

Abstract
The contribution of microenvironment to tumor growth has important implications for optimizing chemotherapeutic response and understanding the biology of recurrent tumors. In this study, we tested the effects of locally administered topotecan on a rat model of glioblastoma that is induced by intracerebral injection of PDGF-IRES-GFP-expressing retrovirus, we treated the tumors by convection-enhanced delivery (CED) of topotecan (136 μM) for 1, 4, or 7 days and then characterized the effects on both the retrovirus-transformed tumor cells (GFP+ cells) as well as the uninfected glial progenitor cells (GFP- cells) that are recruited to the tumor. Topotecan treatment reduced GFP+ cells ~10-fold and recruited progenitors by ~80-fold while providing a significant survival advantage that improved with greater treatment duration. Regions of glial progenitor ablation occurred corresponding to the anatomical distribution of topotecan as predicted by MRI of a surrogate tracer. Histopathologic changes in recurrent tumors point to a decrease in recruitment, most likely due to the chemotherapeutic ablation of the recruitable progenitor pool.
Keywords: Glioma, Glioblastoma, Topotecan, Convection Enhanced Delivery, progenitor
Introduction
Glioblastoma is the most common and most malignant form of primary brain tumor in adults. Understanding the contribution of the tumor microenvironment to glioma growth and response to therapy could lead to novel therapeutic approaches. Most studies concerned with tumor microenvironment have focused on the tumor vasculature. However, glioma cells diffusely infiltrate the brain and intermingle extensively with the surrounding brain tissue. Therefore, the microenvironment of gliomas contains a complex mixture of entrapped and reactive cells. Prominent among these are glial progenitors, which are widely distributed throughout the brain and spinal cord and represent the largest populations of cycling cells in the adult central nervous system [1–4]. Glial progenitors proliferate extensively in response to brain injury [5, 6] and recent studies have suggested that they also proliferate within gliomas and contribute to tumor growth [7–11].
We have developed an animal model of glioblastoma that is induced by selectively targeting adult glial progenitors to overexpress PDGF via stereotactic injection of PDGF-IRES-GFP retrovirus into the subcortical white matter of adult rats [9]. This model has shown remarkable consistency, with 100% of injected animals succumbing to tumor-induced morbidity within 2 to 3 weeks. More importantly, these PDGF-driven tumors recapitulate the cardinal histologic features of human glioblastoma multiforme (GBM, WHO IV) including a high degree of proliferation (~30% Ki67 labeling index), diffuse invasiveness, glomeruloid vascular proliferation, and pseudopalisading necrosis. A compelling finding from this study was that the PDGF-driven tumors are composed of both retrovirally infected cells and uninfected glial progenitors that were “recruited” to participate in tumor formation through paracrine PDGF signaling. The recruited progenitors are highly migratory and proliferative, suggesting that they contribute to tumor growth and dispersion and are not merely entrapped cells [9, 10]. Furthermore, these features are observable clinically as we have shown that human primary glioma cells taken directly from patients as well as from a PDGF-expressing human glioma cell line are similarly capable of recruiting host-derived glial progenitors when xenografted into immunocompromised rats [12].
In this model, the ability to distinguish between retrovirus-infected/tumor-initiating cells and uninfected/recruited progenitors on the basis of GFP expression provides a unique opportunity to compare the effects of chemotherapy on these two cell populations. In this study, topotecan, a camptothecin-class inhibitor of topoisomerase I, was directly infused into the tumor and surrounding brain through the interstitial space via convection enhanced delivery (CED) [13]. Topoisomerase I causes a single-stranded DNA break and forms a “cleavable complex” [14] thus relaxing the DNA supercoil. Topotecan stabilizes the cleavable complex and, when a replication fork meets the complex, an irreversible double-stranded break occurs—the accumulation of which leads to S/G2-phase arrest and apoptosis [15]. The drug is lethal to cells that are undergoing DNA replication and appears to be specific for cells in S-phase [15–20]. Topotecan is a good candidate for administration via CED because it does not cross the blood-brain barrier particularly well [21] and is associated with dose-limiting toxicity when given systemically. Our own previous work with topotecan in an intracranial C6 model showed the ability to safely achieve greater than 1000-fold higher concentrations of topotecan in rat brain when delivered via CED compared to systemic delivery [22].
We predicted that both the proliferative PDGF-producing cells as well as the proliferative recruited glial progenitors in our retroviral tumors would be susceptible to the drug’s activity. We further hypothesized that, because topotecan activity is cell-cycle dependent, prolonging CED of topotecan would significantly decrease the number of residual cells from both cell populations (i.e. infected and recruited) in a time-dependent manner and thus lead to increased survival. Lastly, by analyzing tumors that recurred after treatment, we sought to determine the relative contribution of tumor cells versus recruited glial progenitors in the composition of recurrent tumors.
Materials and Methods
Retrovirus production and stereotactic injections
The PDGF-IRES-GFP retrovirus was produced as previously described [9]. Viral titers were ~10^5 CFU/mL. 5 uL of viral suspension was injected into the right frontal sub-cortical white matter (stereotactic coordinates relative to bregma: 2 mm right, 2 mm rostral, 4 mm ventral) using a 10 uL syringe with a 32G needle (Hamilton, Reno, NV) at a rate of 0.2 uL/min. Electronic syringe pumps were used for the injections (Stoelting, Wood Dale, IL). Two minutes after injection, the needle was slowly retracted and the incision primarily closed with nylon sutures.
Osmotic mini-pump implantation
Osmotic mini-pumps connected to an intracerebral infusion cannula via a catheter (model 2ML1, Brain Infusion Kit 2; Alzet, Wood Dale, IL) were prepared and filled with the desired infusate per manufacturer’s instructions. Pump volume was 2 mL with a flow rate of 10 uL/hr (complete infusion of contents over 7 days). Pumps contained either an isotonic solution of 136 μM topotecan hydrochloride (LKT Labs, St. Paul, MN) or phosphate-buffered saline (PBS) (Gibco, Carlsbad, CA). Topotecan concentration was the maximum tolerated dose determined from preliminary experiments (data not shown). At 7 days post-injection (dpi) of PDGF retrovirus, animals were anesthetized with 2% isoflurane and attached to a stereotactic head frame as above. The previous incision was re-opened and a subcutaneous pocket was formed between the animal’s shoulder blades via blunt dissection. The pump body was inserted into the pocket and the cannula slowly inserted with a probe holder through the same burr hole used for virus injection. The cannula was then secured to the skull with cyanoacrylate glue. The incision was again primarily closed with nylon sutures. Animals underwent an additional surgery also under gas anesthesia for removal of the pump and cannula apparatus.
Survival Studies
Thirty-six animals were used for 2 separate survival studies. In each experiment there were four groups of 3–5 animals each. Control animals received PBS by CED for a total duration of 7 days. Three treatment schedules for topotecan by CED were used in separate groups of animals; 1 day, 4 days, and 7 days. These animals were monitored daily for level of activity, seizure, posturing, and nasal or periorbital hemorrhage. Animals were sacrificed following observation of any of the aforementioned manifestations of tumor morbidity. Survival data analysis was done via the Kaplan-Meier method with statistical significance determined with a post-hoc log-rank test (Prism 4.0). Primary cell cultures of PDGF-driven tumors were generated as previously described [23, 24].
Short-term studies
Immediate post-treatment histologic analyses were performed on 32 tumor-bearing animals (two independent experiments, n=14–18 per experiment). In these experiments, animals received PBS or topotecan via CED for either 1, 4 or 7 days and were immediately sacrificed (n=2–3 per treatment group, per time-point). Also, a study of the regional distribution of PDGFR-α+ glial progenitors was performed in twelve non-tumor-bearing animals (two independent experiments, n=3 untreated animals, n=3 topotecan CED × 7 days per experiment). The regional counts of PDGFR-α+ glial progenitors were also performed on tumor-bearing animals that received PBS via CED and topotecan via CED for 7 days. Mean cell counts were analyzed via one-way ANOVA with post hoc test for linear trend and/or student’s t-test depending on the number of groups being compared (Prism 4.0). All counts are expressed as mean ± SEM.
MRI studies
MRI was performed on six animals to determine the volume of distribution achieved by CED of topotecan. Tumor-bearing animals were implanted with osmotic mini-pumps as above with the addition of gadodiamide (GE Healthcare, Piscataway, NJ; 1:100 dilution, ~5 mM final concentration) into the 136 mM topotecan solution. Animals received a 1-day or 7-day infusion of the gadodiamide/topotecan solution (n=3 per time-point). Mean distribution volume was determined by manual tracing of T1-weighted hyperintense areas. Areas of hyperintensity were thresholded in comparison to the intensity of normal white matter. Areas were multiplied by slice thickness (1 mm) and summated to give a total volume of distribution (Vd). Measurements were performed using Osirix software (Los Angeles, CA). Mean Vds were compared via student’s t-test (Prism 4.0).
Immunohistochemistry
Animals underwent intra-cardiac perfusion of 4% paraformaldehyde prior to brain harvesting. Hematoxylin and eosin (H&E) stains were done on 5 μm paraffin-embedded sections. Immunofluorescence staining was performed on 10 μm cryosections at the genu of the corpus callosum (< 500 mm from catheter site). Antibodies used were: rabbit anti-GFP (1:500; Invitrogen, Eugene, OR), sheep anti-GFP (1:200, AbD Serotec, Raleigh, NC), rabbit anti-Ki67 (1:1000, Dako, Carpinteria, CA), mouse anti-phosphohistone 3 (Abcam, Cambridge, MA), rabbit anti-PDGF receptor alpha (PDGFR-α; a kind gift from William Stallcup, PhD, Burnham Institute for Medical Research, La Jolla, CA), PDGFR-α (Cell Signaling, MA), rabbit anti-activated Caspase-3 (Cell signaling, MA), rabbit anti-Olig2 (Millipore, MA), rabbit anti-NG2 (Millipore, MA), mouse anti-Nestin (Millipore, MA), rabbit anti-sox2 (Millipore, MA), mouse anti smooth muscle actin SMA (Dakocytomation), and the appropriate secondary antibodies. Microscopy was performed with an Eclipse TE-2000 fluorescent microscope (Nikon, Melville, NY) and images taken with Metamorph software (Molecular Devices, Downingtown, PA). Cell counts were performed on 200× or 400× fields and expressed as number of positive cells per high-power field (HPF) and percentage of total cells per field, as determined by nuclear stains with Hoechst 33342 (Molecular Probes, Eugene, OR).
Results
Chronic CED of topotecan increases survival
Median survival of control (PBS) animals was 20 dpi. Animals given 1 day of topotecan via CED had a median survival of 23 dpi. Animals given 4 days of topotecan CED had a median survival of 31 dpi. Seven days of topotecan CED increased median survival to 54 dpi. The survival curves for all three treatment groups were statistically different from the control curve (p<0.05). The survival curve of 7-day treatment was statistically different when compared to 1-day and 4-day curves as well (p<0.05). Figure 1 shows Kaplan-Meier curves of a representative survival study.
Figure 1. Chronic CED of topotecan provides significant survival advantage.
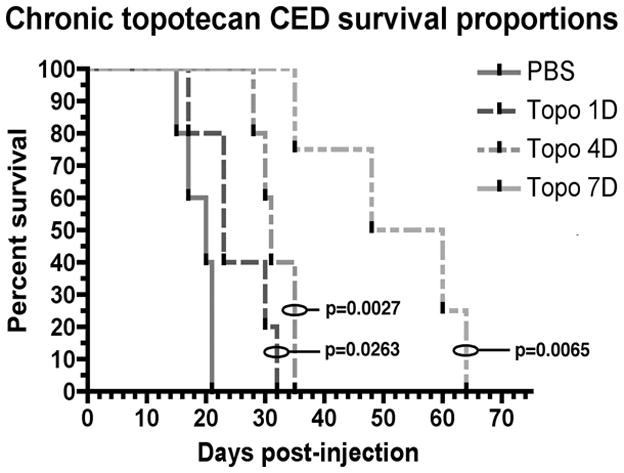
Median survival of PBS-treated animals was 20 dpi. Animals that received 1 day of topotecan CED had a median of survival of 23 dpi. Animals that received 4 days of treatment had a median survival of 31 dpi. Animals that received 7 days of treatment had a median survival of 54 dpi. P-values shown are for comparisons of each curve to the control (log-rank test). Each treatment curve was also statistically different from every other treatment curve (p<0.05).
Chronic CED of topotecan ablates both tumor-initiating cells and recruited glial progenitors
H&E stains of brains harvested immediately post-treatment (i.e. PBS vs. topotecan at 1, 4, and 7 days post-implantation of pumps) show apparent eradication of tumor cells by topotecan treatment after 7 days of CED (Figure 2). However, immunofluorescence staining for the retroviral GFP tag revealed the presence of a few residual tumor cells (Figure 3A).
Figure 2. Seven-day CED of topotecan causes loss of identifiable tumor cells.

A. PDGF retroviral tumor after 7-day CED of PBS. Note the large proliferative lesion with cells invading through the corpus callosum (CC). Thick arrow points to the injection/cannula site. Thin arrows point to areas of pseudopalisading necrosis. B. Topotecan-treated brain (7 days post-treatment) shows the absence of any identifiable tumor cells.
Figure 3. CED of topotecan causes loss of both tumor-initiating cells and recruited glial progenitors.
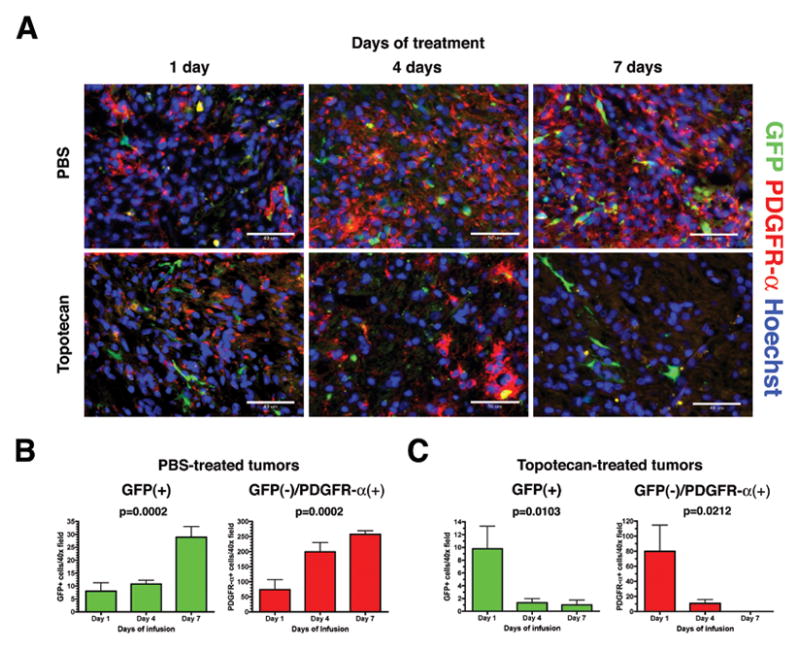
A. Immunohistochemistry for GFP (green) and PDGFR-α (red) shows an increase in both the initially infected PDGF-expressing cells (GFP+) as well as the recruited progenitors (GFP-/PDGFR-α+) over 1, 4, and 7 days when animals are given CED of PBS (upper row). CED of topotecan causes a similarly time-dependent decrease in cells from both populations (lower row). Hoechst nuclear counterstain in blue. B. Increase in GFP+ and GFP-/PDGFR-α+ cells per HPF over time in PBS-treated tumors was statistically significant on one-way ANOVA and also positive for test of linear trend (p=0.0002 and p=0.0002, respectively). C. Decrease in GFP+ and GFP-/PDGFR-α+ cells per HPF over time in topotecan-treated tumors was statistically significant on one-way ANOVA and also positive for test of linear trend (p=0.0103 and p=0.0212, respectively). Fields were taken at 400× magnification (scale bar= 50 μm).
In PBS-treated animals, the number of GFP+ cells as well as PDGFR-α+ cells increased as the tumor grew in a time-dependent manner (Figure 3B). Mean ± SEM GFP+ cells/HPF increased from 8.00 ± 3.28 at day 1 to 10.78 ± 1.43 at day 4 and finally 28.89 ± 4.10 at day 7. Means were statistically different on one-way ANOVA (p=0.0002) and significant on post hoc test for linear trend (p<0.0001). Mean PDGFR-α+ cells/HPF increased from 73.89 ± 32.80 at day 1 to 199.2 ± 30.75 at day 4, and finally to 257.3 ± 11.93 at day 7. Means were statistically different on ANOVA (p=0.0002) and significant for linear trend (p<0.0001).
The opposite effect was observed in topotecan-treated animals (Figure 3C). Mean GFP+ cells/HPF decreased from 9.78 ± 3.52 at day 1 to 1.33 ± 0.65 at day 4, and to 1.00 ± 0.73 at day 7. Means were statistically different (p=0.0103) and significant for linear trend (p=0.0071). Similarly, mean PDGFR-α+ cells/HPF decreased with prolonged treatment from 79.89 ± 34.83 at day 1 to 10.89 ± 4.93 at day 4, and to 0.11 ± 0.11 at day 7. Means were statistically different (p=0.0212) and significant for linear trend (p=0.0105). Similar results were seen when we stained the topotecan-treated and PBS-treated tumors for the glial progenitor marker olig2. As previously reported [9], the tumors were predominantly composed of olig2+ cells, and 7 days of CED caused a nearly complete ablation of the olig2+ population (74.0 ± 6.03 cells/HPF for PBS treated vs 0.167 ± 0.0882 cells/HPF for topotecan-treated, p=0.0003, Figure 2S).
To further elucidate the mechanism by which topotecan is ablating tumor cells, we measured the activated caspase-3 index in tumors treated for 7 days with CED of topotecan versus PBS. The caspase-3 index increased ~8-fold from (1.53 ± 0.491 %cells/field in PBS-treated animals to 11.8 ± 0.536 %cells/field) in topotecan-treated animals (p= 0.001) as shown in Figure 2S. These results demonstrate that topotecan is ablating tumor cells by inducing apoptosis, as previously reported [35, 36].
Next, we tested the effects of topotecan on primary cell cultures generated from PDGF-driven tumors. We first measured topotecan effects on primary culture tumor cells with MTT assay. Incubation of tumor cells with topotecan resulted in a dose-dependent inhibition in tumor cell growth with greater than 80% inhibition demonstrated at 136 μM topotecan (Fig 3SA). We next examined the changes in the total number of tumor cells, GFP+/Olig2+ (tumor-initiating) and GFP-/Olig2+ (recruited) cells due to a 24 hr dose response treatment of topotecan (PBS control, 136 μM, 13 μM, 1.3 μM). The results indicated a statistically significant drop in the number of cells in all cases (one-way ANOVA, Figure 3SB and sup table I). Similarly treating the primary cultures with topotecan (136 μM) caused a nearly complete ablation of PDGFR-α+ cells, including both the GFP+ and the GFP- subpopulations (Fig 3SC).
To further characterize the immunophenotype of cells in the PBS-treated and topotecan-treated brain tumors, we performed immunofluorescence with a panel of antibodies against glial cell and progenitor cell markers (Olig2, NG2, Nestin, GFAP, and Sox2) and proliferation markers (Ki67 and PHH3). In the PBS treated tumors the vast majority of cells expressed PDGFRa (Fig 3), Olig2 (Fig 2SA), NG2 (4SA), Nestin(4SC), and Sox2 (Fig 4SE), and approximately 30% of the cells are Ki67+ (Fig 4SG). Double immunofluorescence analysis with olig2 and PHH3 (which selectively labels cells in G2/M phase of the cell cycle) showed that virtually all proliferating cells were olig2+ (Fig 5SA). These finding are consistent with our previously published results [9, 10]. The Topotecan-treated tumors showed a nearly complete loss of cells expressing glial progenitor markers, PDGFRα (Fig 3), Olig2+ (Fig 2SB), NG2 (Fig 4SB) and cells expressing proliferation markers Ki67 (Fig 4SH) and PHH3 (Fig 5SB). There were a few NG2+ cells which were closely aligned with blood vessels and co-expressed smooth muscle actin (SMA), indicating that they are pericytes (Fig 5SD). There were also some Nestin+/Sox2+ cells scattered around the treatment site. These cells co-expressed GFAP and had the morphological features of reactive astrocytes (Fig. 6SB,D).
Loco-regional ablation of glial progenitors coincides with the distribution of topotecan/gadodiamide solution administered via CED
Mean Vd of topotecan/gadodiamide solution after 1 day of CED was 0.50 mL ± 0.03 and did not statistically differ from 7 days of CED at 0.55 mL ± 0.10 (p=0.4455). At 7 days, more infusate was detected caudal and contralateral (across the corpus callosum) to the cannula site (Figure 4). Knowing the approximate anatomic distribution of drug solution, we then measured the abundance of PDGFR-α+ cells in normal adult rat brains in three separate regions: the lateral white matter on both sides of the brain as well as the midline of the corpus callosum. Mean PDGFR-α indices (# of positive cells/HPF) in normal brain were as follows: left white matter=4.51±0.70, corpus callosum=2.84±0.29, and right white matter=3.72±0.57. These means were not statistically different on one-way ANOVA (p=0.1765). We subsequently measured PDGFR-α indices in the same three regions in animals from 3 different experimental groups: animals without tumors given topotecan via CED, animals with tumors given PBS via CED, and animals with tumors given topotecan via CED. All animals received 7 days of CED, with pump implantation for tumor-bearing animals occurring at 7 days post-injection of PDGF-IRES-GFP retrovirus. Mean regional PDGFR-α indices from each experimental group were then compared to the mean indices from the corresponding regions of non-treated adult brain using the student’s t-test. Results show that CED of topotecan significantly decreases the local PDGFR-α+ glial progenitor population. Decreases in PDGFR-α indices were seen in areas immediately proximal to the infusion site in topotecan-treated vs. control normal brain (0.44±0.76 vs. 3.72±0.98, p<0.05) and tumor-bearing animals (0.07±0.13 vs. 94.82±7.89, p<0.01). Further, this effect was seen at significant distances distally, up to the region of the corpus callosum in topotecan-treated vs. control normal brain (0.27±0.47 vs. 2.84±0.50, p<0.01) and in topotecan-treated vs tumor-bearing animals (0.37±0.40 vs. 53.48±44.21, p<0.01; Figure 5).
Figure 4. Vd of gadodiamide/topotecan CED at 1 day was statistically similar to Vd at 7 days.

A. Heat maps of hyperintense gadolinium signal on T1-weighted MRIs show the anatomic distribution of infusate at 1 day vs. 7 days post-infusion in 3 planes (COR=coronal, AX=axial, SAG=sagittal). At 1 day, the ipsilateral (right) frontal lobe is completely infiltrated with infusate. At 7 days, infusate has extended posteriorly into the ipsilateral parietal lobe and also across the corpus callosum into the contralateral striatum. B. There was a slight increase in mean Vd between 1-day vs. 7-day post-infusion (0.5044±0.0186 mL vs. 0.5566±0.0576 mL respectively) but this was not statistically significant (p=0.4455).
Figure 5. Loco-regional ablation of PDGFR-α+ glial progenitors coincides with anatomic distribution of gadodiamide/topotecan infusate.
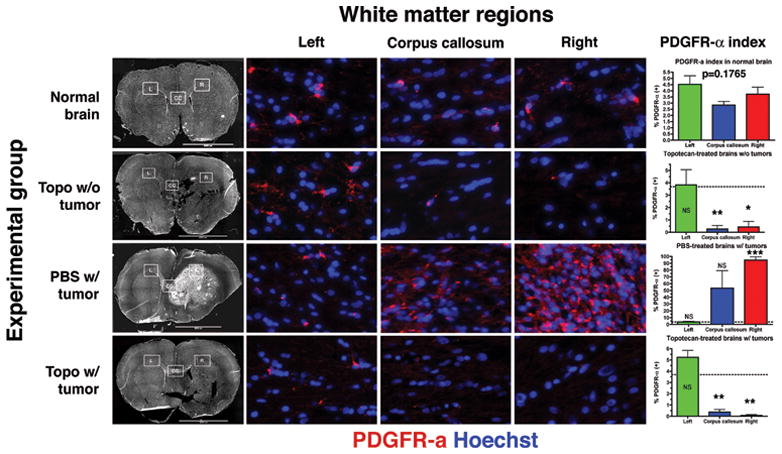
Immunohistochemistry for PDGFR-α (red) and Hoechst counterstain (blue) was done in 3 different anatomic regions of the white matter (inset boxes on whole brain montages in the first column from left; L=left, CC=corpus callosum, R=right). Rows correspond to different experimental groups. Columns 2 to 3 correspond to white matter regions as labeled. High-power images taken at ~800× magnification. Last column shows graphical representation of mean ± SEM PDGFR-α labeling indices. t-tests were performed comparing means from each region in each experimental group with the corresponding region in normal brain. NS=not significant, *p<0.05, **p<0.01, ***p<0.001. Dotted line represents mean PDGFR-α labeling index of all regions of normal white matter in aggregate (3.69%; not statistically different on one-way ANOVA, p=0.1765).
Recurrent tumors contain fewer recruited progenitors
Consistent with our previous work [9], naïve and PBS-treated control tumors were largely composed of uninfected glial progenitors and only ~20% of tumor cells expressed detectable levels of GFP (Figure 6A & 6C). In contrast, tumors that recurred after 7 days of topotecan CED, contained a higher proportion of cells that expressed GFP (Figure 6B & 6D). This was most pronounced in central areas of the recurrent tumors, with some regions approaching 100% GFP+ cells. A more heterogeneous mixture of GFP+ and GFP- cells was seen towards the infiltrative tumor boundary. Just as in naïve tumors, recurrent tumors were composed of PDGFR-α + cells. Histologically, recurrent tumors maintained their GBM-like phenotype except for the presence of larger, more developed tumor-associated vessels (Figure 6E & 6F).
Figure 6. Recurrent tumors are composed of a higher proportion of tumor-initiating GFP+ cells.
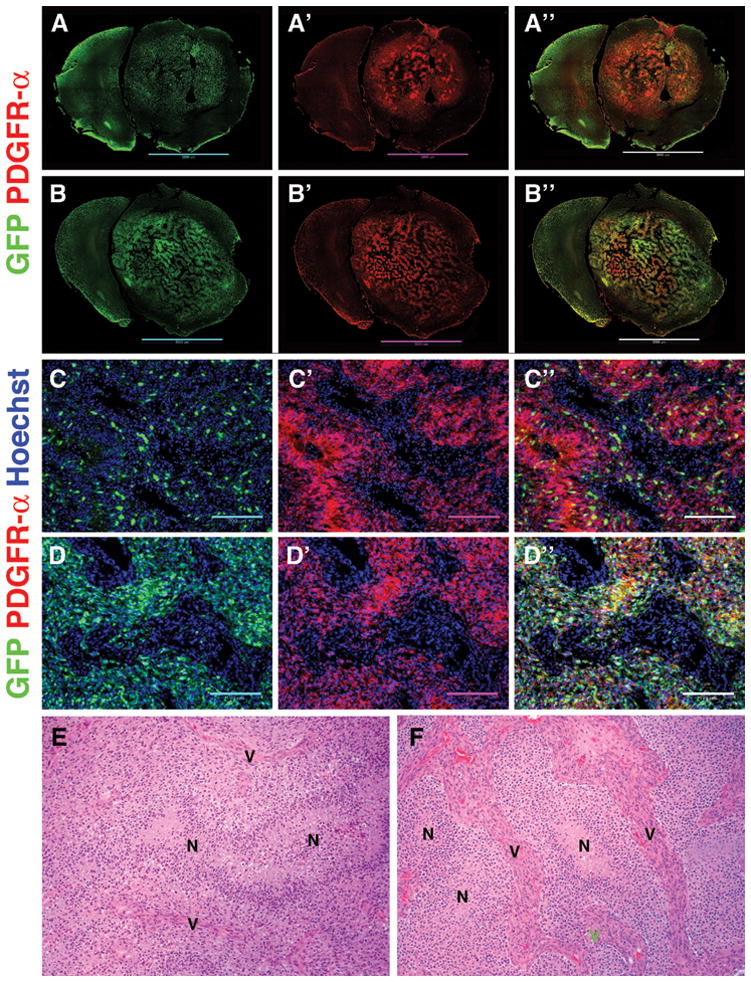
A–A″. Color-separated triptych shows distribution of GFP+ cells (green) in control PBS-treated tumors. The majority of tumor cells are PDGFR-α+ (red) but GFP-. B–B″. Tumors that recurred after topotecan CED (7 days) have a higher proportion of GFP+ cells than naïve tumors. The majority of cells are still PDGFR-α+. C–C″. Color-separated triptych of a naïve tumor showing the distribution and relative abundance of GFP+ cells (100× magnification, scale bar 200 mm). D–D″. Color-separated triptych of a recurrent tumor post-topotecan CED shows increased abundance of GFP+ cells. Hoechst nuclear counterstain in blue. E. H&E stains of a naïve tumor showing vascular proliferation (V) and areas of pseudopalisading necrosis (N). F. H&E stains of a recurrent tumor post-topotecan CED showing similar areas of necrosis (N) but larger tumor vessels (V).
Discussion
By using a growth-factor driven glioma model, which exhibits robust recruitment of endogenous glial progenitors by tumor-initiating cells, we have been able to distinguish, for the first time, the effects of chemotherapy on these two distinct cell populations. Analysis of the initially infected cells and their progeny (i.e. GFP+ cells) as well as the recruited cells (i.e. GFP-/PDGFR-α+) reveals that both populations are significantly decreased by topotecan CED in a time-dependent manner. Even by the 4th day post-treatment, GFP+ cells were decreased 10-fold and recruited cells were decreased 8-fold. This statistically significant trend was continued through 7 days of treatment (Fig. 3B).
Administration of topotecan via CED produced a statistically significant survival advantage at all three tested durations. The magnitude of survival advantage steadily increased in a time-dependent manner as well, with longer treatment times associated with longer survival. It is not surprising that increased duration of topotecan administration increases cytotoxic effects because the proportion of cells cycling through S-phase (either tumor-initiating or recruited) presumably increases over time and topotecan cytotoxicity is specific for cells in S-phase [18–20]. None of the topotecan-treated animals experienced morbidities related to the pump implantation or drug administration over the full 7-day course. The efficacy and safety of prolonged topotecan CED in these animals suggests that chronic CED of topotecan may be a feasible treatment modality for human glioma patients.
MRI analysis following CED of gadodiamide reveals that, even at 1 day post-infusion, the Vd is sufficient to encompass the main tumor mass (Supplementary Figure 1). Although the Vd was not statistically greater after 7 days of infusion, gadodiamide distribution did shift anatomically to more caudal regions as well as more extensively across the corpus callosum to the contralateral side. This implies that steady-state distribution volume was achieved relatively early in treatment. Since gadodiamide distribution is at least an approximate surrogate for topotecan distribution, the MRI scans also provide indirect support that topotecan concentrations were uniformly maintained at high levels throughout the infusion period. Regional analysis of PDGFR-α+ glial progenitor cell distribution compared to gadodiamide distribution on MRI confirms that topotecan was present in concentrations that were toxic to glial progenitors. Nearly complete ablation of PDGFR-α+ cells was observed at the site of infusion and extended to the corpus callosum, while on the side contralateral to treatment, PDGFR-α labeling indices in white matter were statistically similar to normal brain.
While CED of topotecan efficiently killed both the tumor-initiating and recruited cell populations, all animals in the survival studies eventually succumbed to tumor recurrence. Even with 7 days of treatment, rare residual GFP+ cells could be identified through careful immediate post-treatment histological analysis. Consequently, recurrent tumors also contained GFP+ cells, indicating that tumor recurrence was derived from the progeny of retrovirus-infected cells. In recurrent tumors, however, entire regions were seen where nearly 100% of cells were GFP+. As we previously reported [9], only ~20% of cells are GFP+ in the naïve tumors and these cells are widely distributed throughout the entire tumor mass. Solid islands of GFP+ cells were most prominently located within central core areas of recurrent tumor. Towards the infiltrative tumor boundaries, however, increasing numbers of recruited progenitors (i.e. GFP/PDGFR-α+) were seen. This pattern of GFP expression in recurrent tumors is consistent with a marked reduction in the recruitable glial progenitor pool within the zone of treatment, while the small number of residual GFP+ cells surviving treatment continue to proliferate in a cell-autonomous manner, thus forming a central tumor core devoid of recruitment. As the tumor expands and GFP+ cells once again begin to infiltrate the surrounding normal tissue, populations of recruitable PDGF-responsive progenitors that remained after treatment (because they were outside the region of drug distribution) are induced to proliferate and contribute to the growing mass of tumor cells.
At present it is not known what effect the recruited progenitor population may have on tumor growth, survival and time to recurrence. It is possible that the recruited progenitors have an inhibitory effect on the tumor cell population [26]. For example, the recruited progenitors may compete with the bone fide glioma cells for resources such as oxygen or growth factors or may even secrete factors that directly inhibit tumor cell proliferation or survival. If so, topotecan-mediated ablation of the recruited progenitors may actually facilitate tumor re-growth and shorten the time to recurrence.
The increased proportion of GFP+ cells in recurrent tumors (at least regionally) not only points towards a decrease in the recruitable progenitor pool but may also indicate selection for a treatment-resistant phenotype. Perhaps these recurrent cells have cell-cycle lengths that exceed 7 days, such that they never entered S-phase during the treatment period, rendering them insensitive to topotecan. Other mechanisms of resistance to topoisomerase I-inhibition by camptothecin-class drugs have also been reported including decreased enzyme expression [28–31] or decreased enzyme activity [32, 33]. Additionally, we observed that recurrent tumors had much more robust tumor vasculature than naïve tumors and that these recurrent vessels were often found to be surrounding the islands of pure GFP+ tumor cells. It would be of great interest to explore in future studies whether these recurrent tumor cells as well as the recurrent tumor environment have, in fact, been fundamentally changed by treatment. It might also be useful to test treatment modalities that combine CED of traditional cytotoxic drugs (such as topotecan) with targeted anti-angiogenic drugs. Philips et al. have reported, through microarray gene expression profiling, that recurrent tumors are characterized by a shift from a proneural subtype to a more mesenychymal one [34]. Because our treatment regime efficiently and reproducibly reduces tumor cells to a true residual population and does not merely produce a treatment failure, our recurrent tumor model is well suited to study tumor recurrence.
In summary, we have shown that topotecan delivered via CED safely and effectively prolongs survival in our retroviral tumor model with toxicity against both tumor-intiating and recruited cells. Because topotecan is cell cycle-dependent, extended administration of topotecan most likely increases survival in a time-dependent manner by maintaining an effective tissue concentration for a longer duration. Lastly, chemotherapeutic ablation of adult glial progenitors by CED of topotecan is loco-regional and results in recurrent tumors with proportionally more tumor-initiating cells and fewer recruited progenitors. Future efforts will elucidate the relationship of tumor composition to survival as well as the potential cellular changes in recurrent tumors that result from treatment.
Supplementary Material
Acknowledgments
Financial Support:
This work was supported by National Institutes of Health grant RO1 CA 89395 (JNB) and the B.R.A.I.N. Foundation. We thank Ben C. Kennedy for critically reading the manuscript.
Footnotes
There are no conflicts of interest to disclose.
Author Contributions:
KAL, PC and JNB conceived the study, participated in study design, interpreted results and wrote the manuscript. KAL performed the data analysis. KAL, AMT, MCA, and KL conducted the animal studies. JPY and OG conducted the in vitro studies. All authors commented on the manuscript.
References
- 1.Dawson MR, Polito A, Levine JM, Reynolds R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci. 2003;24:476–488. doi: 10.1016/s1044-7431(03)00210-0. [DOI] [PubMed] [Google Scholar]
- 2.Horner PJ, Thallmair M, Gage FH. Defining the NG2-expressing cell of the adult CNS. J Neurocytol. 2002;31:469–480. doi: 10.1023/a:1025739630398. [DOI] [PubMed] [Google Scholar]
- 3.Rhee W, Ray S, Yokoo H, Hoane ME, Lee CC, Mikheev AM, Horner PJ, Rostomily RC. Quantitative analysis of mitotic Olig2 cells in adult human brain and gliomas: implications for glioma histogenesis and biology. Glia. 2009;57:510–523. doi: 10.1002/glia.20780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roy NS, Wang S, Harrison-Restelli C, Benraiss A, Fraser RA, Gravel M, Braun PE, Goldman SA. Identification, isolation, and promoter-defined separation of mitotic oligodendrocyte progenitor cells from the adult human subcortical white matter. J Neurosci. 1999;19:9986–9995. doi: 10.1523/JNEUROSCI.19-22-09986.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alonso G. NG2 proteoglycan-expressing cells of the adult rat brain: possible involvement in the formation of glial scar astrocytes following stab wound. Glia. 2005;49:318–338. doi: 10.1002/glia.20121. [DOI] [PubMed] [Google Scholar]
- 6.Zhao JW, Raha-Chowdhury R, Fawcett JW, Watts C. Astrocytes and oligodendrocytes can be generated from NG2+ progenitors after acute brain injury: intracellular localization of oligodendrocyte transcription factor 2 is associated with their fate choice. Eur J Neurosci. 2009;29:1853–1869. doi: 10.1111/j.1460-9568.2009.06736.x. [DOI] [PubMed] [Google Scholar]
- 7.Shih AH, Dai C, Hu X, Rosenblum MK, Koutcher JA, Holland EC. Dose-dependent effects of platelet-derived growth factor-B on glial tumorigenesis. Cancer Res. 2004;64:4783–4789. doi: 10.1158/0008-5472.CAN-03-3831. [DOI] [PubMed] [Google Scholar]
- 8.Appolloni I, Calzolari F, Tutucci E, Caviglia S, Terrile M, Corte G, Malatesta P. PDGF-B induces a homogeneous class of oligodendrogliomas from embryonic neural progenitors. Int J Cancer. 2009;124:2251–2259. doi: 10.1002/ijc.24206. [DOI] [PubMed] [Google Scholar]
- 9.Assanah M, Lochhead R, Ogden A, Bruce J, Goldman J, Canoll P. Glial progenitors in adult white matter are driven to form malignant gliomas by platelet-derived growth factor-expressing retroviruses. J Neurosci. 2006;26:6781–6790. doi: 10.1523/JNEUROSCI.0514-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Assanah MC, Bruce JN, Suzuki SO, Chen A, Goldman JE, Canoll P. PDGF stimulates the massive expansion of glial progenitors in the neonatal forebrain. Glia. 2009;57:1835–1847. doi: 10.1002/glia.20895. [DOI] [PubMed] [Google Scholar]
- 11.Ogden AT, Waziri AE, Lochhead RA, Fusco D, Lopez K, Ellis JA, Kang J, Assanah M, McKhann GM, Sisti MB, McCormick PC, Canoll P, Bruce JN. Identification of A2B5+CD133- tumor-initiating cells in adult human gliomas. Neurosurgery. 2008;62:505–514. doi: 10.1227/01.neu.0000316019.28421.95. discussion 514-505. [DOI] [PubMed] [Google Scholar]
- 12.Lopez KAAM, Waziri A, Fusco D, Tannebaum A, Linksey K, McKhann GM, Sisti MB, Bruce J, Canoll P. Human Glioma Cells Recruit and Expand Adult Glial Progenitors via Paracrine Platelet-dervied Growth Factor Signaling. Neurosurgery. 2008;62:1424–1425. [Google Scholar]
- 13.Lopez KA, Waziri AE, Canoll PD, Bruce JN. Convection-enhanced delivery in the treatment of malignant glioma. Neurol Res. 2006;28:542–548. doi: 10.1179/016164106X116836. [DOI] [PubMed] [Google Scholar]
- 14.Hsiang YH, Liu LF. Identification of mammalian DNA topoisomerase I as an intracellular target of the anticancer drug camptothecin. Cancer Res. 1988;48:1722–1726. [PubMed] [Google Scholar]
- 15.Hsiang YH, Lihou MG, Liu LF. Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res. 1989;49:5077–5082. [PubMed] [Google Scholar]
- 16.Holm C, Covey JM, Kerrigan D, Pommier Y. Differential requirement of DNA replication for the cytotoxicity of DNA topoisomerase I and II inhibitors in Chinese hamster DC3F cells. Cancer Res. 1989;49:6365–6368. [PubMed] [Google Scholar]
- 17.Pommier Y. Eukaryotic DNA topoisomerase I: genome gatekeeper and its intruders, camptothecins. Semin Oncol. 1996;23:3–10. [PubMed] [Google Scholar]
- 18.D’Arpa P, Beardmore C, Liu LF. Involvement of nucleic acid synthesis in cell killing mechanisms of topoisomerase poisons. Cancer Res. 1990;50:6919–6924. [PubMed] [Google Scholar]
- 19.Drewinko B, Freireich EJ, Gottlieb JA. Lethal activity of camptothecin sodium on human lymphoma cells. Cancer Res. 1974;34:747–750. [PubMed] [Google Scholar]
- 20.Li LH, Fraser TJ, Olin EJ, Bhuyan BK. Action of camptothecin on mammalian cells in culture. Cancer Res. 1972;32:2643–2650. [PubMed] [Google Scholar]
- 21.Herben VM, ten Bokkel Huinink WW, Beijnen JH. Clinical pharmacokinetics of topotecan. Clin Pharmacokinet. 1996;31:85–102. doi: 10.2165/00003088-199631020-00001. [DOI] [PubMed] [Google Scholar]
- 22.Kaiser MG, Parsa AT, Fine RL, Hall JS, Chakrabarti I, Bruce JN. Tissue distribution and antitumor activity of topotecan delivered by intracerebral clysis in a rat glioma model. Neurosurgery. 2000;47:1391–1398. discussion 1398-1399. [PubMed] [Google Scholar]
- 23.Gensert JM, Goldman JE. Heterogeneity of cycling glial progenitors in the adult mammalian cortex and white matter. J Neurobiol. 2001;48:75–86. [PubMed] [Google Scholar]
- 24.Mason JL, Goldman JE. A2B5+ and O4+ Cycling progenitors in the adult forebrain white matter respond differentially to PDGF-AA, FGF-2, and IGF-1. Mol Cell Neurosci. 2002;20:30–42. doi: 10.1006/mcne.2002.1114. [DOI] [PubMed] [Google Scholar]
- 25.Jordan JP, Hand CM, Markowitz RS, Black P. Test for chemotherapeutic sensitivity of cerebral gliomas: use of colorimetric MTT assay. J Neurooncol. 1992;14:19–35. doi: 10.1007/BF00170942. [DOI] [PubMed] [Google Scholar]
- 26.Glass R, Synowitz M, Kronenberg G, Walzlein JH, Markovic DS, Wang LP, Gast D, Kiwit J, Kempermann G, Kettenmann H. Glioblastoma-induced attraction of endogenous neural precursor cells is associated with improved survival. J Neurosci. 2005;25:2637–2646. doi: 10.1523/JNEUROSCI.5118-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishiyama A, Lin XH, Giese N, Heldin CH, Stallcup WB. Co-localization of NG2 proteoglycan and PDGF alpha-receptor on O2A progenitor cells in the developing rat brain. J Neurosci Res. 1996;43:299–314. doi: 10.1002/(SICI)1097-4547(19960201)43:3<299::AID-JNR5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 28.Eng WK, McCabe FL, Tan KB, Mattern MR, Hofmann GA, Woessner RD, Hertzberg RP, Johnson RK. Development of a stable camptothecin-resistant subline of P388 leukemia with reduced topoisomerase I content. Mol Pharmacol. 1990;38:471–480. [PubMed] [Google Scholar]
- 29.Sugimoto Y, Tsukahara S, Oh-hara T, Isoe T, Tsuruo T. Decreased expression of DNA topoisomerase I in camptothecin-resistant tumor cell lines as determined by a monoclonal antibody. Cancer Res. 1990;50:6925–6930. [PubMed] [Google Scholar]
- 30.Woessner RD, Eng WK, Hofmann GA, Rieman DJ, McCabe FL, Hertzberg RP, Mattern MR, Tan KB, Johnson RK. Camptothecin hyper-resistant P388 cells: drug-dependent reduction in topoisomerase I content. Oncol Res. 1992;4:481–488. [PubMed] [Google Scholar]
- 31.Liao Z, Robey RW, Guirouilh-Barbat J, To KK, Polgar O, Bates SE, Pommier Y. Reduced expression of DNA topoisomerase I in SF295 human glioblastoma cells selected for resistance to homocamptothecin and diflomotecan. Mol Pharmacol. 2008;73:490–497. doi: 10.1124/mol.107.041178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta RS, Gupta R, Eng B, Lock RB, Ross WE, Hertzberg RP, Caranfa MJ, Johnson RK. Camptothecin-resistant mutants of Chinese hamster ovary cells containing a resistant form of topoisomerase I. Cancer Res. 1988;48:6404–6410. [PubMed] [Google Scholar]
- 33.Rubin E, Pantazis P, Bharti A, Toppmeyer D, Giovanella B, Kufe D. Identification of a mutant human topoisomerase I with intact catalytic activity and resistance to 9-nitro-camptothecin. J Biol Chem. 1994;269:2433–2439. [PubMed] [Google Scholar]
- 34.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, Aldape K. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 35.Tomicic MT, Christmann M, Kaina B. Topotecan Triggers Apoptosis in p53-Deficient Cells by Forcing Degradation of XIAP and Survivin Thereby Activating Caspase-3-Mediated Bid Cleavage. Pharmacology. 2010;332:316–325. doi: 10.1124/jpet.109.159962. [DOI] [PubMed] [Google Scholar]
- 36.Tomicic MT, Christmann M, Kaina B. Topotecan-triggered degradation of topoisomerase I is p53-dependent and impacts cell survival. Cancer Res. 2005;65:8920–8926. doi: 10.1158/0008-5472.CAN-05-0266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.