Google Scholar]
- 8.Tuck-Muller C.M., Narayan A., Tsien F., Smeets D.F., Sawyer J., Fiala E.S., Sohn O.S., Ehrlich M. DNA hypomethylation and unusual chromosome instability in cell lines from ICF syndrome patients. Cytogenet. Cell Genet. 2000;89:121–128. doi: 10.1159/000015590. [DOI] [PubMed] [Google Scholar]
- 9.Wijmenga C., Hansen R.S., Gimelli G., Björck E.J., Davies E.G., Valentine D., Belohradsky B.H., van Dongen J.J., Smeets D.F., van den Heuvel L.P. Genetic variation in ICF syndrome: Evidence for genetic heterogeneity. Hum. Mutat. 2000;16:509–517. doi: 10.1002/1098-1004(200012)16:6<509::AID-HUMU8>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 10.Jiang Y.L., Rigolet M., Bourc'his D., Nigon F., Bokesoy I., Fryns J.P., Hultén M., Jonveaux P., Maraschio P., Mégarbané A. DNMT3B mutations and DNA methylation defect define two types of ICF syndrome. Hum. Mutat. 2005;25:56–63. doi: 10.1002/humu.20113. [DOI] [PubMed] [Google Scholar]
- 11.Kubota T., Furuumi H., Kamoda T., Iwasaki N., Tobita N., Fujiwara N., Goto Y., Matsui A., Sasaki H., Kajii T. ICF syndrome in a girl with DNA hypomethylation but without detectable DNMT3B mutation. Am. J. Med. Genet. A. 2004;129A:290–293. doi: 10.1002/ajmg.a.30135. [DOI] [PubMed] [Google Scholar]
- 12.Kloeckener-Gruissem B., Betts D.R., Zankl A., Berger W., Güngör T. A new and a reclassified ICF patient without mutations in DNMT3B and its interacting proteins SUMO-1 and UBC9. Am. J. Med. Genet. A. 2005;136:31–37. doi: 10.1002/ajmg.a.30767. [DOI] [PubMed] [Google Scholar]
- 13.Brown D.C., Grace E., Sumner A.T., Edmunds A.T., Ellis P.M. ICF syndrome (immunodeficiency, centromeric instability and facial anomalies): Investigation of heterochromatin abnormalities and review of clinical outcome. Hum. Genet. 1995;96:411–416. doi: 10.1007/BF00191798. [DOI] [PubMed] [Google Scholar]
- 14.Wijmenga C., van den Heuvel L.P., Strengman E., Luyten J.A., van der Burgt I.J., de Groot R., Smeets D.F., Draaisma J.M., van Dongen J.J., De Abreu R.A. Localization of the ICF syndrome to chromosome 20 by homozygosity mapping. Am. J. Hum. Genet. 1998;63:803–809. doi: 10.1086/302021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuetz C., Barbi G., Barth T.F., Hoenig M., Schulz A., Möeller P., Smeets D., de Greef J.C., van der Maarel S.M., Vogel W. ICF syndrome: High variability of the chromosomal phenotype and association with classical Hodgkin lymphoma. Am. J. Med. Genet. A. 2007;143A:2052–2057. doi: 10.1002/ajmg.a.31885. [DOI] [PubMed] [Google Scholar]
- 16.Gennery A.R., Slatter M.A., Bredius R.G., Hagleitner M.M., Weemaes C., Cant A.J., Lankester A.C. Hematopoietic stem cell transplantation corrects the immunologic abnormalities associated with immunodeficiency-centromeric instability-facial dysmorphism syndrome. Pediatrics. 2007;120:e1341–e1344. doi: 10.1542/peds.2007-0640. [DOI] [PubMed] [Google Scholar]
- 17.Pezzolo A., Prigione I., Facchetti P., Castellano E., Viale M., Gimelli G., Pistoia V. T-cell apoptosis in ICF syndrome. J. Allergy Clin. Immunol. 2001;108:310–312. doi: 10.1067/mai.2001.116863. [DOI] [PubMed] [Google Scholar]
- 18.Gimelli G., Varone P., Pezzolo A., Lerone M., Pistoia V. ICF syndrome with variable expression in sibs. J. Med. Genet. 1993;30:429–432. doi: 10.1136/jmg.30.5.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colucci M., Cocito L., Capello E., Mancardi G.L., Serrati C., Cinque P., Schenone A. Progressive multifocal leukoencephalopathy in an adult patient with ICF syndrome. J. Neurol. Sci. 2004;217:107–110. doi: 10.1016/j.jns.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 20.Lander E.S., Botstein D. Homozygosity mapping: A way to map human recessive traits with the DNA of inbred children. Science. 1987;236:1567–1570. doi: 10.1126/science.2884728. [DOI] [PubMed] [Google Scholar]
- 21.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blanco-Betancourt C.E., Moncla A., Milili M., Jiang Y.L., Viegas-Péquignot E.M., Roquelaure B., Thuret I., Schiff C. Defective B-cell-negative selection and terminal differentiation in the ICF syndrome. Blood. 2004;103:2683–2690. doi: 10.1182/blood-2003-08-2632. [DOI] [PubMed] [Google Scholar]
- 25.Bardwell V.J., Treisman R. The POZ domain: A conserved protein-protein interaction motif. Genes Dev. 1994;8:1664–1677. doi: 10.1101/gad.8.14.1664. [DOI] [PubMed] [Google Scholar]
- 26.Deweindt C., Albagli O., Bernardin F., Dhordain P., Quief S., Lantoine D., Kerckaert J.P., Leprince D. The LAZ3/BCL6 oncogene encodes a sequence-specific transcriptional inhibitor: A novel function for the BTB/POZ domain as an autonomous repressing domain. Cell Growth Differ. 1995;6:1495–1503. [PubMed] [Google Scholar]
- 27.Huynh K.D., Bardwell V.J. The BCL-6 POZ domain and other POZ domains interact with the co-repressors N-CoR and SMRT. Oncogene. 1998;17:2473–2484. doi: 10.1038/sj.onc.1202197. [DOI] [PubMed] [Google Scholar]
- 28.Huynh K.D., Fischle W., Verdin E., Bardwell V.J. BCoR, a novel corepressor involved in BCL-6 repression. Genes Dev. 2000;14:1810–1823. [PMC free article] [PubMed] [Google Scholar]
- 29.Reeves R., Beckerbauer L. HMGI/Y proteins: Flexible regulators of transcription and chromatin structure. Biochim. Biophys. Acta. 2001;1519:13–29. doi: 10.1016/s0167-4781(01)00215-9. [DOI] [PubMed] [Google Scholar]
- 30.Stogios P.J., Downs G.S., Jauhal J.J., Nandra S.K., Privé G.G. Sequence and structural analysis of BTB domain proteins. Genome Biol. 2005;6:R82. doi: 10.1186/gb-2005-6-10-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ye B.H., Chaganti S., Chang C.C., Niu H., Corradini P., Chaganti R.S., Dalla-Favera R. Chromosomal translocations cause deregulated BCL6 expression by promoter substitution in B cell lymphoma. EMBO J. 1995;14:6209–6217. doi: 10.1002/j.1460-2075.1995.tb00311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dent A.L., Shaffer A.L., Yu X., Allman D., Staudt L.M. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276:589–592. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- 33.Shaffer A.L., Yu X., He Y., Boldrick J., Chan E.P., Staudt L.M. BCL-6 represses genes that function in lymphocyte differentiation, inflammation, and cell cycle control. Immunity. 2000;13:199–212. doi: 10.1016/s1074-7613(00)00020-0. [DOI] [PubMed] [Google Scholar]
- 34.Phan R.T., Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. 2004;432:635–639. doi: 10.1038/nature03147. [DOI] [PubMed] [Google Scholar]
- 35.Takamori M., Hatano M., Arima M., Sakamoto A., Fujimura L., Hartatik T., Kuriyama T., Tokuhisa T. BAZF is required for activation of naive CD4 T cells by TCR triggering. Int. Immunol. 2004;16:1439–1449. doi: 10.1093/intimm/dxh144. [DOI] [PubMed] [Google Scholar]
- 36.Manders P.M., Hunter P.J., Telaranta A.I., Carr J.M., Marshall J.L., Carrasco M., Murakami Y., Palmowski M.J., Cerundolo V., Kaech S.M. BCL6b mediates the enhanced magnitude of the secondary response of memory CD8+ T lymphocytes. Proc. Natl. Acad. Sci. USA. 2005;102:7418–7425. doi: 10.1073/pnas.0501585102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bilic I., Koesters C., Unger B., Sekimata M., Hertweck A., Maschek R., Wilson C.B., Ellmeier W. Negative regulation of CD8 expression via Cd8 enhancer-mediated recruitment of the zinc finger protein MAZR. Nat. Immunol. 2006;7:392–400. doi: 10.1038/ni1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun G., Liu X., Mercado P., Jenkinson S.R., Kypriotou M., Feigenbaum L., Galéra P., Bosselut R. The zinc finger protein cKrox directs CD4 lineage differentiation during intrathymic T cell positive selection. Nat. Immunol. 2005;6:373–381. doi: 10.1038/ni1183. [DOI] [PubMed] [Google Scholar]
- 39.He X., He X., Dave V.P., Zhang Y., Hua X., Nicolas E., Xu W., Roe B.A., Kappes D.J. The zinc finger transcription factor Th-POK regulates CD4 versus CD8 T-cell lineage commitment. Nature. 2005;433:826–833. doi: 10.1038/nature03338. [DOI] [PubMed] [Google Scholar]
- 40.Piazza F., Costoya J.A., Merghoub T., Hobbs R.M., Pandolfi P.P. Disruption of PLZP in mice leads to increased T-lymphocyte proliferation, cytokine production, and altered hematopoietic stem cell homeostasis. Mol. Cell. Biol. 2004;24:10456–10469. doi: 10.1128/MCB.24.23.10456-10469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kang B.Y., Miaw S.C., Ho I.C. ROG negatively regulates T-cell activation but is dispensable for Th-cell differentiation. Mol. Cell. Biol. 2005;25:554–562. doi: 10.1128/MCB.25.2.554-562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bilic I., Ellmeier W. The role of BTB domain-containing zinc finger proteins in T cell development and function. Immunol. Lett. 2007;108:1–9. doi: 10.1016/j.imlet.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 43.Ehrlich M., Buchanan K.L., Tsien F., Jiang G., Sun B., Uicker W., Weemaes C.M., Smeets D., Sperling K., Belohradsky B.H. DNA methyltransferase 3B mutations linked to the ICF syndrome cause dysregulation of lymphogenesis genes. Hum. Mol. Genet. 2001;10:2917–2931. doi: 10.1093/hmg/10.25.2917. [DOI] [PubMed] [Google Scholar]
- 44.Jin B., Tao Q., Peng J., Soo H.M., Wu W., Ying J., Fields C.R., Delmas A.L., Liu X., Qiu J., Robertson K.D. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum. Mol. Genet. 2008;17:690–709. doi: 10.1093/hmg/ddm341. [DOI] [PubMed] [Google Scholar]
- 45.Jefferson A., Colella S., Moralli D., Wilson N., Yusuf M., Gimelli G., Ragoussis J., Volpi E.V. Altered intra-nuclear organisation of heterochromatin and genes in ICF syndrome. PLoS ONE. 2010;5:e11364. doi: 10.1371/journal.pone.0011364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matarazzo M.R., Boyle S., D'Esposito M., Bickmore W.A. Chromosome territory reorganization in a human disease with altered DNA methylation. Proc. Natl. Acad. Sci. USA. 2007;104:16546–16551. doi: 10.1073/pnas.0702924104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ueda Y., Okano M., Williams C., Chen T., Georgopoulos K., Li E. Roles for Dnmt3b in mammalian development: A mouse model for the ICF syndrome. Development. 2006;133:1183–1192. doi: 10.1242/dev.02293. [DOI] [PubMed] [Google Scholar]
- 48.Miniou P., Jeanpierre M., Bourc'his D., Coutinho Barbosa A.C., Blanquet V., Viegas-Péquignot E. alpha-satellite DNA methylation in normal individuals and in ICF patients: Heterogeneous methylation of constitutive heterochromatin in adult and fetal tissues. Hum. Genet. 1997;99:738–745. doi: 10.1007/s004390050441. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.