

Abstract
Background:
Phenolic constituents were the principle bioactivity compounds exist in Gynura divaricata, little phenolic compounds were reported from the plant previously.
Materials and Methods:
60% ethanol extract from the leaves of Gynura divaricata were isolated and purified by column chromatography of Silica gel, ODS and Sephadex LH-20, the structures of the isolated compounds were identified by UV, 1H-NMR, 13C-NMR and MS spectroscopic techniques. Additionally, a high-performance liquid chromatography-diode array detector-electrospray ionization-mass (HPLC-DAD-ESI-MS) analytical method was developed to identify some minor constituents in the n-butanol fraction of the ethanol extract of Gynura divaricata.
Results:
Six flavonols and one Dicaffeoylquinic acid were isolated from the leaves of Gynura divaricata, and these compounds were identified as follows: quercetin (1), kaempferol (2), kaempferol-3-O-β-D-glucopyranoside (3), quercetin-3-O-rutinoside (4), kaempferol-3,7-di-O-β-D-glucopyranoside (5), kaempferol-3-O-rutinoside-7-O-β-D-glucopyranoside (6), and 3,5-dicaffeoylquinic acid (7). A total of 13 compounds, including 9 flavonol glycosides and 4 phenolic acids, were tentatively identified by comparing their retention time (RT), UV, and MS spectrum values with those that had been identified and the published data.
Conclusion:
This was the first time to use the HPLC-DAD-ESI-MS method to identify the phytochemicals of the genera Gynura. Moreover, compounds (6) and (7) have been isolated for the first time from the genus Gynura.
Keywords: Gynura divaricata DC., HPLC-DAD-ESI-MS, phenolic constituents
INTRODUCTION
Gynura genus belongs to the family Asteraceae, consisting of 12 species in China.[1] Many species are edible medicinal plants and the leaves are also used as a vegetable by the locals in Southwestern China.[2] G. divaricata is a traditional Chinese medicinal plant, which is called “Bai Bei San Qi” in Chinese. It has a long history of use for treatment of diabetes in the folk medicine. The ethanol extract of aerial parts of G. divaricata was reported to demonstrate hypoglycemic activity in vivo, the flavonoid compounds were the active constituents.[3,4] It also has been reported that many constituents with antiproliferation activity exist in G. divaricata.[5,6] The chemical constituents of G. divaricata include flavonols, phenolic acids, cerebrosides, polysaccharides, alkaloids, terpenoids, and sterols.[5–10] Flavonols were the principal constituents of the plant, 4 flavonol compounds, including quercetin, isoquercitrin, rutin, and kaempferol-3-O-rutinoside, have been isolated and identified from the aerial parts of the plant.[9] This article herein describes the isolation and structure elucidation of the flavonol and phenolic acid compounds from the ethanol extract of G. divaricata DC. leaves by NMR and high-performance liquid chromatography-diode array detector-electrospray ionization-mass spectrometry (HPLC-DAD-ESI-MS) methods.
MATERIALS AND METHODS
General
The1H-NMR and13C-NMR spectra were measured with a Bruker Avance-600 FT-NMR spectrometer (Bruker, Coventry, Germany), with TMS internal standard. HPLC-DAD-ESI-MS were recorded on Waters 2995 Series LC and ZQ-4000 Mass spectrometer (Waters Corporation, Milford, MA, USA). Column chromatography was carried out with Silica gel (Qingdao Marine Chemistry Co. Ltd., 200-300 mesh, Qingdao, China), Sephadex LH-20, and Reverse phase octadecylsilyl (RP-ODS) (Pharmacia Co. Ltd., Minnesota, USA). Thin layer chromatography (TLC) was carried out with Silica gel GF254 (Qingdao Marine Chemistry Co. Ltd., Qingdao, China), and the compounds were prepared either by spraying with 10% sulfuric acid ethanol or under UV lamp at 254 nm. HPLC-grade acetonitrile was purchased from Merck Company (Merck, Darmstadt, Germany), other solvents were analytical grade from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China).
Plant material
The Gynura divaricata plant was obtained in 2009 from Guangdong province, China. A voucher specimen (201001) was deposited at the Department of Chemistry, Nanchang University. The leaves of G. divaricata were dried at 40°C in an air oven and finely powdered.
Extraction and isolation
The weighed portion of the crude drug 5 kg was extracted twice with 60% ethanol (v/v) under reflux at 90°C. The extract was evaporated to dryness in vacuo. Extract yield with respect to the dried herb was 25%. The dry extract was suspended in water and subjected to sequential liquid-liquid extraction with chloroform, ethyl acetate (EA), and n-butanol, the yield of those 3 extracts were 31.2, 56.5, and 89.5 g, respectively. The EA fraction was chromatographed using flash column on a Silica gel eluted with chloroform-methanol step-gradient (starting with 100:0 to 4:1), eluted fractions were combined on their TLC pattern to yield 8 fractions. The chloroform-methanol fraction (10:1) was chromatographed on a Sephadex LH-20 column eluted with chloroform-methanol (1:1) to yield compounds 1 and 2. The chloroform-methanol fraction (6:1) chromatographed on a Sephadex LH-20 column eluted with methanol and further chromatographed on an RP-ODS column gradient eluted with methanol-water (40%-60%, v/v) gave compounds 3 and 7. The chloroform-methanol fraction (4:1) chromatographed on a Sephadex LH-20 column eluted with methanol yields compound 4 [Figure 1].
Figure 1.
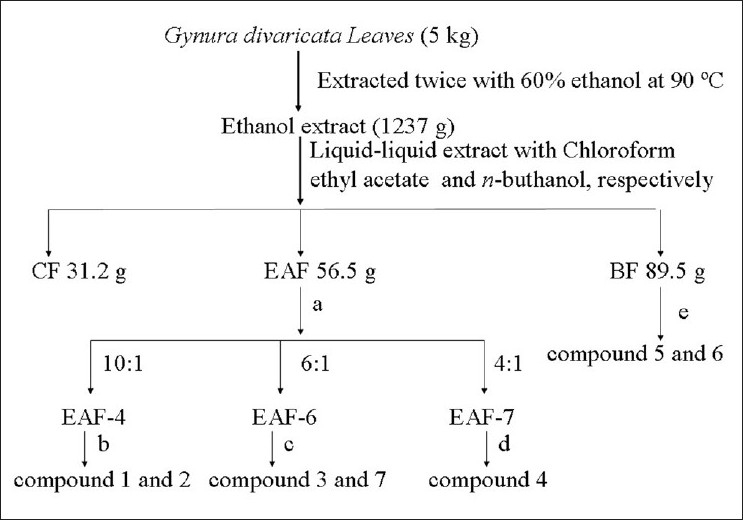
The procedure of extraction and isolation phenolic compounds from G. divaricata extracts. (a) Silica gel chromatograph eluted with a mixture of chloroform and methanol (from 100:0 to 4:1); (b) Sephadex LH-20 chromatograph eluted with a mixture of chloroform and methanol (1:1); (c) Sephadex LH-20 column eluted with methanol coupled with RP-ODS column gradient eluted with methanol-water (from 40% to 60%, v/v); (d) Sephadex LH-20 column eluted with methanol, (e) RP-ODS column gradient eluted with methanol-water (from 10% to 50%, v/v) coupled with RP-ODS column and isocratic eluted with methanol-water (18%, v/v)
The n-butanol fraction was chromatographed using flash RP-ODS column gradient eluted with methanol-water (10%-50%, v/v), and the eluted fractions were combined on their HPLC pattern to yield 4 fractions. The methanol-water fraction (25%, v/v) was further chromatographed using flash RP-ODS column and isocratic eluted with methanol-water (18%, v/v) gave compounds 5 and 6. The other minor constituents of n-butanol extracts were separated and identified by HPLC-DAD-ESI-MS method.
HPLC-MS instrument and conditions
The HPLC-DAD-ESI-MS system consists of a Waters 2995 Series LC and ZQ-4000 Mass spectrometer (Waters, USA), equipped with a vacuum degasser, a quaternary pump, an autosampler, a thermostatted column compartment, a diode array detector (DAD), and an ion-trap mass spectrometer with electrospray ionization interface, controlled by Waters 2995 Series LC/ZQ-4000 Trap Software. Shimadzu shimpack VP-ODS (150 mm × 4.6 mm i.d., 5 μm particle size) was used for separation. Solvents for the mobile phase were water-0.1% acetic acid (A) and acetonitrile (B). The gradient elution was 0-30 min, linear gradient 10%-30% B; 30-40 min, linear gradient 30%-100% B. The flow rate was 0.8 mL/min and the column was operated at 30°C. Peaks were detected with the DAD at 254 nm. The ESI negative and positive ionization (NI and PI) total ion current (TIC) modes were used for MS detection. The m/z values of the monitored ions were from 100 to 800. The other parameters were as follows: capillary voltage, 3.5 kV; cone voltage, 30 V; extraction voltage, 5 V; RF voltage, 0.5 V; source temperature, 90°C; nitrogen gas flow for desolvation, 300 L/h; and temperature of the nitrogen gas for desolvation, 350°C. Samples for assay were dissolved in 45% MeOH as 3 mg/mL solutions and centrifuged at 12,000 rpm (Beckman, USA) for 15 min to remove particles before injection.
RESULTS AND DISCUSSION
The compounds were identified using UV, ESI-MS, and NMR spectral data, and determined as quercetin,[11,12] kaempferol,[13,14] kaempferol-3-O-β-D-glucopyranoside,[15] quercetin-3-O-rutinoside,[15] kaempferol-3-O-rutinoside-7-O-β-D-glucopyranoside,[16,17] kaempferol-3,7-di-O-β-D-glucopyranoside,[16] and 3,5-Dicaffeoylquinic acid.[18]
Compound 1 was obtained as a yellow powder, the ESI-MS yielded a quasi-molecular ion peak [M-H]- at m/z 301 and [M+H]+ at m/z 303. The UV spectrum showed λmax at 256 and 370 nm. The1H-NMR spectrum showed 2 peaks at δ 6.18 (1H, d, J = 2.0 Hz) and 6.40 ppm (1H, d, J = 2.0 Hz) consistent with the meta protons H-6 and H-8 on A-ring and an ABX system at 7.68 (1H, d, J = 2.2 Hz, H-2’), 7.54 (1H, dd, J = 2.0 Hz, 8.4 Hz, H-6’), and 6.88 (1H, d, J = 8.4 Hz, H-5’) corresponding to the catechol protons on B-ring. The13C-NMR spectrum indicated the presence of 15 carbon atoms, the signal at δ 177.9 was attributed to a carbonyl carbon placed at C-4, and the other signals were compatible with those literatures[11,12] on quercetin.
Compound 2 was obtained as a yellow powder, the ESI-MS yielded a quasi-molecular ion peak [M-H]- at m/z 285 and [M+H]+ at m/z 287. The UV spectrum showed λmax at 265 and 366 nm. The1H-NMR spectrum showed 2 peaks at δ 6.17 (1H, d, J = 1.8 Hz) and 6.42 ppm (1H, d, J = 1.8 Hz) consistent with the meta protons H-6 and H-8 on A-ring and an AA’BB’ system at 8.04 (2H, d, J = 8.9 Hz, H-2’, 6’) and 6.93 (2H, d, J = 8.9 Hz, H-3’, 5’) corresponding to the protons on B-ring. The MS and1 H-NMR data were compatible with the literatures[13,14] of kaempferol.
Compound 3 was obtained as a faint yellow powder, the ESI-MS yielded a quasi-molecular ion peak [M-H]- at m/z 447 and [M+H]+ at m/z 449. The UV spectrum showed λmax at 265 and 346 nm. The1 H-NMR spectrum showed 2 peaks at δ 6.21 (1H, d, J =1.8 Hz) and 6.44 ppm (1H, d, J =1.8 Hz) consistent with the meta protons H-6 and H-8 on A-ring and an AA’BB’ system at 8.04 (2H, d, J =8.9 Hz, H-2’, 6’) and 6.89 (2H, d, J =8.9 Hz, H-3’, 5’) corresponding to the protons on B-ring. Compound 3 presented the same aglycone signal patterns of compound 2, but the signal at 5.47 (1H, d, J =7.2 Hz) followed by other characteristic additional signals indicates the presence of a sugar moiety in compound 3. The hexose was determined to be a glucopyranosyl unit bound to the C-3 position of the aglycone by comparison of proton and carbon upfield shift values with the literature data.[15] Therefore, compound 3 was identified as kaempferol-3-O-β-D-glucopyranoside.
Compound 4 was obtained as a faint yellow powder, the ESI-MS yielded a quasi-molecular ion peak [M-H]- at m/z 609 and [M+H]+ at m/z 611. The UV spectrum showed λmax at 258 and 356 nm. The1H-NMR spectrum showed 2 peaks at δ 6.20 (1H, d, J = 2.0 Hz) and 6.40 ppm (1H, d, J = 2.0 Hz) consistent with the meta protons H-6 and H-8 on A-ring and an ABX system at 7.54 (1H, d, J = 2.2 Hz, H-2’), 7.59 (1H, dd, J = 2.0 Hz, 9.0 Hz, H-6’) and 6.85 (1H, d, J = 9.0 Hz, H-5’) corresponding to the catechol protons on B-ring. Compound 4 presented the same aglycone signal patterns of compound 1, two anomeric proton signals at 5.32 (1H, d, J =7.2 Hz) and 4.39 (1H, d, J = 1.6 Hz) were assignable to H-1 of a β-glucosyl proton and to the H-1 of an α-rhamnosyl proton, respectively. A methyl signal 0.99 (3H, d, J =6.2 Hz) in the high-field region was assigned to rhamnose. In the13C-NMR of compound 4, the C-6 signal (68.5) of glucose showed a downfield shift of 7.3 ppm in comparison with the corresponding C-6 signal (61.2) of quercetin-3-O-β-D-glucopyranoside,[15] indicating a 1-6 linkage between the glucose and the rhamnose. Therefore, compound 4 was identified as rutin.
Compound 5 was obtained as a faint yellow powder, the molecular formula C27H30O16 was suggested by a mass spectrum with a quasi-molecular ion peak [M-H]- at m/z 609, further confirmed by the positive mode mass spectral ions: 611 [M+H]+, 449 [M+H-162]+, 287 [M+H-162-162]+. The UV spectrum showed λmax at 264 and 347 nm typical of a kaempferol glycoside derivative.[15,16,19] In the aromatic region of the1 H-NMR spectrum an AA’BB’ system, appearing as two doublets at δ 8.06 (2H, d, J = 8.9 Hz, H-2’, 6’) and 6.90 (2H, d, J = 8.9 Hz, H-3’, 5’), and two meta coupled doublet protons at δ 6.78 and 6.44 were evident. In the saccharide region of the spectrum two anomeric proton signals were present as large doublets at δ 5.48 and 5.08. The coupling constant (J = 7.2 Hz) of the two anomeric protons characteristic for β-configuration. The downfield shift of the H-6 and H-8 proton, as well as downfield shift of the corresponding carbons at δ 99.8 and δ 94.9, with respect to the corresponding signals of aglycone, suggested the linkage with the sugar moiety across the oxygen of the C(7)-OH group.[17] The chemical shift (δ 5.48) suggested that the other sugar moiety is directly attached to the C(3)-OH group, further confirmed by the upfield shift of the signal assigned to C-3 (133.9).[15,16] Acid hydrolysis of compound 5 afforded kaempferol and glucose comparison with the authentic samples on TLC. From the above data, compound 5 was identified as kaempferol-3,7-di-O-β-D-glucopyranoside.
Compound 6 was obtained as a faint yellow powder, the molecular formula C33H40O20 was suggested by a mass spectrum with a quasi-molecular ion peak [M-H]- at m/z 755. The UV and1 H-NMR spectrum of compound 6 was similar to that of 5, suggesting that compound 6 also was a kaempferol glycoside derivative, the only difference being the presence of a methyl signal (δ 0.99) in the high-field region, which was assigned to rhamnose, further confirmed by the doublet proton at δ 4.44, was assigned to the anomeric proton of rhamnose with a coupling constant (J = 1.6 Hz) characteristic for α-linked rhamnose. The13C-NMR spectrum of 6 confirms that compound 6 is a triglycoside of kaempferol [Table 1]. Careful examination of the13C-NMR spectrum of 6 showed that the signal assigned to the glucose C-6 [Table 1] was shifted downfield by appropriately 6 ppm (from 61.3 to 67.3) confirming that the rhamnose moiety linkage to the glucose C-6.[17] From the above data, compound 6 was identified as kaempferol-3-O-rutinoside-7-O-β-D-glucopyranoside.
Table 1.
The 1H-NMR and 13C-NMR spectrum data of kaempferol-3,7-di-O-β-d-glucopyranoside and kaempferol-3-O-rutinoside-7-O-β-d-glucopyranoside (DMSO-d6)
| Atom | Kaempferol-3,7-di-O-β-d-glucoside |
Kaempferol-3-O-rutinoside-7-O-β-d-glucoside |
||||
|---|---|---|---|---|---|---|
|
1H |
13C |
1H |
13C |
|||
| δ(ppm) | J(Hz) | δ(ppm) | δ(ppm) | J(Hz) | δ(ppm) | |
| 2 | 156.5 | 156.5 | ||||
| 3 | 133.9 | 140.0 | ||||
| 4 | 178.1 | 178.1 | ||||
| 4a | 105.9 | 161.4 | ||||
| 5 | 161.3 | 106.1 | ||||
| 6 | 6.44 d | 2.0 | 99.8 | 6.45 d | 2.0 | 99.8 |
| 7 | 163.3 | 163.4 | ||||
| 8 | 6.78 d | 2.0 | 94.9 | 6.76 d | 2.0 | 95.1 |
| 8a | 157.3 | 157.8 | ||||
| 1’ | 121.1 | 121.2 | ||||
| 2’ | 8.06 d | 8.6 | 131.4 | 8.01 d | 8.8 | 131.5 |
| 3’ | 6.90 d | 8.6 | 115.6 | 6.90 d | 8.8 | 115.6 |
| 4’ | 160.6 | 160.6 | ||||
| 5’ | 115.6 | 115.6 | ||||
| 6’ | 131.4 | 131.5 | ||||
| 3-O-Rutinoside | ||||||
| G1 | 5.48 d | 7.2 | 101.2 | 5.35 d | 7.2 | 101.7 |
| G2 | 74.6 | 74.7 | ||||
| G3 | 76.9 | 76.9 | ||||
| G4 | 70.4 | 70.4 | ||||
| G5 | 78 | 77.7 | ||||
| G6 | 61.3 | 67.3 | ||||
| R1 | 4.44 d | 1.6 | 101.2 | |||
| R2 | 70.8 | |||||
| R3 | 71.1 | |||||
| R4 | 72.3 | |||||
| R5 | 68.7 | |||||
| R6 | 0.99 d | 6.2 | 18.2 | |||
| 7-O-Glucoside | ||||||
| G’1 | 5.08 d | 7.2 | 100.2 | 5.08 d | 7.2 | 100.3 |
| G’2 | 73.5 | 73.6 | ||||
| G’3 | 76.9 | 76.3 | ||||
| G’4 | 70.0 | 70.1 | ||||
| G’5 | 77.6 | 76.9 | ||||
| G’6 | 61.1 | 61.1 | ||||
Compound 7 was obtained as amorphous powder, the ESI-MS yielded a quasi-molecular ion peak [M-H]- at m/z 515 and [M+H]+ at m/z 517. The UV spectrum showed λmax 327, 294 (sh), and 248 nm (sh), which were characteristic of caffeic acid derivatives. In the1 H-NMR spectrum, two caffeoyl groups were presented at δ 7.50 (1H, d, J=16.0 Hz, H-7’), 7.43 (1H, d, J=16.0 Hz, H-7”), 7.05 (2H, brs, H-2’, 2”), 7.01 (2H, brd, J=2.0 Hz, H-6, 6”), 6.78 (1H, d, J=8.0 Hz, H-5’), 6.76 (1H, d, J=8.0 Hz, H-5”), 6.26 (1H, dd, J=16.0 Hz, H-8’), 6.14 (1H, dd, J=16.0 Hz, H-8”). A quinic acid moiety was presented at 5.42 (1H, brs, H-3), 5.18 (1H, m, H-5), 3.86 (1H, brs, H-4), 2.20 (2H, m, H-6), 2.01(2H, m, H-2). The1H-NMR data were in agreement with the literature[18] and compound 7 was identified as 3,5-Dicaffeoylquinic acid.
An HPLC-DAD-ESI-MS method was developed to identify the minor phytochemical constituents of n-butanol fraction of G. divaricata extract. The chromatogram of MS TIC in negative mode is shown in Figure 2a. As shown in Figure 2b, 13 major peaks were detected under the HPLC conditions with DAD detection at 254 nm. Peaks of 2, 3, and 11, 12 were co-eluted in the present conditions and unequivocally determined to be kaempferol-3,7-di-O-β-D-glucopyranoside, kaempferol-3-O-rutinoside-7-O-D-glucopyranoside, 3,5-Dicaffeoylquinic acid, and kaempferol-3-O-β-D-glucopyranoside, respectively. And peak 5 was identified as quercetin-3-O-rutinoside. All of those 5 peaks were identified by comparing the retention time (RT), UV [Figure 3], and ESI-MS values with isolation compounds. The other compounds were tentatively identified based on the UV adsorption value, m/z value, and elution order compared with the published data.
Figure 2.
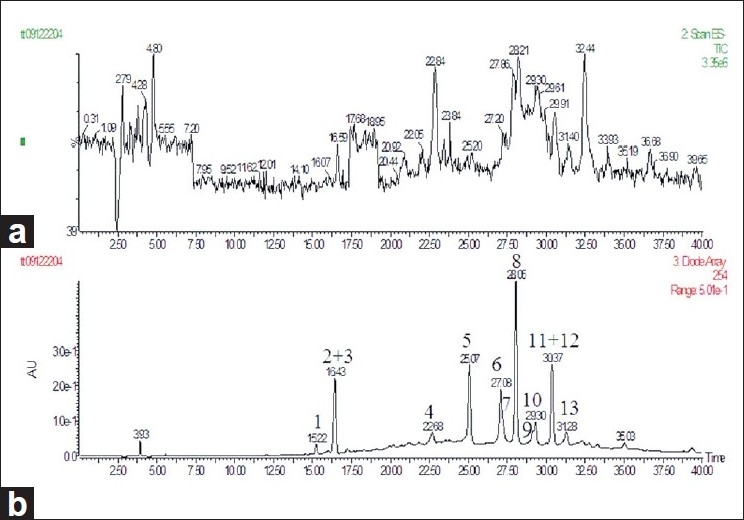
The TIC chromatogram of negative model (a) and HPLC-DAD chromatogram of the n-butanol fraction of G. divaricata extracts (b)
Figure 3.
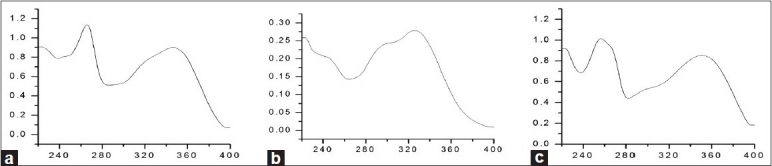
The typical UV spectrum of Kaempferol glucopyranoside derivative (a), Dicaffeoylquinic acid (b), and Quercetin glucopyranoside derivative (c)
Peak 1 was believed to be an unidentified minor flavonol glycoside due to its low concentration in the extract, peak 1 and 3 are a pair of isomers, the UV (λmax) and m/z values [Table 2] were similar to peak 3 (identified as kaempferol-3-O-rutinoside-7-O-β-D-glucopyranoside). The elution order of peak 1 being prior to peak 3 [Table 2] suggested that rutinose of peak 3 was substituted by a robinobiose, and the structure of peak 1 was proposed to be kaempferol-3-O-robinobioside-7-O-β-D-glucopyranoside.[20]
Table 2.
HPLC-DAD-ESI-MS (positive and negative ionization TIC modes) fingerprint of n-butanol fraction of G. divaricata extracts
| Peak No. | tR (min) | λmax(nm) | Product ions (ESI-, m/z) | Product ions (ESI+, m/z) | Identification of compounds |
|---|---|---|---|---|---|
| 1 | 15.22 | 265, 346 | 755 [M-H]- | Kaempferol-3-O-robinobioside-7-O-β-D-glucoside | |
| 2 | 16.43 | 264, 347 | 609 [M-H]- | 611 [M+H]+ 449 [M+H-162]+ 287 [M+H-162-162]+ | Kaempferol-3,7-di-O-β-D-glucoside |
| 3 | 16.51 | 264, 347 | 755 [M-H]- | 757 [M+H]+ 611 [M+H-146]+ 449 [M+H-146-162]+ 287 [M+H-146-162-162]+ | Kaempferol-3-O-rutinoside-7-O-β-D-glucoside |
| 4 | 22.68 | 247, 307 | 337 [M-H]- 191 [M-H-146]- | 339 [M+H]+ 147 [M+H-192]+ | p-Coumaoylquinic acid |
| 5 | 25.07 | 254, 356 | 609 [M-H]- | 611 [M+H]+ 465 [M+H-146]+ 303 [M+H-146-162]+ | Quercetin-3-O-rutinoside |
| 6 | 27.08 | 256, 354 | 463 [M-H]- | 465 [M+H]+ 303 [M+H-162]+ | Quercetin-3-O-β-D-glucoside |
| 7 | 27.26 | 265, 346 | 593 [M-H]- | 595 [M+H]+ 449 [M+H-146]+ 287 [M+H-146-162]+ | Kaempferol-3-O-robinobioside |
| 8 | 28.05 | 265, 347 | 593 [M-H]- | 595 [M+H]+ 449 [M+H-146]+ 287 [M+H-146-162]+ | Kaempferol-3-O-rutinoside |
| 9 | 29.12 | 265, 346 | 447 [M-H]- | 449 [M+H]+ 287 [M+H-162]+ | Kaempferol-3-O-β-D-galacoside |
| 10 | 29.30 | 248, 327 | 515 [M-H]- 353 [M-H-162]. | 499 [M+H-18]+ 163 [M+H-162-192]+ | 3,4-Dicaffeoylquinic acid |
| 11 | 30.37 | 248, 325 | 515 [M-H]- 353 [M-H-162]. | 499 [M+H-18]+ 163 [M+H-162-192]+ | 3,5-Dicaffeoylquinic acid |
| 12 | 30.37 | 265, 347 | 447 [M-H]- | 449 [M+H]+ 287 [M+H-162]+ | Kaempferol-3-O-β-D-glucoside |
| 13 | 31.28 | 248, 325 | 515 [M-H]- 353 [M-H-162]. | 499 [M+H-18]+ 163 [M+H-162-192]+ | 4,5-Dicaffeoylquinic acid |
HPLC-DAD-ESI-MS: High-performance liquid chromatography-diode array detector-electrospray ionization-mass spectrometry, TIC: Total ion current, Identification was supported by comparison with reference standards where available and by correlation with previous literature reports. Peaks 2, 3 and 11, 12 were co-eluted. Peak numbers and retention times (TR) refer to HPLC chromatograms in Figure 2b
Peak 4 yielded a [M-H]- ion at m/z 337, and [M+H]+ ion at m/z 339, [M+H-192]+ ion at m/z 147. The UV spectrum showed λmax> at 307, 293 (sh), and 247 nm (sh), which is characteristic of a Cinnamic acid derivative[19,21]; hence, the structure of peak 4 was proposed to be p-coumaroylquinic acid.[19,21]
Peak 6 yielded a [M-H]- ion at m/z 463, and [M+H]+ ion at m/z 465, [M+H-162]+ ion at m/z 303. The UV spectrum showed λmax at 255 and 356 nm, suggesting that this as a quercetin glycoside.[21] By examining the known flavonol glycoside in the genus Gynura, isoquercitrin was consistent with the above data. And the elution order of isoquercitrin was in agreement with the compound prior toKaempferol-3-O-robinobioside (peak 7) and afterward with rutin (peak 5).[22–24] Thus, peak 6 was tentatively identified as isoquercitrin.
Peak 7 and 8 were a pair of isomers. Both of them gave a [M-H]- ion at m/z 593, and [M+H]+ ion at m/z 595, [M+H-146]+ ion at m/z 449, [M+H-146-162]+ ion at m/z 287. The UV spectrum showed λmax at 265 and 347 nm, which suggested peak 7 and 8 were kaempferol glycoside derivatives.[15–17,19] By examining the known kaempferol glycoside in the genus Gynura, Kaempferol-3-O-robinobioside and kaempferol-3-O-rutinoside were consistent with the above data.[25] The elution order in HPLC of Kaempferol-3-O-robinobioside being prior to kaempferol-3-O-rutinoside has been reported by many in the literature.[26,27] Thus, peak 7 and 8 were identified as Kaempferol-3-O-robinobioside and kaempferol-3-O-rutinoside, respectively.
Peak 9 yielded a [M-H]- ion at m/z 447, and [M+H]+ ion at m/z 449, [M+H-162]+ ion at m/z 287. The UV spectrum showed λmax at 265 and 346 nm, suggesting this as a kaempferol glycoside. So peak 9 is an isomer of kaempferol-3-O-β-D-glucopyranoside (peak 12). Thus, peak 9 was tentatively identified as kaempferol-3-O-β-D-galacopyranoside.
Peak 10, 11, and 13 are isomers. Both of them gave a [M-H]- ion at m/z 515, [M-H-162]- ion at m/z 353, and [M+H]+ ion at m/z 517, [M+H-18]+ ion at m/z 499, [M+H-162-192]+ ion at m/z 163. The 3 compounds also had similar UV absorptions with maxima at 327, 294 (sh), and 248 nm (sh), which is characteristic of caffeic acid derivatives.[28–31] Peak 11 was isolated by the chromatography column and identified as 3,5-Dicaffeoylquinic acid by the NMR and ESI-MS spectrum data. According to the elution order in HPLC of Dicaffeoylquinic acid reported in the literature,[31–33] 3,4-Dicaffeoylquinic acid is prior to 3,5-Dicaffeoylquinic acid, which is prior to 4,5-Dicaffeoylquinic acid, in a sequence. Thus, peak 10 and 13 were tentatively identified as 3,4-Dicaffeoylquinic acid and 4,5-Dicaffeoylquinic acid, respectively.
The flavonoid and phenolic acid compounds were affected by the concentration of extraction ethanol. The single-factor experiment showed that 60% ethanol was suitable to extract the phenolic constituents from the plant. The levels of phenolic contents were decreased as the concentration of ethanol increased. Chloroform was used to remove the nonpolar constituents, while little extracts were obtained using diethyl ether and petroleum ether. The ethyl acetate extracts showed powerful antioxidant activity and highest total phenolic content. HPLC analysis showed that ethyl acetate extracts only shared 3 principal peaks, and the kaempferol-3-O-β-D-glucopyranoside was the major constituent. However, n-butanol extract shared numerous flavonoid compounds, while the total phenolic was lower. In order to fully elaborate the phenolic compounds of the extract from G. divaricata, the extracts of ethyl acetate and n-butanol were isolated using chromatograph column and HPLC-DAD-ESI-MS method. To our best knowledge, the present study is the first report of the isolation and identification of triglycoside of kaempferol and Dicaffeoylquinic acid from the leaves of G. divaricata. And we also developed a HPLC-DAD-ESI-MS method to separate and identify the minor constituents of the n-butanol extracts. The bioactive evaluation of the isolated compounds and the crude drug deserved further research.
CONCLUSION
Seven phenolic compounds were isolated and identified from the leaves of G. divaricata, and the structures were fully elucidated by the spectrum methods. HPLC-DAD-ESI-MS method was used to identify the other 8 minor phenolic constituents of the n-butanol extracts. This was the first time to use the HPLC-DAD-ESI-MS method to identify the phytochemicals of the genera Gynura, and kaempferol-3-O-rutinoside-7-O-β-D-glucopyranoside and 3,5-Dicaffeoylquinic acid were identified for the first time from the genus Gynura.
Acknowledgments
This project was supported by the National Natural Sciences Foundation of China (No.20662008).
Footnotes
Source of Support: National Natural Sciences Foundation of China (No.20662008)
Conflict of Interest: None declared
REFERENCES
- 1.Chen YL. Flora of China. Beijing: Science Press; 1999. p. 309. [Google Scholar]
- 2.Yang X, Guo JX. Assessment on nutritive composition and nutrition value of main wild vegetables in South China. Shipin Kexue. 2002;23:121–5. [Google Scholar]
- 3.Akowuah GA, Sadikun A, Mariam A. Flavonoid identification and hypoglycemic studies of the butanol fraction from Gynura procumbens. Pharm Biol. 2002;40:405–10. [Google Scholar]
- 4.Zhang XF, Tan BK. Effects of an ethanolic extract of Gynura procumbens on serum glucose, cholesterol and triglyceride levels in normal and streptozotocin-induced diabetic rats. Singapore Med J. 2000;41:9–13. [PubMed] [Google Scholar]
- 5.Chen L, Wang JJ, Song HT, Zhang GG, Qin LP. New cytotoxic cerebroside from Gynura divaricata. Chinese Chem Lett. 2009;20:1091–3. [Google Scholar]
- 6.Chen SC, Hong LL, Chang CY, Chen CJ, Hsu MH, Huang YC, et al. Antiproliferative constituents from Gynura divaricata subsp.formosana. Chin Pharm J. 2003;55:109–19. [Google Scholar]
- 7.Chen L, Wang JJ, Zhang GG, Song HT, Qin LP. A new cerebroside from Gynura divaricata. Nat Prod Res. 2009;23:1330–6. doi: 10.1080/14786410902836677. [DOI] [PubMed] [Google Scholar]
- 8.Chen L, Li H, Song H, Zhang G. A new cerebroside from Gynura divaricata. Fitoterapia. 2009;80:517–20. doi: 10.1016/j.fitote.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 9.Hu Y, Li WL, Lin HW, Zhou M, Ren BR. Method for separation and identification of chemical constituents of Gynura divaricata. Chin J Nat Med. 2006;4:156–8. [Google Scholar]
- 10.Roeder E, Eckert A, Wiedenfeld H. Pyrrolizidine alkaloids from Gynura divaricata. Planta Med. 1996;62:386. doi: 10.1055/s-2006-957921. [DOI] [PubMed] [Google Scholar]
- 11.Dhasan PB, Jegadeesan M, Kavimani S. Cucurbitacins isolated from the fruits of Momordica cymbalaria Hook f. Pharmacogn Mag. 2008;4:96–101. [Google Scholar]
- 12.Abdel-sattar E, Abdel-Monem, Sleem AA. Biological and chemical study of Cleome paradoxa B. Pharmacogn Res. 2009;1:175–8. [Google Scholar]
- 13.Adebayo AH, Tan NH, Akindahunsi AA, Zeng GZ, Zhang YM. Anticancer and antiradical scavenging activity of Ageratum conyzoides L.(Asteraceae) Pharmacogn Mag. 2010;6:62–6. doi: 10.4103/0973-1296.59968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thakur A, Jain V, Hingorani L, Laddha KS. Phytochemical studies on Cissus quadrangularis Linn. Pharmacogn Res. 2009;1:213–5. [Google Scholar]
- 15.Ebada SS, Ayoub NA, Singab ANB, Al-azizi MM. Phytophenolics from Peltophorum africanum sond.(Fabaceae) with promising hepatoprotective activity. Pharmacogn Mag. 2008;4:287–93. [Google Scholar]
- 16.Le Gall G, Dupont MS, Mellon FA, Davis AL, Collins GJ, Verhoeyen ME, et al. Characterization and content of flavonoid glycosides in genetically modified tomato (Lycopersicon esculentum) fruits. J Agric Food Chem. 2003;51:2438–46. doi: 10.1021/jf025995e. [DOI] [PubMed] [Google Scholar]
- 17.Fiorentino A, D’abrosca B, Pacifico S, Golino A, Mastellone C, Oriano P, Monaco P. Reactive oxygen species scavenging activity of flavone glycosides from Melilotus neapolitana. Molecules. 2007;12:263–70. doi: 10.3390/12020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thuong PT, Su ND, Ngoc TM, Hung TM, Dang NH, Thuan ND, et al. Antioxidant activity and principles of Vietnam bitter tea Ilex kudingcha. Food Chem. 2009;113:139–45. [Google Scholar]
- 19.Lin LZ, Harnly JM. Phenolic compounds and chromatographic profiles of pear skins (Pyrus spp.) J Agric Food Chem. 2008;56:9094–101. doi: 10.1021/jf8013487. [DOI] [PubMed] [Google Scholar]
- 20.Ferreres F, Pereira DM, Valentão P, Andrade PB, Seabra RM, Sottomayor M. New phenolic compounds and antioxidant potential of Catharanthus roseus. J Agric Food Chem. 2008;56:9967–74. doi: 10.1021/jf8022723. [DOI] [PubMed] [Google Scholar]
- 21.Lin LZ, Harnly JM. A screening method for the identification of glycosylated flavonoids and other phenolic compounds using a standard analytical approach for all plant materials. J Agric Food Chem. 2007;55:1084–96. doi: 10.1021/jf062431s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang RZ, Wei XL, Gao FF, Wang LS, Zhang HJ, Xu YJ, et al. Simultaneous analysis of anthocyanins and flavonols in petals of lotus (Nelumbo) cultivars by high-performance liquid chromatography-photodiode array detection/electrospray ionization mass spectrometry. J Chromatogr A. 2009;1216:106–12. doi: 10.1016/j.chroma.2008.11.046. [DOI] [PubMed] [Google Scholar]
- 23.Pawlowska AM, Camangi F, Bader A, Braca A. Flavonoids of Zizyphus jujuba L.and Zizyphus spina-christi (L) Willd (Rhamnaceae) fruits. Food Chem. 2009;112:858–62. [Google Scholar]
- 24.Slimestad R, Torskangerpoll K, Nateland HS, Johannessen T, Giske NH. Flavonoids from black chokeberries, Aronia melanocarpa. J Food Compos Anal. 2004;18:61–8. [Google Scholar]
- 25.Hou WC, Lin RD, Lee TH, Huang YH, Hsu FL, Lee MH. The phenolic constituents and free radical scavenging activities of Gynura formosana Kiamnra. J Sci Food Agric. 2005;85:615–21. [Google Scholar]
- 26.Apáti P, Houghton PJ, Kéry A. HPLC investigation of antioxidant components in Solidaginis herba. Acta Pharm Hung. 2004;74:223–31. [PubMed] [Google Scholar]
- 27.Pietta P, Gardana C, Mauri P, Zecca L. High-performance liquid chromatographic analysis of flavonol glycosides of Solidago virgaurea. J Chromatogr. 1991;558:296–301. [Google Scholar]
- 28.Lin LZ, Harnly JM. Identification of the phenolic components of chrysanthemum flower (Chrysanthemum morifolium Ramat) Food Chem. 2010;120:319–26. [Google Scholar]
- 29.Inbaraj BS, Lu H, Kao TH, Chen BH. Simultaneous determination of phenolic acids and flavonoids in Lycium barbarum Linnaeus by HPLC-DAD-ESI-MS. J Pharm Biomed Anal. 2010;51:549–56. doi: 10.1016/j.jpba.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 30.Nugroho A, Kim KH, Lee KR, Alam MB, Choi JS, Kim WB, et al. Qualitative and quantitative determination of the caffeoylquinic acids on the Korean mountainous vegetables used for chwinamul and their peroxynitrite-scavenging effect. Arch Pharm Res. 2009;32:1361–7. doi: 10.1007/s12272-009-2003-6. [DOI] [PubMed] [Google Scholar]
- 31.Dugo P, Cacciola F, Donato P, Jacques RA, Caramão EB, Mondello L. High efficiency liquid chromatography techniques coupled to mass spectrometry for the characterization of mate extracts. J Chromatogr A. 2009;1216:7213–21. doi: 10.1016/j.chroma.2009.08.030. [DOI] [PubMed] [Google Scholar]
- 32.Clifford MN, Kirkpatrick J, Kuhnert N, Roozendaal H, Salgado PR. LC-MSn analysis of the cis-isomers of chlorogenic acids. Food Chem. 2008;106:379–85. [Google Scholar]
- 33.Zheng W, Clifford MN. Profiling the chlorogenic acids of sweet potato (Ipomoea batatas) from China. Food Chem. 2008;106:147–52. [Google Scholar]