

Abstract
It is well recognized that masticatory muscle function helps determine morphology, although the extent of function on final form is still debated. GDF-8 (myostatin), a transcription factor is a negative regulator of skeletal muscle growth. A recent study has shown that mice homozygous for the myostatin mutation had increased muscle mass and craniofacial dysmorphology in adulthood. However, it is unclear whether such dysmorphology is present at birth. This study examines the onset and relationship between hypermuscularity and craniofacial morphology in neonatal and adult mice with GDF-8 deficiency.
Fifteen (8 wild-type and 7 GDF-8 −/−), 1 day old and 16 (9 wt and 7 GDF-8 −/−), 180 day old male CD-1 mice were used. Standardized radiographs were taken of each head, scanned, traced, and cephalometric landmarks identified. Significant mean differences were assessed using a group × age, two-way ANOVA.
Myostatin-deficient mice had significantly (p<0.01) smaller body and masseter muscle weights and craniofacial skeletons at 1 day of age and significantly greater body and masseter muscle weights at 180 days of age compared to controls. Myostatin-deficient mice showed significantly (p<0.001) longer and “rocker-shaped” mandibles and shorter and wider crania compared to controls at 180 days. Significant correlations were noted between masseter muscle weight and all cephalometric measurements in 180 day old Myostatin-deficient mice.
Results suggest in this mouse model, there may be both early systemic skeletal growth deficiencies and later compensatory changes from hypermuscularity. These findings reiterate the role that masticatory muscle function plays on the ontogeny of the cranial vault, base, and most notably the mandible.
Keywords: Morphology, Myostatin, GDF-8, Mice, Craniofacial Growth
INTRODUCTION
Myostatin is a negative regulator of muscle growth. Inactivation of myostatin has been useful in producing livestock with dramatic increases in muscle mass, as evidenced by the Belgian Blue and Piedmontese muscle doubled cattle breeds. An 11-bp deletion in the coding region of the gene produces a frame-shift mutation which alters one of the hallmark protein residues common to the TGF-β superfamily. The myostatin knockout mouse, which shares phenotypic characteristics with these cattle, has a similar mutation in this area of the gene (Kambadur et al., 1997). The gene is highly conserved across species, and a spontaneous gene mutation associated with increased muscle mass has been identified in humans (Schuelke et al., 2004). Transcripts are found early in development and continue in adulthood where it may play a homeostatic role, since increases in myostatin in adult mice leads to profound cachexia of muscle tissue (Zimmers et al., 2002). It therefore functions to inhibit excessive growth of muscle in order to maintain an overall balance of this tissue relative to adipose tissue.
Myostatin, GDF-8, is a member of the TGF-β domain and utilizes the TGFβr pathway for signaling. In humans, GDF-8 maps to chromosome 2 (2q 32.2). Orthologs for this gene have been found in Canis familiaris, Pan troglodytes, Gallus gallus, and most importantly for research purposes Mus musculus (chromosome 1 27.80cM) (NCBI, 2008). Several isoforms have been identified and are expressed in both embryonic and mature skeletal muscle. GDF-8 is a 375- amino acid polypeptide with an N-terminal pro-peptide region and C-terminal signaling peptide proteolytic processing at the conserved RSRR site resulting in a 12.5 kDa C-terminal mature peptide which can form a GDF-8 active homodimer (Phillip et al., 2005). GDF-8 has functions in local cytokine and growth factor activity as well as protein, receptor, and calcium ion bindings (NCBI, 2008).
Myostatin appears early in development, and as somite condensations become topographically organized it becomes restricted to the myotome layer. In fetal stages transcripts are found throughout developing muscle. Here myostatin has a profound effect on the regulation of differentiation pathways for mesenchymal cells. When myostatin activity is blocked, more cells are committed to myoblast and myotube formation which results in significantly elevated numbers of muscle fibers in adult muscle. In some muscles this increase in fiber number approximates 90 percent (McPherron et al., 1997). Average fiber diameter of cells is also increased, demonstrating that both hypertrophy and hyperplasia are responsible for increased muscle mass. Fiber type distributions are also altered. Knockout mice have decreased numbers of type I fibers and increased numbers of type II fibers, especially the type IIB, glycolytic fast-contracting fiber (Girgenrath et al., 2005; Salerno et al., 2004). These adaptations however, do not necessarily lead to functionally superior muscle. When limb muscles from knockout and wild-type mice are compared, the myostatin-deficient muscle, although larger, has no relative increase in maximum tetanic force generation. And when force is calculated per unit area in limb muscles sampled so far, the myostatin-deficient muscle is actually weaker. This may be related to irregularities found in the IIB fibers from the knockouts, which have cytoplasmic inclusions containing tubular aggregates and diminished mitochondrial to nuclear ratios, suggesting mitochondrial depletion problems during development (Amthor et al., 2007)
The myostatin knockout mouse is of interest to craniofacial biology investigation since its masticatory muscles have been demonstrated to be significantly larger. The masseter muscle weighs about 80% more (Vecchione et al., 2007), and the temporalis muscle is approximately 40% larger than wild-type controls (Byron et al., 2006). Unlike limb muscles there is an increase in the proportion of type IIA and IIX fibers in temporalis muscle, but the mean fiber areas are similar between wild-type and knockout. Bite forces are significantly increased, but when controlled for overall muscle size, the bite force/unit area is similar to that of the wild-type (Byron et al., 2006). These findings indicate that a consistent amount of force is applied over a much greater area of the craniofacial skeleton in the myostatin knockout, and this should in turn, lead to altered biomechanical stress and bony morphology. This is indeed the case. In previous studies (Byron et al., 2006; Vecchione et al., 2007), we reported that adult myostatin knockout mice were generally more brachycephalic, had smaller cranial vault and maxillary lengths, and exhibited significantly different mandibular shapes with the mandible longer, and shortened in the vertical dimension compared to wild-type controls. However, it was still unclear if these craniofacial skeletal abnormalities were related to the GDF-8 knockout condition in the pre- or neonatal period or were simply a result of postnatal compensatory changes? In this follow-up study we describe craniofacial morphology from the neonatal developmental stage through adulthood in order to further typify the interactions between muscle and bone in this model.
MATERIAL & METHODS
Fifteen (8 wild-type control and 7 GDF-8 −/− myostatin-deficient), one day old, and 16 (9 wild-type control and 7 GDF-8 −/− myostatin-deficient), 180 day old, male CD-1 mice were used in this study. Myostatin-deficient mice were produced by deletion of the C-terminal region of the myostatin gene in embryonic stem cells as described by McPherron et al. (1997). All mice were housed together and given food (Harlan TekLad hard rodent chow) and water ad libitum. Wild-type control and GDF-8 −/− myostatin-deficient mice were bred and maintained in the Department of Cellular Biology & Anatomy Medical College of Georgia. Mice were euthanized at one day postnatal or 180 days of age at skeletal maturity by CO2 overdose according to the protocol approved by the Medical College of Georgia Institutional Animal Care and Use Committee (IACUC) and body weights were immediately recorded.
Mice heads were disarticulated and the masseter muscles (Figure 1) were then dissected and weighed wet to the nearest 0.001g on a Mettler vacumn balance. Heads were then fixed in 10% neutral buffered formalin for 24 hours, and then transferred to 70% ethanol for radiographic analysis. Lateral and dorso-ventral radiographs were taken using a Faxitron MX-20 (Faxitron X-Ray Corporation) at 35kV for 250 seconds at 5× magnification with X-OMAT V diagnostic film (Kodak). Radiographs were scanned on an AGFA DuoScan using AGFA FotoLook 3.2 software.
Figure 1.

Inferior view of masseter muscles and mandibles from wild-type (WT) control and myostatin (GDF-8−/−) deficient mice at 180 days of age. Note the extremely large masticatory musculature in the myostatin-deficient mice (scale in mm).
Twelve cephalometric landmarks were identified on the scanned images and a number of measurements reflecting various craniofacial dimensions were made using Dolphin Cephalometric Software (Vecchione et al., 2007). Lateral cephalometric landmarks were identified as follows: Pa (parietal), the most posterior point of the parietal bone; Na (nasion), the junction of the frontonasal and internasal sutures; Rh (rhinion), the most anterior point of the nasal bones in the midline; Os (occipito-sphenale), the middle of the sphenoocciptial synchrondrosis; Fp (fronto-parietal suture), the vertex of the skull at bregma; Ps (pre-sphenoid), the middle of the presphenoethmoidal synchrondrosis; Ra (ramus), the most posterior point of the body of the ramus just superior to the angular process; Go (gonion), the most inferior point of mandibular angular process; Co (condylion), most postero-superior point of condylar process; I (incisor), the most prominent point between incisal edges of lower incisors; Op (opisthion), the most posterior rim of foramen magnum; Ba (basion), the most posterior point of occipital bone anterior to the foramen magnum. Dorso-ventral cephalometric landmarks were identified as follows: Rh (rhinion), the most anterior point of the nasal bones in the midline; and Op (opisthion), the most posterior rim of foramen magnum. Cephalometric measurements that were calculated included: Cranial vault height (Fp-Os); Cranial vault length (Pa-Na); Craniofacial length (Op-Rh); Cranial base length (Ba-Ps); Mandibular body length (Ra-I); and Mandibular shape index ((Co-Go/Ra-I)*100). Landmark identification and measurements were made twice on 20% of the sample and intraobserver error was assessed by correlation and paired design. Intraobserver reliability was r=0.90, p<0.001 and there was no significant differences between mean measurement, t=1.662, df=87, p>0.05.
Mean somatic and cephalometric data were calculated and compared between groups using a two-way (Group × Age) Analysis of Variance (ANOVA). The relationship between masseter muscle weight and various cephalometric measurements were assessed using a Pearson Product Moment Correlation. All data were analyzed using SPSS 15.0 for windows (SPSS, Inc., Chicago, IL). Differences were considered significant if p < 0.05.
RESULTS
Myostatin-deficient mice had smaller body weights than wild-type controls (by 12.38%) at 1 day of age and greater body weights (by 14.81%) than wild-type controls at 180 days of age (Figure 2). Body weight in the myostatin-deficient mice was significantly less (Group F = 17.39; p<0.001) at 1 day of age and increased significantly more from 1 to 180 days of age (Group × Age F = 6.95; p<0.01) compared to wild-type controls (asterisks - Figure 2). Myostatin-deficient mice also had smaller masseter muscle weights (by 21.15%) at 1 day of age and dramatically greater masseter muscle weights (by 43.55%) at 180 days of age compared to wild-type controls (Figures 1 and 2). Masseter muscle weight in the myostatin-deficient mice was significantly less (Group F = 18.79; p<0.001) at 1 day of age and increased significantly more (Group × Age F = 86.48; p<0.001) from 1 to 180 days of age compared to wild-type controls (asterisks - Figure 2).
Figure 2.

Mean (+/− S.E.) body weight (top) and masseter muscle weight (bottom) by age and group and the results of statistical analysis. In both graphs, note the significantly lower weights at 1 day of age (asterisks) and the significantly greater weights in the myostatin (GDF-8−/−) -deficient mice at 180 days of age compared to the wild-type control mice.
Qualitatively, as seen on the dorso-ventral and lateral head radiographs (Figures 3 and 4), myostatin knock-out mice were more brachycephalic, both in the cranial vault and maxilla, compared to wild-type controls. These differences were more pronounced at 180 days of age (Figures 3 and 4). Myostatin-deficient mice also showed significantly remodeled mandibular rami, mandibular bodies, and coronoid processes resulting in a “rocker-shaped” mandibular morphology compared to wild-type controls at 180 days of age (Figure 4).
Figure 3.

Dorsoventral radiographs from wild-type control (left column) and Myostatin-deficient (right column) 1 day (top row) and 180 day old (bottom row) mice. Note the shortened cranial vault (CV) and maxilla (MX) in the 180 day old myostatin-deficient skull compared to the wild-type skull producing brachycephaly in the adult myostatin-deficient skull.
Figure 4.
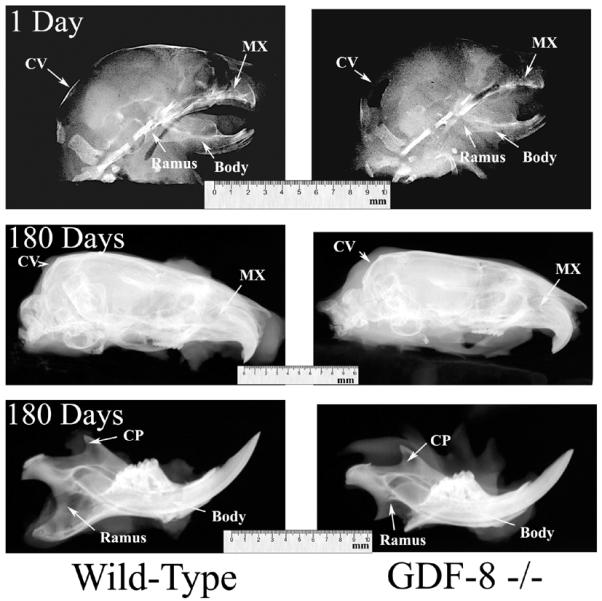
Lateral radiographs from wild-type control (left column) and myostatin-deficient (right column) 1 day old (top row) and 180 day old (middle and bottom rows) mice. Note the shortened cranial vault (CV) and maxilla (MX) in the 180 day old myostatin-deficient skull compared to the wild-type skull producing brachycephaly in the adult myostatin-deficient skull. Also note the dramatically altered ramus and coranoid process (CP), and the elongated and rounded body in the adult myostatin-deficient mandible compared to the wild-type mandible, producing a “rocker-type” mandibular morphology.
Cephalometric analysis revealed quantitative differences in all six measurements between the two groups. Wild-type control mice had greater cranial vault height (Fp-Os) and cranial vault length (Pa-Na) dimensions at both 1 and 180 days of age compared to the myostatin-deficient mice (Figure 5). Significant group mean differences were noted for the cranial vault height (F = 3.74; p <0.05) and cranial vault length (F = 16.69; p <0.001) dimensions (asterisks - Figure 5). Wild-type control mice also had greater craniofacial length (Op-Rh) at both 1 and 180 days of age compared to the myostatin-deficient mice (Figure 6). Significant group mean differences were noted for the craniofacial length (F = 5.09; p <0.05) dimension (asterisks - Figure 6). In contrast, cranial base length (Ba-Ps) (Figure 6) was greater in wild-type control mice at 1 day of age and greater in myostatin-deficient mice by 180 days of age (Figure 6). A significant mean group by age interaction was noted for the cranial base length dimension (F = 5.00; p <0.05) (asterisks - Figure 6).
Figure 5.

Mean (+/− S.E.) cranial vault height (top) and cranial vault length (bottom) by age and group and the results of statistical analysis. Note the significantly (asterisks) shorter cranial vault height and length in the myostatin (GDF-8−/−) -deficient mice compared to the wild-type control mice at both 1 and 180 days of age.
Figure 6.

Mean (+/− S.E.) craniofacial length (top) and cranial base length (bottom) by age and group and the results of statistical analysis. Note the significantly (asterisks) shorter craniofacial length in the myostatin (GDF-8−/−) -deficient mice compared to the wild-type control mice at both 1 and 180 days of age. In contrast, note the significantly (asterisks) shorter cranial base length at 1 day of age and significantly greater cranial base length at 180 days of age in the myostatin (GDF-8−/−) -deficient mice compared to the wild-type control mice.
Wild-type control mice had greater mandibular body length (Ra-I) and mandibular shape index ((Co-Go/Ra-I)*100) at 1 day of age compared to myostatin-deficient mice (Figure 7). However, by 180 days of age, both mandibular body length (Ra-I) and mandibular shape index ((Co-Go/Ra-I)*100) were greater in myostatin-deficient mice compared to the wild-type control mice (Figure 7). Significant mean group by age interactions were noted for both the mandibular body length (F = 20.35; p <0.001) and the mandibular shape index (F = 10.02; p <0.01) dimensions (asterisks - Figure 7).
Figure 7.

Mean (+/− S.E.) mandibular body length (top) and mandibular shape index (bottom) by age and group and the results of statistical analysis. In both dimensions, note the significantly (asterisks) smaller means at 1 day of age and significantly greater means at 180 days of age in the myostatin (GDF-8−/−) -deficient mice compared to the wild-type control mice.
The relationship between masseter muscle mass and the various cephalometric measurements for both groups and ages were assessed using a Pearson Product Moment Correlation (Table 1). At 1 day of age, no significant relationships were noted between masseter muscle weight and any of the craniofacial measures in the wild-type control mice. In contrast, 1 day old myostatin-deficient mice showed a significant positive correlation coefficient between masseter muscle weight and cranial base length (Ba-Ps), and a significant negative correlation coefficient between masseter muscle weight and cranial vault height (Fp-Os) (table). At 180 days of age, a significant positive correlation coefficient was noted only between masseter muscle weight and the mandibular shape index ((Co-Go/Ra-I)*100) in wild-type control mice. In contrast, 180 day old myostatin-deficient mice showed significant positive correlation coefficients between masseter muscle weight and cranial vault height (Fp-Os), cranial vault length (Pa-Na), cranial base length (Ba-Ps), and mandibular ramal height (Ra-I). A significant negative correlation coefficient was found between masseter muscle weight and mandibular shape index ((Co-Go/Ra-I)*100) (Table 1).
Table 1.
Correlation coefficients between masseter muscle weight and various craniofacial dimensions by Group and Age.
| Dimension\Group | WT 1 Day |
GDF-8 −/− 1 Day |
WT 180 Day |
GDF-8 −/− 180 Day |
|---|---|---|---|---|
| Cranial Vault Height |
−.604 | −.975*** | −.653 | .763* |
| Cranial Vault Length |
−.636 | −.482 | −.335 | .828* |
| Craniofacial Length |
−.395 | .522 | .101 | .268 |
| Cranial Base Length |
.360 | .972*** | −.543 | .923*** |
| Mandibular Ramal Height |
.557 | .121 | −.401 | .903** |
| Mandibular Shape Index |
.224 | .572 | .709* | −.730* |
= p<0.05;
= p<0.01;
= p<0.001
DISCUSSION
Results from the present study demonstrated that myostatin-deficient mice were significantly different in all dimensions measured compared to wild-type controls mice at 180 days of age. Although all animals were timed-pregnant and harvested on the same day, 1 day old myostatin-deficient mice had slightly, but significantly smaller body weights, masseter muscle weights, and skeletal sizes than age-matched controls. In contrast, 180 day old myostatin-deficient mice had dramatically and significantly greater body and masseter muscle weights, surprisingly, however they still had shorter craniofacial length measures but longer cranial base and mandibular length measures. These results are similar to those reported by Byron et al., (2008) who showed that adult myostatin-deficient mice had significantly shortened and compressed (in an anterior-posterior dimension) temporal bones and a smaller squamousal suture bevel compared to wild-type controls. These findings suggest that in this GDF-8 −/− myostatin-deficient mouse model there may be both early systemic skeletal growth deficiencies and later compensatory changes from hypermuscularity.
There were several developmental differences evidenced in the neonates with myostatin deficiency in comparison to wild-type controls. During this stage both the body weight and the masseter muscle weight were significantly less in knockouts. In embryonic and early fetal stages of muscle development tissue hyperplasia predominates. Cells are committed to a myoblast lineage and their fusion produces increasing numbers of myotubes (Buckingham et al., 2003). In the late fetal and early neonatal periods these myotubes mature into individual myofibers, and begin production of large volumes of myofibrillar proteins which assemble into sarcomeric units. The late fetal and early neonatal period is characterized by tissue hypertrophy, increasing the size of muscle fibers, with relatively little increase in fiber number. With myostatin deficiency, the hyperplastic period of muscle development is most likely lengthened, allowing for increased proliferation of myoblast and myotube populations. With the hypertrophic stage delayed, the knockout mice had smaller muscle weights at birth, but with enhanced growth potential due to increased numbers of myofibers. At birth, masseter muscle fibers from wild-type mice were probably producing greater amounts of contractile proteins and sarcomeric units, eventually making them heavier. This also resulted in significant differences in whole body weights, due to overall differences in muscle tissue maturation. Significant differences in craniofacial dimensions were also identified. With the loss of myostatin, the delicate signaling balances between cell proliferation, protein synthesis and tissue interactions and skeletal growth are probably thrown out of balance in early developmental stages.
Like other members of the TGF-β superfamily GDF-8 primarily uses the TGFβr signaling pathway in which it elicits function by binding to a family of serine/theronine kinase receptors followed by activiation of the receptor’s SMAD proteins, which when transolacted to the cell’s nucleus regulates transcription of target genes, and its receptor Acvr IIB within muscle. However, many members of the TGF-β superfamily, including GDF-8, also access other signaling pathways including MAPK, ALK, ERK, JNK, and p38 stress response pathways to either induce the TGF-βr pathway, i.e. ALK-2 induced activation of SMAD2/3 phosphorylation, or access different pathways associated with those signaling receptors, which has complicated the biochemical research concerning the myostatin pathway (Rebbapragada et al., 2003; Tsuchida et al., 2008). Recent research suggests a possible antagonistic relationship or at least a competing relationship for binding sites, serine/threonine kinase receptors with at least one isoform of bone morphogenetic protein, BMP-7, suggesting that although myostatin’s receptor Acvr IIB, is not generally indicated in bone, it still may have an effect on bone development at the cellular level (Rebbapragada et al., 2003).
Myoblasts, chondrocytes, osteoblasts and adipocytes develop from a common pool of skeletal mesenchymal stem cells (Owen, 1988). Recruitment of stem cells through developmental pathways leading to cell differentiation is a complex phenomenon involving tightly regulated signaling and binding of growth factors. The TGF-β superfamily of growth factors is pivotal in cell migration, proliferation, tissue differentiation and morphogenesis. Of these, the bone morphogenic protein subfamily (composed of at least 15 molecules, Kingsley, 1994), are essential for cranial base (Dewulf et al., 1995; Kettunen et al., 2006), and limb morphogenesis (Yi et al., 2000). They also have a direct role in regulation of protein synthesis in muscle development (Verschueren et al., 1995). The rate of protein synthesis in skeletal muscle at birth is very high, and declines approximately 50% during the first three weeks of life (Davis et al., 1989). This decline is influenced in part by the rapidly increasing levels of BMP2, BMP7 and myostatin which all act as negative regulators of transcription (Suryawan et al., 2006). Myostatin also has the ability to prevent BMP7 from binding to its receptor, and therefore acts as a competitive inhibitor for BMP7 (Rebbapragada et al., 2003). These and perhaps many other signaling paths are altered. The net results of which are subtle changes in the size of the cranial vault, the cranial base and mandible. These changes most likely occur through modification of skeletal mesenchymal stem cell recruitment at key periods of differentiation and proliferation. This would leave some tissue primordia slightly larger or smaller than would be normal for a particular stage of development. For all six of the craniofacial dimensions, the myostatin-deficient mice were significantly smaller. This was probably the end result of increased myoblast recruitment from the skeletal mesenchymal stem cell pools.
Cranial vault height, cranial vault length, and craniofacial length were all smaller in the knockout mice at birth compared to wild-type mice, and these differences were maintained throughout life. These areas encompass the cranial vault and nasal capsule where the principal joints articulating the skeleton are sutures. Over time, increased forces generated from muscles like the temporalis and masseter in the knockout mice, are not necessarily seen as an overall change in cranial morphology. Rather these sutures have inherent ability to buffer excessive force through the process of increased interdigitation (Jaslow, 1990). This results in increased bony surface area at the joints, increased extracellular matrix to buffer forces and increases in bending strength and toughness (Herring and Mucci, 1991). In fact sutures from our mice have been found to be more anatomically interdigitated, less stiff, and undergo more displacement under tension than in wild-type sutures (Byron et al., 2004; Nicholson et al., 2006). So the head and face of myostatin-deficient mice were smaller, and maintained their relative size through allometric growth. Increased masticatory forces were also evidenced by changes in the microanatomic complexity of facial and cranial sutural joints. This adaptation allowed dissipation of increased chewing forces without overall changes in growth pattern.
Other craniofacial dimensions however did change in relation to their wild-type counterparts over time. The cranial base in the knockout mice was shorter than in the wild-types at birth, but the knockout mice obtained a significantly larger size when growth was completed. The same was true for mandibular length, being shorter at birth and longer in adulthood in the knockout mice. These changes were most likely influenced by differences in muscle force during growth, especially since the greatest changes occurred between 80 to 120 days of maturation, after the muscles had reached full maturity. The cartilaginous tissues producing the majority of growth in the cranial base and mandible responded to increased muscle force by growth in a more sagittal rather than vertical direction, making their end shapes longer than in wild-type animals. This is in agreement with most reports of facial growth which conclude that increased forces result in shortened faces (Schendel et al., 1980) and deep bites (Sassouni, 1969). Further, as reported in an earlier study (Vecchione et al., 2007), the overall shape of the 180 day old myostatin-deficient mouse mandible, compared to the wild-type control mandible, resembles a unique human jaw morphology described previously in Polynesian populations (Houghton, 1978; Schendel et al., 1980; Kean and Houghton, 1990). The effect of hypermuscularity is thought to have resulted in a characteristic mandibular shape described as a “rocker mandible” (Marshall and Snow, 1956). A rocker mandible lacks an antegonial notch and the angle of the mandible is convex which results in rocking when placed on a flat surface. These features of the Polynesian mandible occur at puberty (Houghton, 1977) when bone shape is altered by local muscular forces (Enlow, 1975). The Inuit (i.e., Eskimo) skull is also characterized by a large mandible, larger muscle attachments, and palatal and mandibular tori. It has also been suggested that the distinctive shape of the Inuit skull is a result of vigorous chewing (Collins’ “hard chewing hypothesis”, Hylander, 1977). Hylander (1977) also suggested that the Inuit skull is adapted to generate and dissipate large vertical and biting forces and the masseter muscles are also positioned more anteriorly which may help generate larger bite forces. These observations also suggest that hypermuscularity is an important determinant of craniofacial morphology.
These results suggest that postnatal function activates muscle enlargement, not prenatal gene expression. This may be viewed as a discordant result given the fact that heterozygote and homozygote Belgian Blue Cattle are so large at birth that they have to be delivered via c-section (Casas et al., 2004; Grobet et al., 1997). There are several possible explanations for why these mice were smaller at birth: 1) There may be a species differentiation in myostatin even though this pathway is conserved. However, it has been shown that myosin expression and fiber size in Myostatin-deficient cattle were found to be developmentally delayed compared to normal muscle development (Martyn et al., 2004). Also, evidence from a human case (Schuelke et al., 2004) showed that even though the infant was in the 75 percentile for weight and was hypermuscular, a normal birthing process was achieved. 2) Bellinge et al., 2005 suggest there are at least 6 mutations that cause inactivation of GDF-8 causing hypermuscularity or more specifically the double muscling phenotype of cattle, which suggests genetic heterogeneity between the murine and bovine models. Single nucleotide polymorphisms have also been implicated (Pan et al., 2007; Tantia et al., 2006). The resulting phenotypes of GDF-8 inactivation have also been described as heterogeneous (Bellinge et al., 2005; Potts et al., 2003; Grobet et al., 1997); 3) It is also unknown if differences in timing of expression may be due to regulation of genes, cytokines, or other epigenetic factors associated with the GDF-8 gene, or the TGF-β pathway in general, i.e. Follistatin regulation (Tellegren et al. 2004); or SMAD (Cassar-Malek et al., 2007; Forbes et al., 2006); or 4) Finally there may be a question of hyperplasia versus hypertrophy. Evidence indicates that GDF-8 inactivation phenotypes are hypermuscular mostly due to a hyperplasia response, with some hypertrophic action (Cassar-Malek et al., 2007; Pan et al., 2007; Martyn et al., 2004). It may be the case that the murine model displays primarily a resulting hypertrophic phenotype which may necessitate postnatal function for realization of muscularity. This point should be further investigated.
Ultimately myostatin-deficient mice had significantly greater body and muscle weight compared to wild-type controls. Myostatin deficiency produced brachycephalic craniofacial dimensions, including smaller cranial dimensions, foreshortened midface and a lengthened mandible which was shorter in vertical dimension. Only some of these differences occurred as a result of increased muscle force since the cranial and midfacial dimensions were already evident at birth. Without investigating these earlier stages the developmental influences on craniofacial morphology could not be ascertained. Hypermuscularity does have a significant influence over adult skeletal phenotype, but at least for craniofacial growth, early changes in cellular growth and differentiation have an equal impact on final form.
ACKNOWLEDGEMENTS
This investigation was supported by research grant AR 049717 from the National Institutes of Health, Bethesda, MD 20892. This work was reviewed and approved by the Medical College of Georgia Institutional Animal Care and Use Committee (IACUC). This paper was presented in part at the 85th General Session of the International Association of Dental Research (IADR/AADR), New Orleans, March, 2007.
Grant Support: Grant sponsor: National Institute of Health ; Grant number: AR 049717
Contributor Information
Lisa Vecchione, Pittsburgh Cleft-Craniofacial Research Center, Department of Surgery, Division of Plastic Surgery and Department of Orthodontics & Dentofacial Orthopedics, University of Pittsburgh, Pittsburgh, PA 15260.
Jeffrey Miller, Division of Oral and Maxillofacial Surgery, University of Connecticut, Farmington CT 06030
Craig Byron, Department of Biology, Mercer University, Macon GA 31207
Gregory M. Cooper, Department of Surgery, Division of Plastic Surgery, Department of Orthopedic Surgery, and Department of Oral Biology, University of Pittsburgh, Pittsburgh PA 15260
Timothy Barbano, Department of Anthropology, University of Pittsburgh, Pittsburgh PA 15260.
James Cray, Jr., Department of Anthropology, University of Pittsburgh, Pittsburgh PA 15260
Joseph E. Losee, Pittsburgh Cleft-Craniofacial Research Center, Department of Surgery, Division of Plastic Surgery, University of Pittsburgh, Pittsburgh, PA 15260
Mark W. Hamrick, Department of Cellular Biology & Anatomy, Medical College of Georgia, Augusta, GA, 30912
James J. Sciote, Department of Orthodontics, Temple University, Philadelphia, PA 19140
Mark P. Mooney, Department of Oral Biology, Departments of Anthropology, Department of Surgery, Division of Plastic Surgery, and Department of Orthodontics, University of Pittsburgh, Pittsburgh PA 15260
REFERENCES
- Amthor H, Macharia R, Navarrete R, Schuelke M, Brown SC, Otto A, Voit T, Muntoni F, Vrbova G, Partridge T, Zammit P, Bunger L, Patel K. Lack of myostatin results in excessive muscle growth but impaired force generation. PNAS. 2007;104:1835–1840. doi: 10.1073/pnas.0604893104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinge RHS, Liberles DA, Iaschi SPA, O’Brien PA, Tay GK. Myostatin and its implications on animal breeding: a review. Animal Genetics. 2005;36:1–6. doi: 10.1111/j.1365-2052.2004.01229.x. [DOI] [PubMed] [Google Scholar]
- Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, Montarras D, Rocancourt D, Relaix F. The formation of skeletal muscle: from somite to limb. J Anat. 2003;202:59–68. doi: 10.1046/j.1469-7580.2003.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byron CD, Borke J, Yu, Pashley D, Wingard CJ, Hamrick M. Effects of increased muscle mass on mouse sagittal suture morphology and mechanics. Anat Rec. 2004;279:676–684. doi: 10.1002/ar.a.20055. [DOI] [PubMed] [Google Scholar]
- Byron CD, Hamrick MW, Wingard CJ. Alterations of temporalis muscle contractile force and histological content from the myostatin and Mdx deficient mouse. Arch Oral Biol. 2006;51:396–405. doi: 10.1016/j.archoralbio.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Byron CD, Maness H, Yu JC, Hamrick MW. Enlargement of the temporalis muscle and alterations in the lateral cranial vault. Integr Compar Biol. 2008 doi: 10.1093/icb/icn020. in press. [DOI] [PubMed] [Google Scholar]
- Casas E, Bennet GL, Smith TPL, Cundiff LV. Association of myostatin on early calf mortality, growth, and carcass composition traits in crossbred cattle. J Anim Sci. 2004;82:2913–2918. doi: 10.2527/2004.82102913x. [DOI] [PubMed] [Google Scholar]
- Cassar-Malek I, Passelaigue F, Bernard C, Leger J, Hocquette JF. Target genes of myostatin loss-of-function in muscles of late bovine fetuses. BMC Genomics. 2007;8:63. doi: 10.1186/1471-2164-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis TA, Fiorotto ML, Nguyen HV, Reeds PJ. Protein turnover in skeletal muscle of suckling rats. Amer J Physiol. 1989;257:R1141–1146. doi: 10.1152/ajpregu.1989.257.5.R1141. [DOI] [PubMed] [Google Scholar]
- Dewulf N, Verschueren K, Lonnoy O, Moren A, Grimsby S, Vande SK, Miyazono K, Huylebroeck D, Ten Dijke P. Distinct spatial and temporal expression patterns of two type I receptors for bon morphogenic proteins during mouse embryogenesis. Endocrinology. 1995;136:2652–2663. doi: 10.1210/endo.136.6.7750489. [DOI] [PubMed] [Google Scholar]
- Enlow DH. Handbook of Facial Growth. W. B. Saunders Co.; Philadelphia: 1975. [Google Scholar]
- Forbes D, Jackman M, Bishop A, Thimas M, Kambadur R, Sharma M. Myostatin auto-regulates its expression by feedback loop through Smad7 dependent mechanism. J Cell Phys. 2006;206:264–272. doi: 10.1002/jcp.20477. [DOI] [PubMed] [Google Scholar]
- Girgenrath S, Song K, Whittemore L-A. Loss of myostation expression alters fiber-type distribution and expression of myosin heavy chain isoforms in slow- and fast-type skeletal muscle. Musc Nerv. 2005;31:34–40. doi: 10.1002/mus.20175. [DOI] [PubMed] [Google Scholar]
- Grobet L, Martin LJ, Poncelet D, Pirottin D, Brouwers B, Riquet J, Schoeberlein A, Dunner S, Meissier F, Massabanda J, Fries R, Hanset R, Georges M. A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat Genet. 1997;17:71–74. doi: 10.1038/ng0997-71. [DOI] [PubMed] [Google Scholar]
- Herring S, Mucci RJ. In vivo strain in cranial sutures: the zygomatic arch. J Morphol. 1991;207:225–239. doi: 10.1002/jmor.1052070302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton P. Rocker jaws. Amer J Phys Anthrop. 1977;47:365–370. doi: 10.1002/ajpa.1330470303. [DOI] [PubMed] [Google Scholar]
- Houghton P. Polynesian mandibles. J Anat. 1978;2:251–260. [PMC free article] [PubMed] [Google Scholar]
- Hylander WL. In vivo bone strain in the mandible of Galago crassicaudatus. Amer J Phys Anthrop. 1977;46:309–26. doi: 10.1002/ajpa.1330460212. [DOI] [PubMed] [Google Scholar]
- Jaslow CR. Mechanical properties of cranial sutures. J Biomech. 1990;23:313–321. doi: 10.1016/0021-9290(90)90059-c. [DOI] [PubMed] [Google Scholar]
- Kambadur R, Sharma M, Smith TP, Bass JJ. Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res. 1997;7:910–916. doi: 10.1101/gr.7.9.910. [DOI] [PubMed] [Google Scholar]
- Kean MR, Houghton P. Polynesian face and dentition: functional perspective. Amer J Phys Anthropol. 1990;82:361–369. doi: 10.1002/ajpa.1330820311. [DOI] [PubMed] [Google Scholar]
- Kettunen P, Nie X, Kvinnsland IH, Luukko K. Histological development and dynamic expression of Bmp2-6 mRNAs in the embryonic and postnatal mouse cranial base. Anat Rec. 2006;288:1250–1258. doi: 10.1002/ar.a.20402. [DOI] [PubMed] [Google Scholar]
- Kingsley DM. The TGF-β superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 1994;8:133–146. doi: 10.1101/gad.8.2.133. [DOI] [PubMed] [Google Scholar]
- Marshall DS, Snow CE. An evaluation of Polynesian craniology. Amer J Phys Anthropol. 1956;14:405–427. doi: 10.1002/ajpa.1330140316. [DOI] [PubMed] [Google Scholar]
- Martyn JK, Bass JJ, Oldham JM. Skeletal muscle development in normal and double-muscled cattle. Anat Rec. 2004;281:1363–1371. doi: 10.1002/ar.a.20140. [DOI] [PubMed] [Google Scholar]
- McPherron AC, Lawler AM, Lee S-J. Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- National Center for Biotechnology Information, National Institute of Health [Accessed April 23]. 2008.
- Nicholson EK, Stock SR, Hamrick MW, Ravosa MJ. Biomineralization and adaptive plasticity of the temporomandibular joint in myostatin knockout mice. Arch Oral Biol. 2006;51:37–49. doi: 10.1016/j.archoralbio.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Owen M. Marrow stromal stem cells. J Cell Sci. 1988;S10:63–76. doi: 10.1242/jcs.1988.supplement_10.5. [DOI] [PubMed] [Google Scholar]
- Pan J, Wang X, Song W, Chen J, Li C, Zhao Molecular cloning and expression pattern of myostatin gene in yellow catfish (Pelteobagrus fulvidraco) DNA Seq. 2007;18:279–287. doi: 10.1080/10425170701243492. [DOI] [PubMed] [Google Scholar]
- Philip B, Lu Z, Gao Y. Regulation of GDF-8 signaling by the p38 MAPK. Cell Signal. 2005;17:365–75. doi: 10.1016/j.cellsig.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Potts JK, Echternkamp SE, Smith TPL, Reecy JM. Characterization of gene expression in double-muscled and normal-muscled bovine embryos. Animal Genetics. 2003;34:438–444. doi: 10.1046/j.0268-9146.2003.01055.x. [DOI] [PubMed] [Google Scholar]
- Rebbapragada A, Benchabane H, Wrana JL, Celeste AJ, Attisano L. Myostatin signals through a transforming growth factor β-like signaling pathway to block adipogenesis. Molec and Cell Biol. 2003;23:7230–7242. doi: 10.1128/MCB.23.20.7230-7242.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salerno MS, Thomas M, Forbes D, Watson T, Kambadur R, Sharma M. Molecular analysis of fiber type-specific expression of murine myostatin promoter. Amer J Physiol Cell Physiol. 2004;287:C1031–C1040. doi: 10.1152/ajpcell.00492.2003. [DOI] [PubMed] [Google Scholar]
- Sassouni V. A classification of skeletal facial types. Amer J Orthod. 1969;55:109–123. doi: 10.1016/0002-9416(69)90122-5. [DOI] [PubMed] [Google Scholar]
- Schendel SA, Walker G, Kamisugi A. Hawaiian craniofacial morphometrics: average Mokapuan skell, artificial cranial deformation, and the “rocker’ mandible. Amer J Phys Anthropol. 1980;52:491–500. doi: 10.1002/ajpa.1330520406. [DOI] [PubMed] [Google Scholar]
- Schuelke M, Wagner KR, Sholtz LE, Hubner C, Riebel T, Komen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- Suryawan A, Frank JA, Nguyen HV, Davis TA. Expression of the TGF-beta family of ligands is developmentally regulated in skeletal muscle of neonatal rats. Ped Res. 2006;59:175–179. doi: 10.1203/01.pdr.0000196718.47935.6e. [DOI] [PubMed] [Google Scholar]
- Tantia MS, Vijh RK, Kumar STB, Mishra B, Reecy JM. Comparative analysis of GDF 8 (myostatin) in Bos indicus and Bos taurus. DNA Sequence. 2006;17:311–313. doi: 10.1080/10425170600807603. [DOI] [PubMed] [Google Scholar]
- Tellgren A, Berglund AC, Savolainen P, Janis CM, Liberles DA. Myostatin rapid sequence evolution in ruminants predates domestication. Mol Phyl and Evol. 2004;33:782–790. doi: 10.1016/j.ympev.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Tsuchida K, Nakatani M, Uezumi A, Murakami T, Cui X. Signal transduction pathway through activin receptors as a therapeutic target of musculoskeletal diseases and cancer. Endocr J. 2008;55:11–21. doi: 10.1507/endocrj.kr-110. [DOI] [PubMed] [Google Scholar]
- Vecchione L, Byron C, Cooper GM, Barbano T, Hamrick MW, Sciote JJ, Mooney MP. Craniofacial morphology in myostatin-deficient mice. J Dent Res. 2007;86:1068–1072. doi: 10.1177/154405910708601109. [DOI] [PubMed] [Google Scholar]
- Verschueren K, Dewulf N, Goumans MJ, Lonnoy O, Feijen A, Grimsby S. Expression of type I and type IB receptors for activin in midgestation mouse embryos suggests distinct functions in organogenesis. Mech Dev. 1995;52:109–213. doi: 10.1016/0925-4773(95)00395-h. [DOI] [PubMed] [Google Scholar]
- Yi SE, Daluiski A, Pederson r, Rosen V, Lyons KM. The type I BMP receptor BMPRIB is required for chondrogenesis in the mouse limb. Development. 2000;127:621–630. doi: 10.1242/dev.127.3.621. [DOI] [PubMed] [Google Scholar]
- Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, McPherron AC, Wolfman NM, Lee S-J. Induction of cachexia in mice by systemically administered myostatin. Sci. 2002;296:1486–1488. doi: 10.1126/science.1069525. [DOI] [PubMed] [Google Scholar]