

Abstract
The striatum has important roles in motor control and action learning and, like many brain regions, receives multiple monoaminergic inputs. We have examined serotonergic modulation of rat and mouse corticostriatal neurotransmission and find that serotonin (5-HT) activates 5-HT1b receptors resulting in a long-term depression (LTD) of glutamate release and striatal output that we have termed 5-HT–LTD. 5-HT–LTD is presynaptically mediated, cAMP pathway dependent, and inducible by endogenous striatal 5-HT, as revealed by application of a selective 5-HT reuptake inhibitor. 5-HT–LTD is mutually occlusive with dopamine/endocannabinoid-dependent LTD, suggesting that these two forms of LTD act on the same corticostriatal terminals. Thus, serotonergic and dopaminergic mechanisms exist that may interact to persistently sculpt corticostriatal circuits, potentially influencing action learning and striatal-based disorders.
Introduction
A single monoamine neurotransmitter, such as serotonin (5-HT) or dopamine, can potently modulate the strength of various synapse types within a nucleus. A common theme in brain, however, is the convergent release of multiple monoamine neurotransmitters to a given region. This configuration requires an understanding of how multiple monoamine-dependent mechanisms may interact to modulate synaptic potentials to ultimately dictate the output of a given nucleus.
The dorsal striatum, a basal ganglia structure necessary for action selection and habit formation, receives dual monoaminergic input; dopaminergic innervation originating from the substantia nigra pars compacta and serotonergic innervation arising from the dorsal raphe nucleus (DRN) (Steinbusch et al., 1981). Striatal dopamine modulates the primary excitatory input to striatum: glutamatergic cortical fibers that abundantly synapse onto the dendritic spine heads of GABAergic medium spiny projection neurons (MSNs). Dopamine signaling through the D2 dopamine receptor is necessary for the mobilization of endocannabinoids (eCBs) from MSNs that act retrogradely to induce a long-term depression (eCB–LTD) of glutamatergic synaptic transmission (Gerdeman et al., 2002; Kreitzer and Malenka, 2005). This form of plasticity at the corticostriatal synapse is implicated in normal voluntary movement and skill learning and is lost in parkinsonism (Kreitzer and Malenka, 2007).
Although important aspects of dopaminergic modulation of the corticostriatal synapse have been illuminated, serotonergic effects on this synapse are not well understood. 5-HT fibers predominately terminate as nonjunctioning axo-axonic varicosities but also form axospinous or axodendritic asymmetric synapses on presumed MSNs (Soghomonian et al., 1989). Several 5-HT receptor subtypes are localized to the dorsal striatum, including 5-HT1a, 5-HT1b, 5-HT1d, 5-HT1e, 5-HT2a, 5-HT1c, 5-HT3a, 5-HT4, 5-HT5a, 5-HT6, and 5-HT7 (Di Matteo et al., 2008). This ultrastructural configuration and receptor expression profile, coupled with findings showing that subsets of DRN serotonergic cells fire coincidently with the initiation and termination of selected voluntary movements (Fornal et al., 1996), suggest a role for direct serotonergic modulation of synaptic transmission onto striatal MSNs.
To explore this possibility and potential interactions between serotonergic effects and dopamine-dependent plasticity, we examined the effect of 5-HT on glutamatergic signaling onto MSNs and net striatal output in rodent corticostriatal slices. We found that application of 5-HT, or selective 5-HT transporter reuptake blockade, induces LTD of glutamatergic signaling at corticostriatal synapses (5-HT–LTD) that reciprocally occludes eCB–LTD. These findings reveal a unique monoaminergic mechanism controlling presynaptic inhibition of corticostriatal glutamate release, a phenomenon that offers new insight into how dopamine and 5-HT systems may interact to influence striatum-dependent processes.
Materials and Methods
Brain slice preparation.
After anesthetization of P14–P18 Sprague Dawley rats or D1 enhanced green fluorescent protein (GFP) BAC transgenic mice (GENSAT) (Heintz, 2004), brains were removed, and 300-μm-thick (whole-cell recordings) or 350-μm-thick (field potential recordings) coronal sections through the striatum were prepared using a vibratome (Leica Microsystems). Sections were equilibrated for 30 min (slices for field potential recordings) or 1 h (slices for whole-cell recordings) at 33°C in 95% CO2/5% O2 bubbled artificial CSF (aCSF) containing the following (in mm): 124 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 glucose. Sections were subsequently incubated in the same solution at 24°C.
Whole-cell voltage-clamp recordings.
Electrodes of 2–5 mΩ resistance were pulled from borosilicate glass on a Flaming/Brown micropipette puller (Sutter Instruments). Pipettes were filled with a CsMeSO3-based solution of 295–300 mOsm (in mm: 120 CsMeSO3, 5 NaCl, 10 tetraethylammonium-Cl, 10 HEPES, 5 QX-314, 1.1 EGTA, 0.3 Na-GTP, and 4 Mg-ATP for ESPC recordings) and a CsCl-based solution of 295–300 mOsm (in mm: 150 CsCl, 10 HEPES, 2 MgCl2, 0.3 Na-GTP, 5 QX-314, 3 Mg-ATP, and 0.2 BAPTA for IPSC recordings). All recordings were performed between 29°C and 31°C and in the presence of picrotoxin (50 μm). Striatal MSNs were held at −60 mV, and nucleus accumbens core MSNs were held between −70 and −75 mV, using a Multiclamp 700A amplifier (Molecular Devices). A bipolar concentric stimulating electrode (World Precision Instruments) was used to elicit EPSCs with amplitudes between 300 and 600 pA every 20 s. The stimulating electrode was placed at the border of the white matter of the external capsule and the dorsolateral striatum for dorsolateral striatum recordings. For the dorsomedial striatum, recordings were performed from MSNs just lateral to the dorsolateral-most point of the lateral ventricle, whereas the stimulating electrode was placed at the border of the overlying corpus callosum and striatum. For nucleus accumbens recordings, the recording electrode was placed just lateral to the anterior commisure fiber bundle, whereas the stimulating electrode was situated just lateral to the recording electrode; both the recording and stimulating electrodes were within the core of the nucleus accumbens. Recordings of EPSCs or IPSCs were filtered at 2 kHz and digitized at 10 kHz. Data were acquired using Clampex 9.2 (Molecular Devices). Experiments were discarded if series resistance varied by >15% or increased over 22 MΩ. The sensitivity to detect changes in ESPC amplitude was not affected across the ranges of baseline EPSC amplitude and series resistances used.
Extracellular recordings.
Extracellular field potential recordings were performed using recording electrodes of 2–5 mΩ resistance filled with 0.9% NaCl. Slice preparation and recording chamber parameters followed whole-cell methods above. A bipolar twisted tungsten electrode delivered test pulses every 30 s (0.03 Hz) to elicit population spikes (PSs). All other protocols were performed as described previously (Yin et al., 2007).
Two-photon laser scanning microscopy and glutamate uncaging.
A combined Two-photon laser scanning microscopy (2PLSM) and glutamate uncaging (2PLSU) microscope was used to uncage 4-methoxy-7-nitroindolinyl-caged l-glutamate (MNI-glutamate) and image dendritic spines of striatal MSNs filled with Alexa Fluor-594 (20 μm) through the patch pipette from 200 μm rat coronal hemisections superfused with aCSF maintained at 29°C. One titanium:sapphire laser (Chameleon Ultra II; Coherent) was tuned to 720 nm to photoactivate bath-applied MNI-glutamate (2.5 mm), whereas a second identical laser was tuned to 810 nm to excite Alexa Fluor-594 for imaging cellular morphology. All other instrumentation was used as described previously (Carter and Sabatini, 2004). Alexa Fluor-594 was dissolved in the CsMeSO3-based solution described above and 10–15 min passed to allow for dye equilibration. 2PLSU pulses at 500 μs, electrical stimulation, and 2PLSM image acquisitions were executed sequentially, with one sequence defined as the following: electrical stimulation delivery, 10 s pause, 2PLSM imaging, 10 s pause, 2PLSU pulse delivery, 20 s pause.
Statistical analysis.
Mean amplitude of EPSCs, IPSCs, and PSs were measured in Igor Pro, Clampex 9.2, or Clampfit 9.2, and the data were analyzed in GraphPad Prism (GraphPad Software). PS amplitude was defined as the relative difference between the peak of the early positivity and the peak negativity. EPSC, IPSC, and PS amplitudes were averaged per minute, expressed as percentage of average baseline amplitude, and compared using a two-tailed Student's t test. A one-way ANOVA followed by post hoc Dunn's test was used to analyze selected sets of spontaneous EPSC (sEPSC) and miniature EPSC (mEPSC) data.
Drugs.
5-HT, picrotoxin, and QX-314 were purchased from Sigma-Aldrich. Cell-impermeant BAPTA, tetracesium salt (molecular weight of 1004.03) was purchased from Invitrogen. All other drugs were purchased from Tocris Bioscience.
Results
Exogenous 5-HT–LTD
Effects of 5-HT on glutamatergic transmission onto MSNs in the dorsolateral or dorsomedial striatum were examined by measuring evoked EPSC (eEPSC) amplitudes in each subregion. The stimulating and recording electrode locations for both whole-cell and field potential recordings in the dorsolateral striatum are depicted in Figure 1a. After acquisition of a baseline recording, a 10 min bath application of 5 μm 5-HT resulted in a significant depression of EPSC amplitude (5-HT–LTD) (EPSC amplitude at t = 70 min, 54.08 ± 9.9% of baseline; n = 6; p < 0.0001) (Fig. 1a). The observed depression was not a result of an increase in series resistance (Fig. 1b). The EPSC amplitude decrease was long lasting and did not recover even 55 min after the cessation of 5-HT application, prompting us to designate this depression as 5-HT–LTD. The amplitude of EPSCs was stable in the absence of drug application (Fig. 1a). Recording MSNs in the dorsomedial striatum, we found that 5 μm 5-HT also significantly depressed EPSC amplitude (EPSC amplitude at t = 25 min, 65.3 ± 4.0% of baseline; n = 8; p < 0.0001). 5-HT–LTD was observed in 26 of 28 dorsolateral cells and 8 of 8 dorsomedial MSNs.
Figure 1.
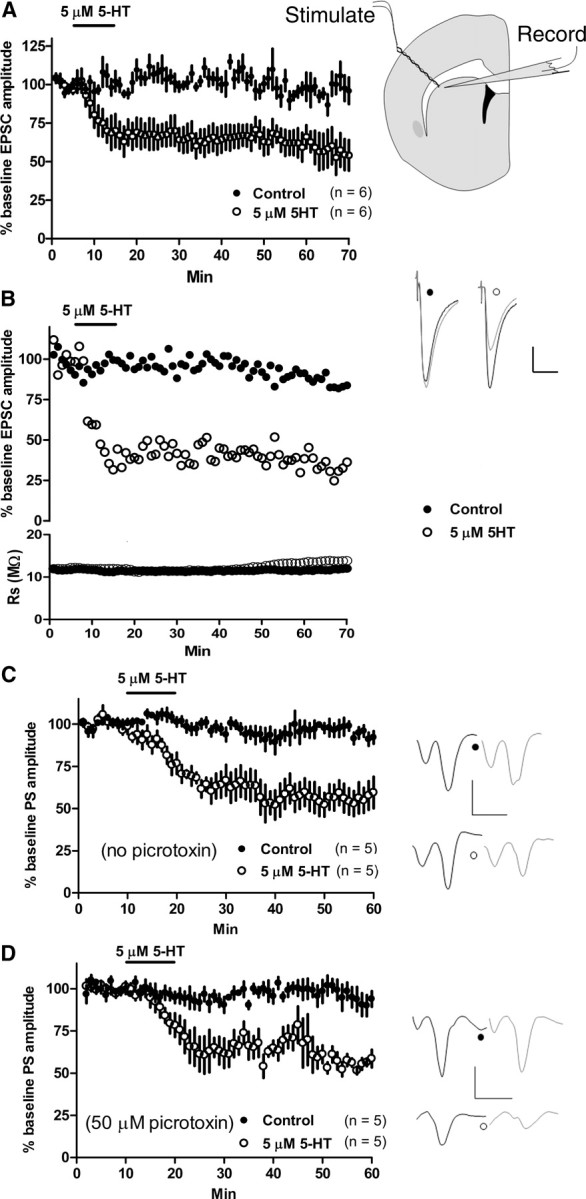
5-HT induces glutamatergic long-term depression. A, Right, The stimulating electrode was placed at the border of the dorsolateral striatum and the overlying white matter of a striatal hemisection, whereas a recording electrode measuring striatal PSs or MSN EPSCs was placed within ∼100–200 μm of the stimulating electrode tip. Left, Application of 5-HT (5 μm) for 10 min induced LTD EPSC amplitude recorded from MSNs in the dorsolateral striatum. No alteration in EPSC amplitude was observed in the absence of drug (control). Example traces show baseline (black) and post-5-HT application (gray) EPSCs. B, The 5-HT-induced depression of EPSC amplitude was not a result of an increase in series resistance (bottom). C, D, 5-HT (5 μm, 10 min) persistently depressed PS amplitude whether in the absence (C) or presence (D) of picrotoxin (50 μm). Response amplitude was stable in the absence of drug (control). Fiber volley amplitude was not depressed after 5-HT application. Calibration: B, 100 pA, 20 ms; C, D, 0.5 mV, 4 ms. All traces are averages of 15 sweeps, and all data are expressed as the mean ± SEM.
The whole-cell data suggest that 5-HT–LTD may suppress striatal output. We examined effects of 5-HT application on net striatal output in acute corticostriatal slices using field potential recording. 5-HT (5 μm, 10 min) application resulted in a depression of PS amplitude (PS amplitude at t = 60 min, 59.5 ± 9.4% of baseline; n = 5; p = 0.003) that persisted after 5-HT removal (Fig. 1c). 5-HT is also known to excite GABAergic striatal fast-spiking interneurons (Blomeley and Bracci, 2009), cells that potently inhibit MSN firing by directly innervating MSN somata (Tepper et al., 2008). To determine whether increased GABAA receptor signaling contributes to 5-HT-induced PS depression, which primarily derives from MSN responses, we applied 5-HT (5 μm, 10 min) in the constant presence of picrotoxin (50 μm). A lasting depression of PS amplitude was observed (PS amplitude at t = 60 min, 58.6 ± 5.2% of baseline; n = 5; p < 0.0001) (Fig. 1d). The PS amplitude was stable in the presence or absence of picrotoxin (Fig. 1c,d).
5-HT–LTD is mediated by 5-HT1b receptors and presynaptically expressed
We next sought to determine the 5-HT receptor subtype mediating 5-HT–LTD. Application of the 5-HT1b receptor-selective agonist CP 93129 (2 μm, 10 min) induced LTD of EPSC amplitude in whole-cell recordings from the dorsolateral striatum (EPSC amplitude at t = 40–45 min, 61.2 ± 3.1% of baseline; p < 0.0001; n = 5) (Fig. 2a). CP 93129-induced depression was subsequently partially reversed by the 5-HT1b-specific antagonist NAS-181 (20 μm) compared with the NAS-181-free control [t = 40–45 min, 76.3 ± 2.1% (NAS-181) of baseline; p = 0.004; n = 5]. Despite this partial reversal, EPSC amplitude remained depressed compared with baseline (t = 40–45 min; p < 0.0001; n = 5) (Fig. 2a), indicating that CP 93129-induced lasting depression is not a result of lingering 5-HT1b activation. The 5-HT1a agonist 8-hydroxy-2(di-n-propylamino)tetralin (20 μm) did not alter responses (EPSC amplitude at t = 25 min, 102.2 ± 13.8% of baseline; p = 0.9; n = 3). To rule out involvement of other 5-HT receptor subtypes in 5-HT–LTD, we applied 5-HT (5 μm, 10 min) in the continuous presence of NAS-181 (20 μm). This treatment completely blocked 5-HT–LTD (vs baseline at t = 25 min, 102.6 ± 10.3%; p = 0.8; n = 5) (Fig. 2b). Application of the mixed 5-HT1b and 5-HT2 receptor agonist TCB-2 (2 μm, 10 min) (Best and Regehr, 2008) also persistently depressed EPSC amplitude (vs baseline at t = 25 min, 72.8 ± 5.6%; p = 0.001; n = 5), and this effect was blocked in the continuous presence of NAS-181 (5 μm) (vs baseline at t = 25 min, 94.7 ± 8.2%; p = 0.5; n = 4). Thus, 5-HT1b appears to be the receptor subtype responsible for 5-HT–LTD.
Figure 2.
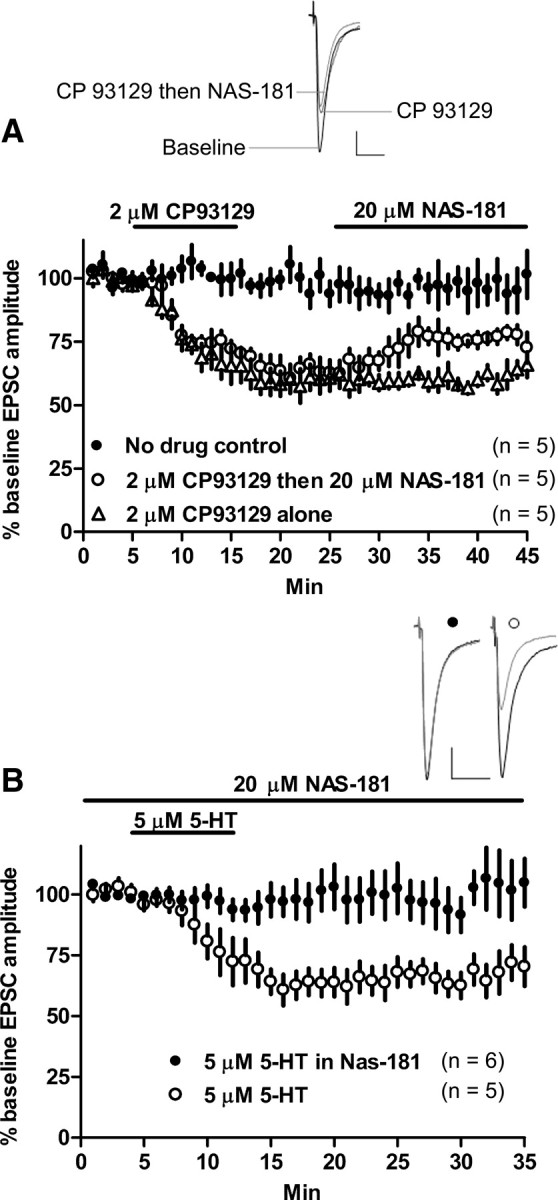
5-HT–LTD is 5-HT1b mediated. A, The 5-HT1b receptor agonist CP 93129 (2 μm, 10 min) produced persistent depression of EPSC amplitude. The 5-HT1b antagonist NAS-181 (20 μm) was unable to completely reverse the established CP 93129-induced depression. B, NAS-181 (20 μm) blocked 5-HT (5 μm, 10 min)-induced EPSC amplitude depression. Calibration: 100 pA, 20 ms.
Next, we examined whether 5-HT–LTD is expressed presynaptically or postsynaptically. We first examined whether CP 93129 depressed EPSC amplitude evoked by 2PLSU of MNI-glutamate (uEPSCs) at single MSN dendritic spine heads, which were visualized using 2PLSM. Stimulating electrode eEPSCs (alternated with uEPSCs) were used as a positive control. CP 93129 application did not depress uEPSC amplitude (vs baseline at t = 15 min, 91.6 ± 10%; p = 0.4; n = 7) but depressed eEPSC amplitude onto the same cell (vs baseline at t = 10–15 min, 43.2 ± 8.5%; p < 0.0001; n = 7) (Fig. 3a). eEPSC amplitude was significantly depressed compared with uEPSC amplitude (t = 10–15 min vs uEPSC; p = 0.0026; n = 7), suggesting that 5-HT–LTD is presynaptically expressed. Next, we delivered paired-pulse stimulation at a 50 ms interpulse interval every 20 s and applied CP 93129 (2 μm). CP 93129 (2 μm, 10 min) increased the paired-pulse ratio (PPR) (EPSC2/EPSC1) compared with baseline (mean EPSC2/EPSC1 amplitude: baseline, 1.22 ± 0.06 vs CP 93129, 1.42 ± 0.09; p = 0.02; n = 8) (Fig. 3b), suggesting a decrease in transmitter release probability. Presynaptically expressed LTD should also affect NMDA receptor-dependent EPSCs (NMDAR–EPSCs), and we examined this by applying the AMPAR antagonist NBQX (5 μm) and recording at a −40 mV holding potential. Bath application of CP 93129 (2 μm) persistently depressed NMDAR–EPSC amplitude (t = 25–30 min, 21.4 ± 1.0% of baseline; p < 0.0001; n = 5) (Fig. 3c). To further examine potential postsynaptic mechanisms in 5-HT–LTD, we loaded the postsynaptic cell with the calcium chelator BAPTA (20 mm) through the recording pipette. Postsynaptic BAPTA did not block 5-HT actions (t = 25 min, 48.8 ± 7.6% of baseline; p = 0.0005; n = 5) (Fig. 3d).
Figure 3.
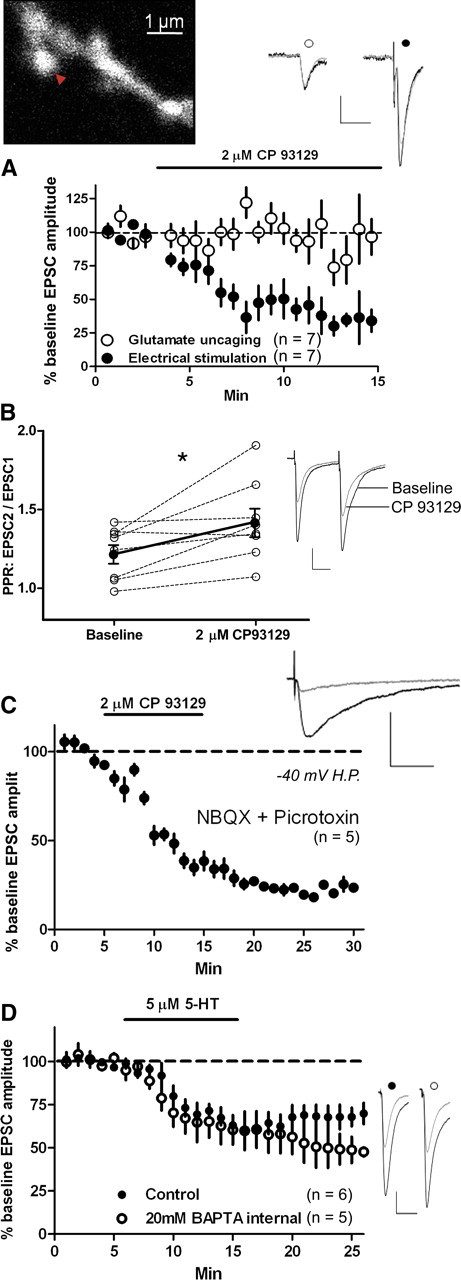
5-HT–LTD is presynaptically expressed. A, Top left, 2PLSM image of a spiny dendrite filled with Alexa Fluor-594. Red arrow, Location of MNI-caged glutamate (2.5 mm) focal photolysis by a 500 μs, 720 nm 2PLSU pulse. Bottom, CP 93129 (2 μm) depressed electrical stimulation-evoked EPSC amplitude (filled circles) but not MNI-glutamate uncaging-evoked EPSC amplitude (open circles). B, CP 93129 increased the PPR over predrug baseline (*p = 0.02). C, Bath application of CP 93129 (2 μm) persistently depressed NMDAR-mediated EPSC amplitude. The cell was held at −40 mV and NBQX (5 μm) was present to block AMPAR-mediated currents. H.P., Holding potential. D, Intracellular loading of the calcium buffering agent BAPTA (20 mm) did not block the depression of EPSC amplitude by bath application of 5-HT (5 μm) for 10 min. Calibration: A, 20 pA, 20 ms; B–D, 100 pA, 20 ms.
We next examined the effect of CP 93129 (2 μm, 10 min) on the frequency and amplitude of TTX-insensitive mEPSCs and sEPSCs before (baseline), during (CP 93129), and 20–25 min after (wash) CP 93129 application (Fig. 4a). CP 93129 significantly increased mEPSC (Fig. 4b) and sEPSC interevent interval (IEI) over baseline values (mEPSC: baseline, 0.47 ± 0.08 s vs CP 93129, 0.76 ± 0.1 s, n = 8, p < 0.05; sEPSC: baseline, 0.33 ± 0.04 s vs CP 93129, 0.60 ± 0.13 s, n = 6, p < 0.05). Despite 20–25 min of drug washout, the average IEI remained significantly increased over baseline (mESPC: wash, 0.84 ± 0.17 s, n = 8, p < 0.05; sEPSC: wash, 0.82 ± 0.2 s, n = 6, p < 0.01). mEPSC and sEPSC IEI averages were not significantly different between drug and washout conditions (p > 0.05). The average mEPSC (Fig. 4c) and sEPSC amplitude in either the drug or washout conditions did not differ significantly from baseline values (mEPSC: baseline, 26.3 ± 0.7 pA vs CP 93129, 27 ± 0.7 pA; or baseline vs washout, 24.8 ± 1.2 pA, n = 8, p > 0.05; sEPSC: baseline, 26.9 ± 3 pA vs CP 93129, 25.3 ± 2.4 pA; or baseline vs washout, 23.6 ± 2.5 pA, n = 6, p > 0.05). Importantly, application or washout of CP 93129 did not change postsynaptic holding current (Fig. 4d).
Figure 4.
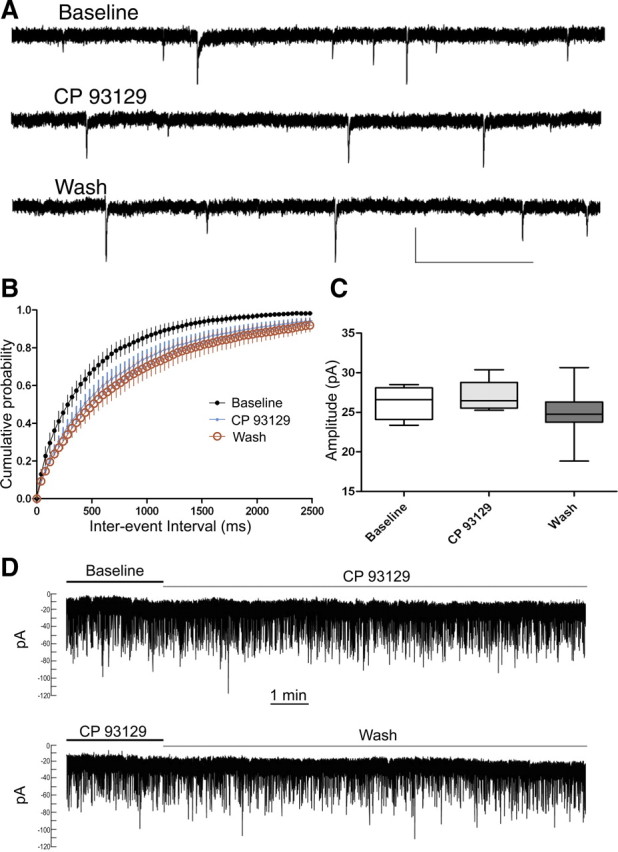
5-HT–LTD involves changes in mEPSC frequency but not amplitude. A, mEPSC traces during baseline, CP 93129 (2 μm), and 20–25 min of washout. B, CP 93129 (blue dots) increased the IEI of TTX-insensitive mEPSCs over baseline (black dots). After 20 min of drug washout (red circles), the IEI remained significantly greater than baseline (wash) (n = 6). C, mEPSC amplitude was not significantly different across conditions (n = 6). D, Traces of example striatal MSN recordings show that the holding current did not change during bath application of CP 93129 (2 μm) (top) or by washout of CP 93129 (2 μm) (bottom). Calibration: A, 30 pA, 1 s.
5-HT–LTD is independent of cannabinoid receptor 1 activation
The classical form of LTD at striatal glutamatergic synapses requires retrograde eCB signaling (Gerdeman et al., 2002), and this system can be co-opted by neurotensin in the striatum in a similar manner to 5-HT-induced eCB release in the olivary nucleus (Best and Regehr, 2008; Yin et al., 2008). We, therefore, tested whether 5-HT–LTD is mediated by cannabinoid receptor 1 (CB1) activation. As a positive control for the actions of the CB1 antagonist AM251, we examined the ability of the antagonist to block CB1-agonist-induced synaptic depression. After a 5 min baseline period, we applied the CB1 agonist WIN 55,212-2 for 10 min with or without AM251 (3 μm) present during the entire experiment. The WIN 55,212-2-induced depression of EPSC amplitude (vs baseline at t = 15 min, 72.6 ± 4.4%; p < 0.0001; n = 5) was completely blocked by AM251 (vs baseline at t = 15 min, 96.7 ± 1.9, p = 0.2, n = 5; vs WIN 55, 212-2 alone at t = 15 min, p = 0.001) (Fig. 5a). To test whether 5-HT–LTD is indeed mediated by CB1, we applied 5-HT (5 μm) or CP 93129 (2 μm) in the presence of the CB1 antagonist AM251 (3 μm). CB1 activation was unnecessary for 5-HT- or CP 93129-induced depression of EPSC amplitude (t = 25 min: 5-HT, 70.4 ± 4.9% of baseline, p = 0.0009, n = 5; CP 93129, 57.6 ± 6% of baseline, p < 0.0001, n = 6) (Fig. 5b,c).
Figure 5.
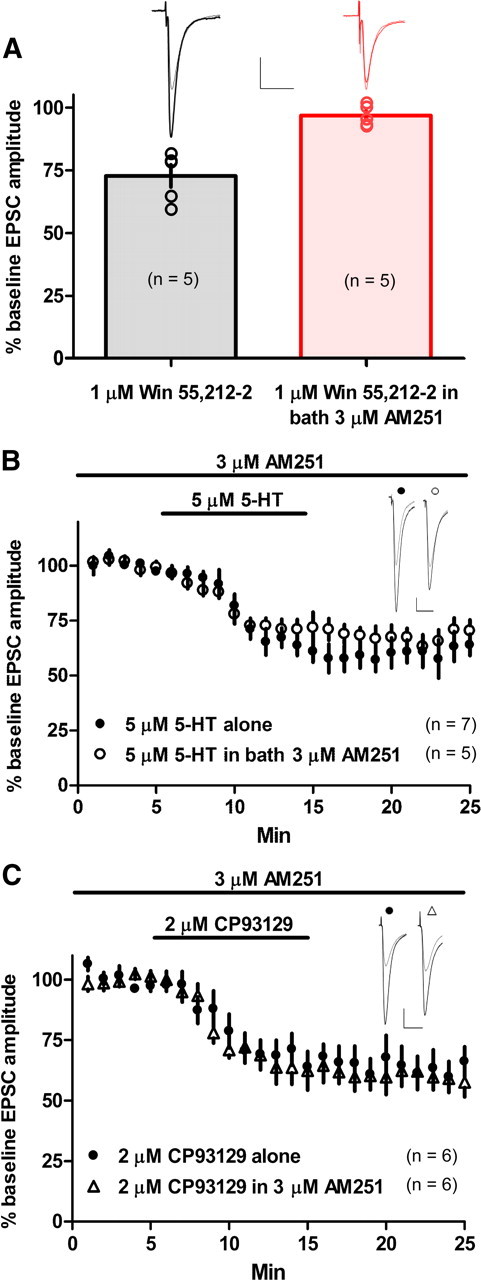
5-HT–LTD does not require CB1 activation. A, Positive control for the CB1 antagonist AM251 showing that AM251 (3 μm) completely blocks WIN 55,212-2 (1 μm)-induced depression of EPSC amplitude. B, C, Bath application of 5-HT (5 μm) (B) or the 5-HT1b-specific agonist CP 93129 (2 μm) (C) induced a depression of EPSC amplitude in the presence of AM251 (3 μm). Calibration: 100 pA, 20 ms.
5-HT–LTD at other synapses
The striatum heavily expresses 5-HT1b transcript (Maroteaux et al., 1992), and the 5-HT1b receptor protein was shown to be present on MSN axon terminals (Boschert et al., 1994). However, no reduction in GABAergic IPSC amplitude in dorsolateral MSNs was observed after the same 10 min application of 5 μm 5-HT (IPSC amplitude at t = 25 min, 90.4 ± 10.8% of baseline; n = 7; p = 0.4) (Fig. 6a). Because MSN axon collaterals impinge on other MSNs, it was interesting that we did not observe 5-HT–LTD at GABAergic synapses on MSNs after 5-HT application. Therefore, we tested whether selective 5-HT1b activation, presumably on MSN collateral terminals, could induce LTD. CP 93129 (2 μm, 10 min) application depressed IPSC amplitude recorded in the dorsolateral striatum (vs baseline t = 30–35 min, 59.2 ± 9.1%; p = 0.002; n = 6) (Fig. 6b).
Figure 6.
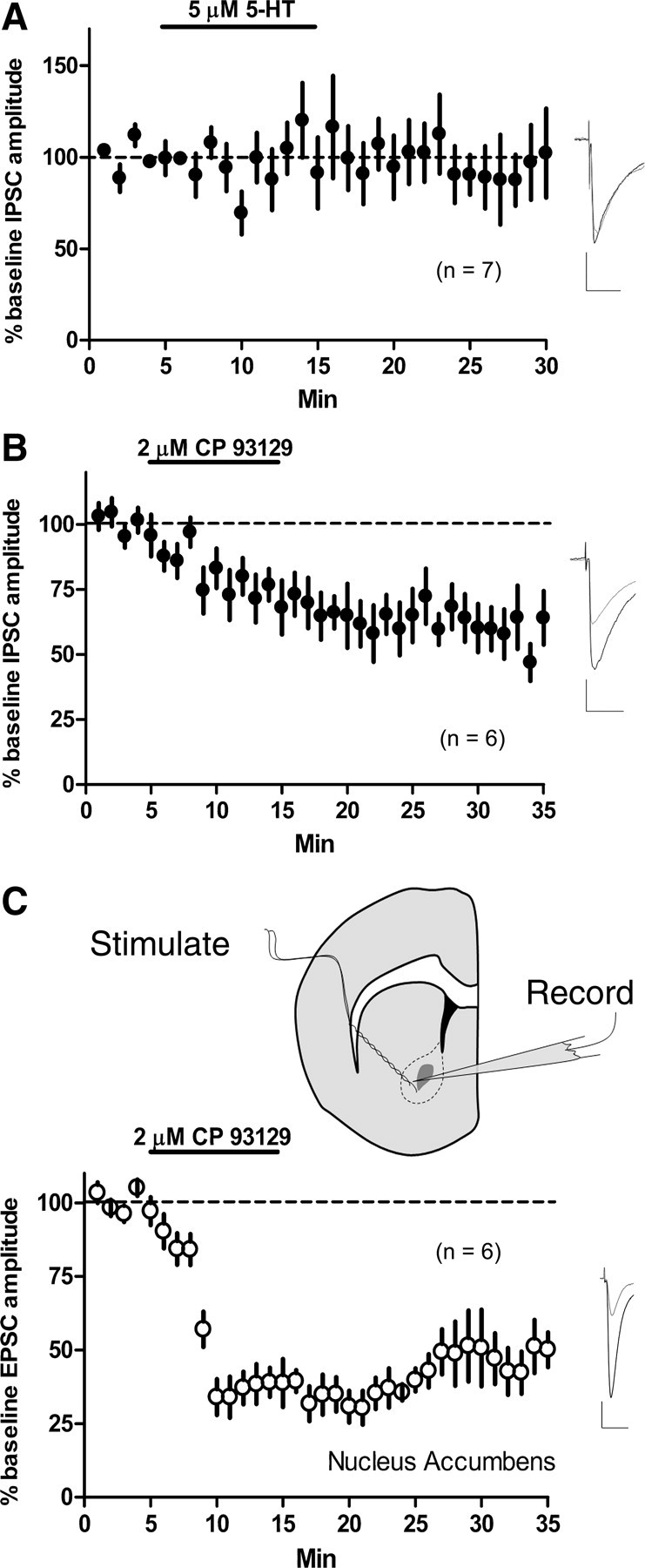
5-HT1b activation, but not 5-HT application, depresses GABAergic transmission onto MSNs. 5-HT–LTD in nucleus accumbens. A, Bath application of 5-HT (5 μm) for 10 min did not induce a depression of evoked IPSCs recorded from MSNs of the dorsolateral striatum. B, However, a 10 min bath application of the 5-HT1b-selective agonist CP 93129 (2 μm) resulted in persistent depression of IPSC amplitude. C, Top, Both stimulating and recording electrodes were situated within the core of the nucleus accumbens (approximately delineated by dashed line). Bath applying CP 93129 (2 μm) for 10 min resulted in a lasting depression of evoked EPSCs recorded from MSNs of the nucleus accumbens core. Calibration: 100 pA, 20 ms.
Because 5-HT application has been demonstrated to result in a reversible inhibition of glutamate release in the nucleus accumbens (Muramatsu et al., 1998), we examined the effects of CP 93129 on evoked EPSCs recorded from MSNs of the nucleus accumbens core. CP 93129 (2 μm, 10 min) resulted in a rapid and lasting depression of EPSC amplitude (vs baseline at t = 30–35 min, 47.3 ± 8.3%; p = 0.0002; n = 6) (Fig. 6c).
Endogenous 5-HT–LTD
To determine whether 5-HT–LTD is inducible by endogenous 5-HT in the acute slice preparation, we bath applied the selective 5-HT reuptake inhibitor (SSRI) citalopram (4 μm) for 10 min. Citalopram significantly depressed EPSC amplitude (vs baseline at 30–35 min, 61.8 ± 8.5%; p = 0.002; n = 5) (Fig. 7a). When examining the individual cell data, three of five cells displayed LTD, whereas two showed no depression (Fig. 7a). To promote a more reliable SSRI-induced LTD for experiments examining receptor dependence, we applied citalopram (4 μm) along with a burst stimulation protocol consisting of six pulses (50 ms interpulse interval) every 20 s for 10 min in an attempt to nonselectively activate serotonergic fibers through current spread from the stimulating electrode. This stimulation protocol began 3 min after the onset of citalopram application and ended 3 min after beginning drug washout (membrane potential was maintained at −60 mV). The same stimulating electrode was used to elicit test pulses and burst stimulation. We chose this burst stimulation frequency because responses fully recovered to baseline levels 5–10 min after cessation of burst stimulation alone (vs baseline at t = 30–35 min, 93 ± 5.8%; p = 0.3; n = 6) (Fig. 7b). Thus, this pattern of stimulation by itself is not sufficient to induce any form of LTD. Citalopram coupled with the burst stimulation protocol (Cit-Burst) consistently and persistently depressed EPSC amplitude (six of six cells) (t = 35 min, 73 ± 3.2% of baseline; p < 0.0001; n = 6) (Fig. 7b). The CB1 antagonist AM251 (3 μm) was present throughout the experiment to exclude possible CB1-mediated effects. In subsequent experiments we used the Cit-Burst paradigm, as opposed to citalopram alone, to ensure that LTD was consistently induced.
Figure 7.
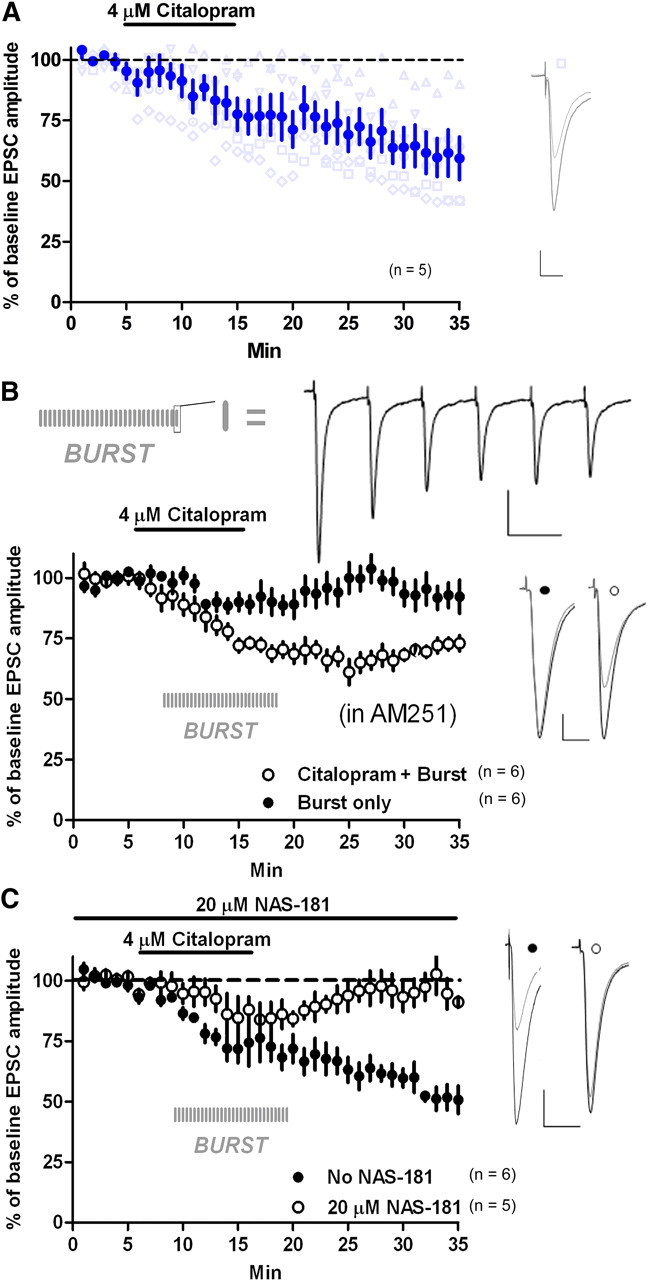
Endogenous 5-HT–LTD. A, Citalopram (4 μm, 10 min) induced LTD (blue circles). Three of five cells showed LTD, whereas the remaining neurons showed little to no depression of EPSC amplitude (gray triangles). B, Cit-Burst induced LTD. AM251 (3 μm) was present throughout. Each vertical, gray line indicates when a six-pulse burst was delivered. At top is a representative trace of a six-pulse burst. C, NAS-181 (20 μm) blocked Cit-Burst-induced depression. AM251 was not present. Calibration: B, six-pulse traces, 100 pA, 50 ms; all other traces; 100 pA, 20 ms.
To determine whether the 5-HT1b receptor also mediated the Cit-Burst-induced LTD, we applied Cit-Burst in the presence of the 5-HT1b antagonist NAS-181 (20 μm). This endogenous 5-HT–LTD was completely blocked by NAS-181 (vs baseline at t = 30–35 min, 93.3 ± 5.2%, p = 0.1, n = 5; vs no NAS-181 at t = 30–35 min, 58.5 ± 5.2%, p = 0.002, n = 5), indicating that 5-HT1b mediates this effect (Fig. 7c). To ensure that blockade of 5-HT–LTD was achieved by 5-HT1b antagonism alone, AM251 was not applied. To further rule out involvement of calcium-mediated postsynaptic eCB release in the expression of endogenous 5-HT–LTD, we loaded the postsynaptic cell with BAPTA (20 mm). Cit-Burst produced persistent depression of EPSC amplitude under this condition (vs baseline at t = 25 min, 56.6 ± 14.8%; p = 0.02; n = 6).
5-HT–LTD is not pathway specific
To assess whether 5-HT–LTD is specific to either the direct or indirect output pathways of the striatum, we recorded in slices prepared from BAC-transgenic mice expressing GFP under control of the D1 dopamine receptor promoter. CP 93129 (2 μm, 10 min) persistently depressed EPSC amplitude in D1-containing (GFP-positive) (vs baseline at t = 35 min, 62.2 ± 6.9%; p < 0.0001; n = 10) and putative D2-containing (GFP-negative) cells (vs baseline at t = 35 min, 66.2 ± 8.3%; p < 0.0001; n = 10) (Fig. 8a). Cit-Burst also persistently depressed averaged EPSC amplitude in both D1-containing (vs baseline at t = 35 min, 71.2 ± 7.9%; p < 0.0001; n = 10) and putative D2-containing (vs baseline at t = 35 min, 69.7 ± 6.8%; p < 0.0001; n = 10) MSNs (Fig. 8b). Interestingly, three D1-containing cells and two putative D2-containing cells exhibited a transient depression of EPSC amplitude.
Figure 8.

5-HT–LTD is not pathway specific. A, CP 93129 (2 μm, 10 min) persistently depressed EPSC amplitude in D1-containing and putative D2-containing MSNs. B, Cit-Burst also induced a lasting depression of EPSC amplitude in both D1-containing and putative D2-containing MSNs. Calibration: 100 pA, 20 ms.
5-HT–LTD is cAMP pathway dependent
5-HT1b, a Gi/Go-coupled receptor, is reported to negatively couple to adenylyl cyclase (AC) (Bouhelal et al., 1988). To explore whether 5-HT–LTD is indeed AC dependent, we applied forskolin (10–20 μm), an activator of AC. Forskolin application alone increased EPSC amplitude (vs baseline at t = 25 min, 126.8 ± 9.1%; p = 0.03; n = 6) (Fig. 9a). After incubating slices for at least 15 min in 10–20 μm forskolin, we applied CP 93129 (2 μm) for 4 min and observed initial, but not persistent, depression of EPSC amplitude (t = 25 min, 95.4 ± 5% of baseline; p = 0.4; n = 6) (Fig. 9b), whereas CP 93129 (2 μm, 4 min) without forskolin persistently depressed EPSC amplitude (t = 25 min, 59.3 ± 3.6% of baseline; p < 0.0001; n = 5). Because AC catalyzes the conversion of ATP to cAMP, we examined whether cAMP saturation might prevent 5-HT–LTD. Bath application of 8-Br-cAMP (80 μm), a cell-permeable cAMP analog, blocked CP 93129-induced LTD (vs baseline at t = 25 min, 90.8 ± 8%; p = 0.35; n = 5), suggesting that 5-HT–LTD works through the cAMP-dependent pathway (Fig. 9c).
Figure 9.
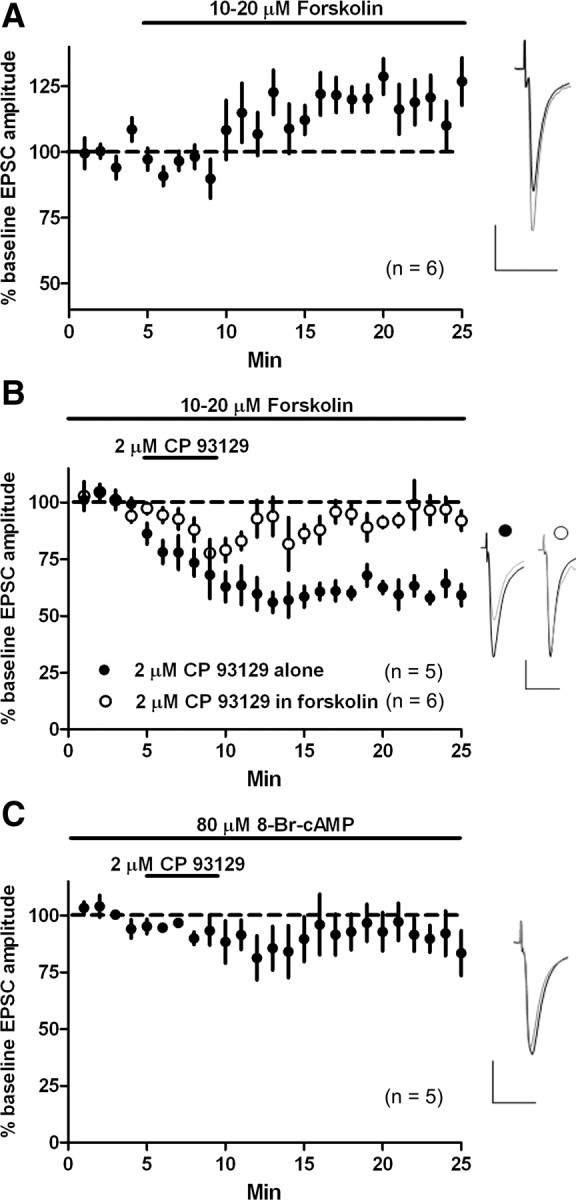
5-HT–LTD is cAMP pathway dependent. A, Forskolin (10–20 μm) potentiated EPSC amplitude. B, CP 93129 (2 μm, 4 min) persistently depressed EPSC amplitude. Forskolin blocked CP 93129-induced LTD of EPSC amplitude, but transient EPSC depression was observed during agonist application. C, In the presence of 8-Br-cAMP (80 μm), CP 93129 did not depress EPSC amplitude. Calibration: 100 pA, 20 ms.
5-HT–LTD and eCB–LTD are mutually occlusive
Thus far, 5-HT–LTD and eCB–LTD both appear to activate presynaptic Gi/Go-coupled receptors (5-HT1b and CB1). Because both 5-HT1b and CB1 engage similar post-receptor signaling pathways (Bouhelal et al., 1988; Davis et al., 2003), we reasoned that if 5-HT1b and CB1 were on the same population of corticostriatal terminals then eCB–LTD and 5-HT–LTD may be mutually occlusive. To examine this, we first induced eCB–LTD with a high-frequency stimulation protocol (HFS) consisting of four 100 pulse trains (every 10 s) with an interpulse interval of 10 ms (100 Hz for 1 s) while depolarizing the cell to a 0 mV holding potential. HFS induced a depression of EPSC amplitude (t = 14 min, 74.4 ± 6.6% of baseline; p = 0.005; n = 5) (Fig. 10a). During stabilization of EPSC amplitude after HFS, we then applied Cit-Burst to induce 5-HT–LTD. Cit-Burst induced a minor, additional depression that was not significantly different from EPSC amplitude after HFS alone (EPSC amplitude at t = 14 min vs t = 35 min, 59.5 ± 8.9% of the original pre-HFS baseline; p = 0.2; n = 5).
Figure 10.
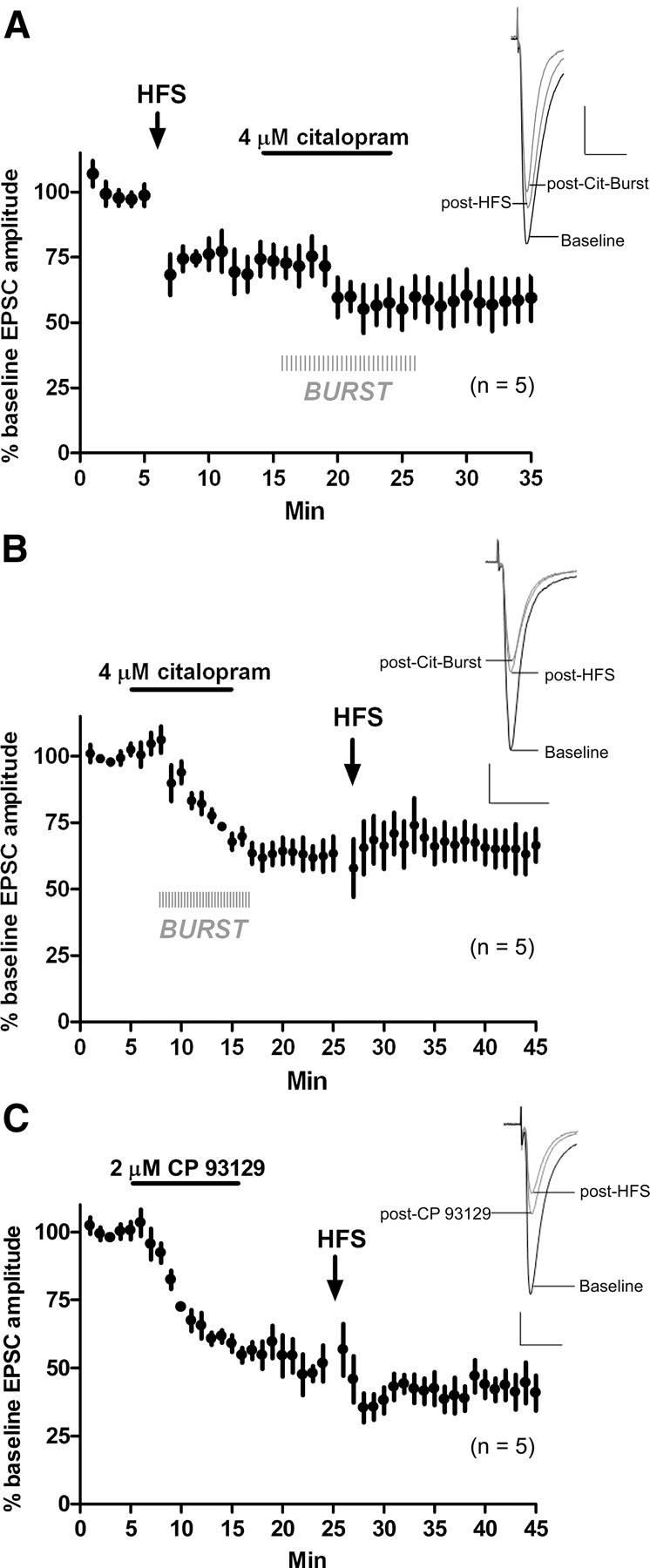
5-HT–LTD and eCB–LTD are mutually occlusive. A, Delivery of HFS induced LTD. After a 7 min stabilization period, Cit-Burst did not significantly further depress EPSC amplitude. B, After stabilization of Cit-Burst-induced depression, ESPC amplitude was not further depressed by HFS. C, After stabilization of CP 93129-induced depression, ESPC amplitude was not further depressed by HFS. Calibration: 100 pA, 20 ms.
To examine whether 5-HT–LTD occludes eCB–LTD, we applied Cit-Burst, which again induced LTD of EPSC amplitude compared with baseline (t = 25 min, 63.5 ± 6.4%; p = 0.0005; n = 5) (Fig. 10b). After stabilization of EPSC amplitude, we then delivered HFS. Compared with EPSC amplitude after Cit-Burst, HFS did not induce an additional depression (EPSC amplitude t = 25 min vs t = 45 min, 67.1 ± 4.9%; p = 0.7; n = 5), showing eCB–LTD and 5-HT–LTD to be mutually occlusive. It is possible that some eCBs are released by the burst component of Cit-Burst, and this could decrease the ability of HFS to induce an additional eCB-dependent depression after Cit-Burst. Thus, we bath applied CP 93129 (2 μm), waited for stabilization of the EPSC amplitude, and then delivered HFS. CP 93129 (2 μm) alone depressed the EPSC amplitude (vs baseline at t = 24 min, 51.7 ± 6.6%; p < 0.0001; n = 5). HFS did not further depress EPSC amplitude after CP 93129 application (EPSC amplitude t = 24 min vs t = 45 min, 40.8 ± 6.5%; p = 0.3; n = 5) (Fig. 10c), showing that this route to 5-HT–LTD also occludes eCB–LTD.
Discussion
We show that exogenously applied or endogenous 5-HT induces a presynaptically expressed, 5-HT1b-dependent LTD of glutamatergic transmission (5-HT–LTD) in the dorsal striatum that culminates in a depression of net striatal output. 5-HT–LTD is cAMP pathway dependent and, within the striatum, represents a novel pathway to lasting depression that parallels and occludes the dopamine-dependent, eCB-mediated LTD.
Mutual occlusion between 5-HT–LTD and eCB–LTD, increased PPR, and decreased sEPSC and mEPSC frequencies indicate a presynaptic mechanism of 5-HT–LTD expression, perhaps involving a decrease in probability of glutamate release. In contrast, mEPSC and uEPSC amplitudes were not affected, and postsynaptic manipulations, particularly BAPTA loading, did not prevent 5-HT–LTD induction. This evidence for presynaptic 5-HT1b localization is supported by electron microscopic, electrophysiological, and neurochemical studies that revealed that 5-HT1b receptors are localized to presynaptic terminals and act as autoceptors or heteroceptors in several brain regions (Sari et al., 1999; Morikawa et al., 2000). Considering that application of 5-HT, a 5-HT1b agonist, and citalopram all induce LTD, the most parsimonious explanation of these findings is that 5-HT–LTD induction involves 5-HT acting heterosynaptically on 5-HT1b receptors localized on corticostriatal terminals.
It should be noted that our glutamate uncaging experiment could be confounded by sample bias through inadvertent examination of 5-HT1b-unresponsive synapses on spines. However, it is unlikely that we would have consistently observed no change in uEPSCs given the strong 5-HT1b agonist inhibition of eEPSCs. To minimize eEPSC and uEPSC amplitude disparity, and thus synapse selection disparity, we used electrical stimulation parameters that evoked the smallest-amplitude eEPSCs that showed acceptable consistency. Because all corticostriatal synapses are on MSN spines and a band of 5-HT1b mRNA-positive cell bodies is visible in layer V of mouse cortex whereas no signal is apparent in the thalamic intralaminar nuclei (Allen Brain Atlas; http://www.brain-map.org/), we would have expected to observe depression in most, if not all, of the uEPSCs. Thus, although we cannot fully rule out an effect of sample bias, we feel that it is unlikely to account for our findings.
Reversible inhibition of glutamate release has been demonstrated during 5-HT application in the nucleus accumbens (Muramatsu et al., 1998). We showed that 5-HT–LTD is not a consequence of residual agonism by applying the 5-HT1b antagonist NAS-181 after induction and stabilization of EPSC amplitude depression. We also observed 5-HT–LTD at inhibitory synapses onto dorsolateral MSNs and at glutamatergic synapses onto MSNs of the nucleus accumbens core. Interestingly, we observed a reversible inhibition during Cit-Burst delivery in a few D1-containing and putative D2-containing cells recorded from mouse slices and in all cells when CP 93129 was applied in the presence of forskolin. The reversible effect was not seen in mouse slices when we applied CP 93129 alone. This suggests that the duration and concentration of 5-HT1b activation, and coupling to downstream AC, may be critical in governing the transition from short- to long-term inhibition of glutamate release.
Although 5-HT–LTD and eCB–LTD share the common feature of presynaptic Gi/Go-coupled signaling and depression of neurotransmitter release, 5-HT–LTD is a unique form of plasticity in brain because of its distinct requirements for induction. Unlike eCB–LTD, which is mediated by retrograde eCB signaling (Gerdeman et al., 2002), our postsynaptic manipulation data indicate that retrograde signaling is not involved in the induction of 5-HT–LTD. This is important because both presynaptic activity and postsynaptic calcium influx appear to be required for eCB–LTD (Singla et al., 2007; Adermark et al., 2009), indicating synapse specificity, and, thus, a “Hebbian” form of plasticity. In the contrasting case of 5-HT–LTD, our data suggest that presynaptic corticostriatal action potential firing is unnecessary for 5-HT–LTD induction. Because all of our experiments involved a constant holding potential during recordings and a sodium channel blocker (QX-314) in a cesium-based internal solution, we can assume that postsynaptic activity is not necessary for 5-HT–LTD induction. Without the need for presynaptic or postsynaptic activity, 5-HT–LTD appears to be non-Hebbian.
DRN firing is implicated in the control of actions (Fornal et al., 1996; Jacobs and Fornal, 1999). Single-unit recordings from freely moving cats demonstrated significant firing increases in subsets of serotonergic DRN neurons preceding initiation of oral-buccal movements, resumption of normal firing rates during termination of motor sequences (Fornal et al., 1996), and abrupt decreases in DRN activity to levels at or below baseline that are tightly correlated with spontaneous pauses in motor sequences. The notion of striatal 5-HT influencing action control is consistent with the clinical use of SSRIs for the treatment of obsessive-compulsive disorder (Goddard et al., 2008). Genetic deletion of the corticostriatal synapse-specific protein SAPAP3 results in an obsessive-compulsive disorder-like behavioral phenotype that is alleviated by SSRI treatment (Welch et al., 2007). In the context of the present findings, the dynamic interaction of striatal 5-HT–LTD and eCB–LTD is a candidate mechanism for the long-term filtering of corticostriatal input enabling proper action selection that may also represent a point of perturbation by therapeutic and illicit drugs (Rocha et al., 1998).
Footnotes
The authors declare no competing financial interests.
References
- Adermark L, Talani G, Lovinger DM. Endocannabinoid-dependent plasticity at GABAergic and glutamatergic synapses in the striatum is regulated by synaptic activity. Eur J Neurosci. 2009;29:32–41. doi: 10.1111/j.1460-9568.2008.06551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best AR, Regehr WG. Serotonin evokes endocannabinoid release and retrogradely suppresses excitatory synapses. J Neurosci. 2008;28:6508–6515. doi: 10.1523/JNEUROSCI.0678-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomeley CP, Bracci E. Serotonin excites fast-spiking interneurons in the striatum. Eur J Neurosci. 2009;29:1604–1614. doi: 10.1111/j.1460-9568.2009.06725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boschert U, Amara DA, Segu L, Hen R. The mouse 5-hydroxytryptamine1B receptor is localized predominantly on axon terminals. Neuroscience. 1994;58:167–182. doi: 10.1016/0306-4522(94)90164-3. [DOI] [PubMed] [Google Scholar]
- Bouhelal R, Smounya L, Bockaert J. 5-HT1B receptors are negatively coupled with adenylate cyclase in rat substantia nigra. Eur J Pharmacol. 1988;151:189–196. doi: 10.1016/0014-2999(88)90799-6. [DOI] [PubMed] [Google Scholar]
- Carter AG, Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron. 2004;44:483–493. doi: 10.1016/j.neuron.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Davis MI, Ronesi J, Lovinger DM. A predominant role for inhibition of the adenylate cyclase/protein kinase A pathway in ERK activation by cannabinoid receptor 1 in N1E-115 neuroblastoma cells. J Biol Chem. 2003;278:48973–48980. doi: 10.1074/jbc.M305697200. [DOI] [PubMed] [Google Scholar]
- Di Matteo V, Pierucci M, Esposito E, Crescimanno G, Benigno A, Di Giovanni G. Serotonin modulation of the basal ganglia circuitry: therapeutic implication for Parkinson's disease and other motor disorders. Prog Brain Res. 2008;172:423–463. doi: 10.1016/S0079-6123(08)00921-7. [DOI] [PubMed] [Google Scholar]
- Fornal CA, Metzler CW, Marrosu F, Ribiero-do-Valle LE, Jacobs BL. A subgroup of dorsal raphe serotonergic neurons in the cat is strongly activated during oral-buccal movements. Brain Res. 1996;716:123–133. doi: 10.1016/0006-8993(96)00006-6. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Goddard AW, Shekhar A, Whiteman AF, McDougle CJ. Serotonin mechanisms in the treatment of obsessive-compulsive disorder. Drug Discov Today. 2008;13:325–332. doi: 10.1016/j.drudis.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Heintz N. Gene expression nervous system atlas (GENSAT) Nat Neurosci. 2004;7:483. doi: 10.1038/nn0504-483. [DOI] [PubMed] [Google Scholar]
- Jacobs BL, Fornal CA. Activity of serotonergic neurons in behaving animals. Neuropsychopharmacology. 1999;21:9S–15S. doi: 10.1016/S0893-133X(99)00012-3. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Maroteaux L, Saudou F, Amlaiky N, Boschert U, Plassat JL, Hen R. Mouse 5HT1B serotonin receptor: cloning, functional expression, and localization in motor control centers. Proc Natl Acad Sci U S A. 1992;89:3020–3024. doi: 10.1073/pnas.89.7.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa H, Manzoni OJ, Crabbe JC, Williams JT. Regulation of central synaptic transmission by 5-HT(1B) auto- and heteroreceptors. Mol Pharmacol. 2000;58:1271–1278. doi: 10.1124/mol.58.6.1271. [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Lapiz MD, Tanaka E, Grenhoff J. Serotonin inhibits synaptic glutamate currents in rat nucleus accumbens neurons via presynaptic 5-HT1B receptors. Eur J Neurosci. 1998;10:2371–2379. doi: 10.1046/j.1460-9568.1998.00248.x. [DOI] [PubMed] [Google Scholar]
- Rocha BA, Scearce-Levie K, Lucas JJ, Hiroi N, Castanon N, Crabbe JC, Nestler EJ, Hen R. Increased vulnerability to cocaine in mice lacking the serotonin-1B receptor. Nature. 1998;393:175–178. doi: 10.1038/30259. [DOI] [PubMed] [Google Scholar]
- Sari Y, Miquel MC, Brisorgueil MJ, Ruiz G, Doucet E, Hamon M, Vergé D. Cellular and subcellular localization of 5-hydroxytryptamine1b receptors in the rat central nervous system: immunocytochemical, autoradiographic and lesion studies. Neuroscience. 1999;88:899–915. doi: 10.1016/s0306-4522(98)00256-5. [DOI] [PubMed] [Google Scholar]
- Singla S, Kreitzer AC, Malenka RC. Mechanisms for synapse specificity during striatal long-term depression. J Neurosci. 2007;27:5260–5264. doi: 10.1523/JNEUROSCI.0018-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soghomonian JJ, Descarries L, Watkins KC. Serotonin innervation in adult rat neostriatum. II. Ultrastructural features: a radioautographic and immunocytochemical study. Brain Res. 1989;481:67–86. doi: 10.1016/0006-8993(89)90486-1. [DOI] [PubMed] [Google Scholar]
- Steinbusch HW, Nieuwenhuys R, Verhofstad AA, Van der Kooy D. The nucleus raphe dorsalis of the rat and its projection upon the caudatoputamen. A combined cytoarchitectonic, immunohistochemical and retrograde transport study. J Physiol (Paris) 1981;77:157–174. [PubMed] [Google Scholar]
- Tepper JM, Wilson CJ, Koós T. Feedforward and feedback inhibition in neostriatal GABAergic spiny neurons. Brain Res Rev. 2008;58:272–281. doi: 10.1016/j.brainresrev.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JM, Lu J, Rodriguiz RM, Trotta NC, Peca J, Ding JD, Feliciano C, Chen M, Adams JP, Luo J, Dudek SM, Weinberg RJ, Calakos N, Wetsel WC, Feng G. Cortico-striatal synaptic defects and OCD-like behaviours in SAPAP3-mutant mice. Nature. 2007;448:894–900. doi: 10.1038/nature06104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Park BS, Adermark L, Lovinger DM. Ethanol reverses the direction of long-term synaptic plasticity in the dorsomedial striatum. Eur J Neurosci. 2007;25:3226–3232. doi: 10.1111/j.1460-9568.2007.05606.x. [DOI] [PubMed] [Google Scholar]
- Yin HH, Adermark L, Lovinger DM. Neurotensin reduces glutamatergic transmission in the dorsolateral striatum via retrograde endocannabinoid signaling. Neuropharmacology. 2008;54:79–86. doi: 10.1016/j.neuropharm.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]