

Abstract
The extracellular signal-regulated kinases (ERKs) are members of the mitogen-activated protein kinase (MAPK) superfamily of enzymes and have recently garnered considerable attention in the field of learning and memory. ERK activation has been shown to be required for the induction of long-term potentiation (LTP) in the rat hippocampus and for the formation of associative and spatial memories in both the rat and the mouse. However, the individual roles for the two isoforms of ERK have yet to be deciphered. To investigate the specific contribution of the ERK1 (p44) isoform of MAPK to mammalian learning, we performed a general behavioral and physiological characterization of mice lacking the ERK1 gene. The ERK1-null animals demonstrated significantly higher levels of activity in the open field test. However, we observed no other discernible deficits in the ERK1 knockout mice in our behavioral testing. Specifically, no differences were observed in the acquisition or retention (24 h and 2 wk after training) of either contextual or cue fear conditioning between the ERK1−/− and their wild-type littermate controls. In addition, no learning phenotype was observed in the passive avoidance test. When hippocampal slices were analyzed, we found no deficits in baseline synaptic transmission or in tetanus-induced LTP in hippocampal area CA1. We found no apparent compensatory changes in the expression of ERK2 (p42 MAPK). We conclude that hippocampus- and amygdala-dependent emotional learning does not depend critically on the activity of ERK1.
The cellular processes underlying learning and the formation of memory involve the regulation of synaptic strength as well as the establishment of new synaptic connections. The mechanisms by which neuronal activity is translated into changes in synaptic organization have received considerable attention. The recognition that intracellular signaling pathways involving protein kinases are essential intermediates in the induction of long-term changes in synaptic strength represents a significant advance in our understanding of the molecular mechanisms of learning and memory. The MAP kinases have been shown to be critically involved in the formation of long-term memory (Kornhauser and Greenberg 1997). Originally discovered as regulators of cell division and differentiation, mitogen-activated protein kinases (MAPKs) are abundantly expressed in neurons in the mature central nervous system. Recently, their role in these nondividing, terminally differentiated neurons has come under investigation.
A number of previous studies have demonstrated a major role for MAPK in mammalian associative learning. Our laboratory has previously shown activation of extracellular signal-regulated kinase (ERK) isoforms of MAPK in the rat hippocampus following a cue and contextual fear-conditioning paradigm. In addition, intraperitoneal injection of an inhibitor of MAPK kinase (MEK), the upstream activator of MAPK, blocked fear conditioning in these animals (Atkins et al. 1998). Preventing MAPK activation with a MEK inhibitor also impaired fear learning in the mouse (Selcher et al. 1999). In a further refinement, Schafe et al. (1999) demonstrated selective blockade of long-term, but not short-term, fear conditioning following intraventricular infusion of a MEK inhibitor. Together with reports utilizing other associative learning paradigms including conditioned taste aversion (Berman et al. 1998) and step-down inhibitory avoidance (Walz et al. 1999), these fear-conditioning studies solidly implicate MAPK in the formation of associative memories.
Regulation of the MAPK family of enzymes involves a core cascade of three protein kinases. Signal transduction through this pathway begins with the activation of ras and the physical interaction of this small G protein with the serine/threonine kinases Raf-1 or B-Raf, resulting in their activation. The Raf kinases then phosphorylate and activate MEK (or MAPKK). MEK1 and MEK2 are dedicated dual-specificity kinases that activate both ERK1 (p44 MAPK) and ERK2 (p42 MAPK) by phosphorylating these enzymes on threonine and tyrosine residues (e.g., Thr 183 and Tyr 185 for ERK2), provoking their enzymatic activation. On activation, the ERKs are translocated to the nucleus, where they phosphorylate transcription factors, leading to changes in gene expression (English et al. 1999).
Although a role for the ERKs in a number of behavioral paradigms has been established, it is unclear which of the two ERK isoforms contributes to learning. The inhibitors of the MAPK cascade used in previous learning studies (PD098059, U0126, and SL327) are quite selective in their inhibition of MEK but, unfortunately, do not differentiate between the two ERK isoforms. Previous studies using knockout and transgenic mice have elucidated the specific role of individual isoforms in other protein kinase families known to be critical for synaptic plasticity and learning (Silva et al. 1992; Abeliovich et al. 1993; Abel et al. 1997; Weeber et al. 2000). Therefore, to investigate the specific function of the ERK isoforms in learning and memory, we employed mice in which one ERK isoform was genetically inactivated. Specifically, we characterized mice lacking the ERK1 isoform of MAPK in an attempt to determine the behavioral contribution of this protein kinase.
RESULTS
Normal Development in ERK1-Null Animals
ERK1-null animals were generated by homologous recombination in embryonic stem cells (T. Nekrasova and G.E. Landreth, in prep.). Homozygous ERK1−/− mice were viable and fertile and developed normally. Histological analysis of the brains of the ERK1−/− animals did not reveal any gross differences from the wild-type littermates (Fig. 1A, and data not shown).
Figure 1.
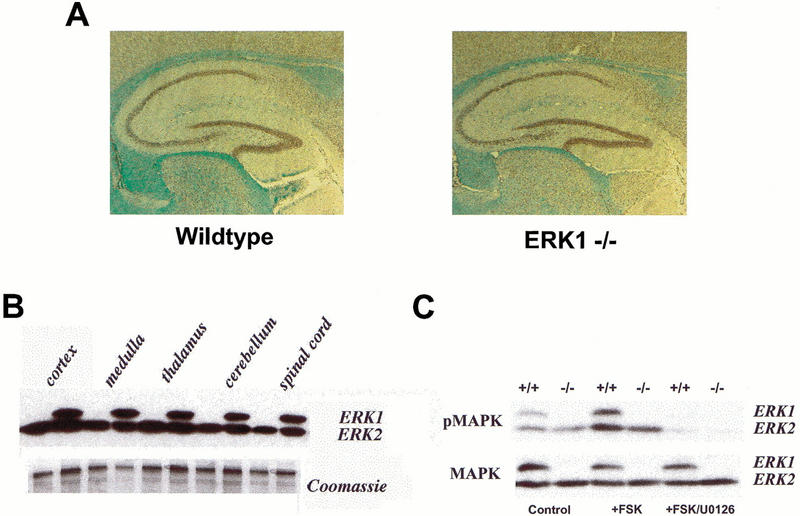
ERK1-null animals are developmentally normal. (A) Sagittal sections through the brains of adult wild-type and ERK1−/− mice at 25x magnification. (B) Representative Western blots showing ERK protein expression in tissue from various regions of the CNS from ERK1 knockout and wild-type mice. (C) Western blots showing phospho-ERK (top) and total ERK (bottom) protein expression in 400–μm-thick hippocampal slices prepared from ERK1-null mice and littermate wildtypes. The first two lanes show tissue from control slices. Hippocampal tissue in the middle two lanes received a 10 min exposure to 50 μM forskolin (+FSK), while tissue in the last two lanes were exposed to 50 μM forskolin following a 1 h pre-incubation with 20 μM U0126 (+FSK/U0126).
ERK expression was evaluated in both knockout and wild-type mice by Western blot analyses of ERK protein expression. Similar to wild-type controls, tissue from various areas of the CNS of ERK1-null mice revealed abundant ERK2 labeling. As expected, the knockout animals displayed no immunoreactivity to ERK1 (Fig. 1B,C). Phospho-ERK staining was also assessed in hippocampal slices prepared from ERK1 knockout and wild-type mice. Both basal and forskolin-stimulated activation of ERK2 appeared normal in the ERK1 knockout mice (Fig. 1C). In addition, endogenous activation of receptor-coupled ERK2 is also normal in the mutant mice. ERK2 activation was found to be similar in ERK1 null mice as in wild types following potassium depolarization, which elicits ERK activation via endogenous neurotransmitters and calcium influx (data not shown). For ERK1 knockout mice as for controls, application of the MEK inhibitor U0126 significantly attenuated ERK2 phosphorylation. These results demonstrate that ERK1 knockout mice show no perceptible up-regulation of ERK2 expression or activity.
Hyperactivity in ERK1−/− Mice
We began our behavioral characterization by monitoring ERK1−/− and littermates in the open field test. The open field test evaluated locomotor activity by recording movements in both the horizontal and vertical planes. This test also measured an index of anxiety-related response by calculating the ratio between the distance traveled in the center of the open field and the total distance traveled. For example, an increase in anxiety would correspond with a decrease in this ratio, as anxious animals spend more of their time in the perceived safety of the corners and along the edges of the open field arena.
The ERK1 knockout mice had a tendency to be more active in the open field than their wild-type littermates. These knockout mice showed significantly higher levels of horizontal activity measured both by the number of beam breaks (Fig. 2A) and by the total distance traveled (Fig. 2B) than did control mice [horizontal (beam breaks), F(1,24) = 4.54, P = 0.043; horizontal (total distance), F(1,14) = 6.10, P = 0.021]. For both of these measures, there was also a significant change in activity across time [horizontal (beam breaks), F(14,336) = 39.64, P < 0.0001; horizontal (total distance), F(14,336) = 23.12, P < 0.0001] but no significant interaction between genotype and time. There was no significant effect of genotype in vertical activity in the open field (Fig. 2C), although there was a significant change in activity across time [F(14,336) = 2.22, P = 0.007; no significant interaction effect]. The center distance to total distance ratio for ERK1−/− and wild-type mice is shown in Figure 2D. The two sets of mice showed no difference in this measure, suggesting that the lack of ERK1 has no effect on baseline anxiety levels. We conclude from these data that ERK1 knockout mice are mildly hyperactive.
Figure 2.

ERK1 knockout mice show increased horizontal activity in the open field. (A,B) Horizontal activity measured by the number of beam breaks and total distance respectively for ERK1−/− (open box; n = 14) versus wild-type (filled circle; n = 12) mice. (C) Vertical activity was not different between ERK1−/− and wild-type mice. (D) No difference in center distance-to-total distance ratio for ERK1−/− and wild-type controls. Data are represented as the mean (± s.e.m.).
ERK1−/− Mice Exhibit Normal Motor and Sensory Performance
The motor abilities of both ERK1−/− and wild-type littermates were analyzed using the rotarod test. The amount of time an animal can remain balanced on a rotating rod was used as an index of the animal's motor coordination. In addition, increases in proficiency at this task are indicative of normal motor learning.
ERK1 knockout mice and their wild-type controls performed comparably in all trials on the accelerating rotarod (Fig. 3). Two-way ANOVA revealed no significant effect of genotype. Not surprisingly, there was a significant effect of trial number, indicating that the amount of time spent on the rotarod increased with training [F(7,112) = 23.19, P < 0.0001]. There was no significant interaction between genotype and trial number, suggesting that both sets of mice showed similar improvements over time. Therefore, these mutant mice were not discernibly impaired in terms of motor coordination or motor learning.
Figure 3.

Mice deficient in ERK1 show no impairment on the rotarod task. There was no significant difference between the amount of time ERK1−/− (open box; n = 10) and wild-type (filled circle; n = 8) mice spent balanced on the accelerating rotarod. Performance in both sets of mice improved significantly over the course of eight trials.
Acoustic startle and prepulse inhibition tests were conducted. These tests are sensory tasks designed to measure an animal's responsiveness to auditory stimuli. Normal mice exhibit a startle response to a loud noise. The presentation of a modest prepulse of sound immediately before this loud noise produces an attenuation of the startle response, a phenomenon known as prepulse inhibition. Prepulse inhibition is believed to be a good assay of sensorimotor gating and can be used as an indicator of hearing.
There was no difference in the amount of acoustic startle produced by a 120-dB stimulus between mutant mice and littermate controls (Fig. 4A). This acoustic startle was inhibited by delivery of prepulses of varying intensities with no difference in the level of inhibition between the two genotypes (Fig. 4B). As expected, levels of prepulse inhibition were enhanced as the intensity of the prepulse increased [F(4,96) = 44.23, P < 0.0001]. There was also no significant interaction effect between genotype and prepulse intensity level. These data indicate that ERK1-null mice are not impaired in terms of basic auditory function.
Figure 4.
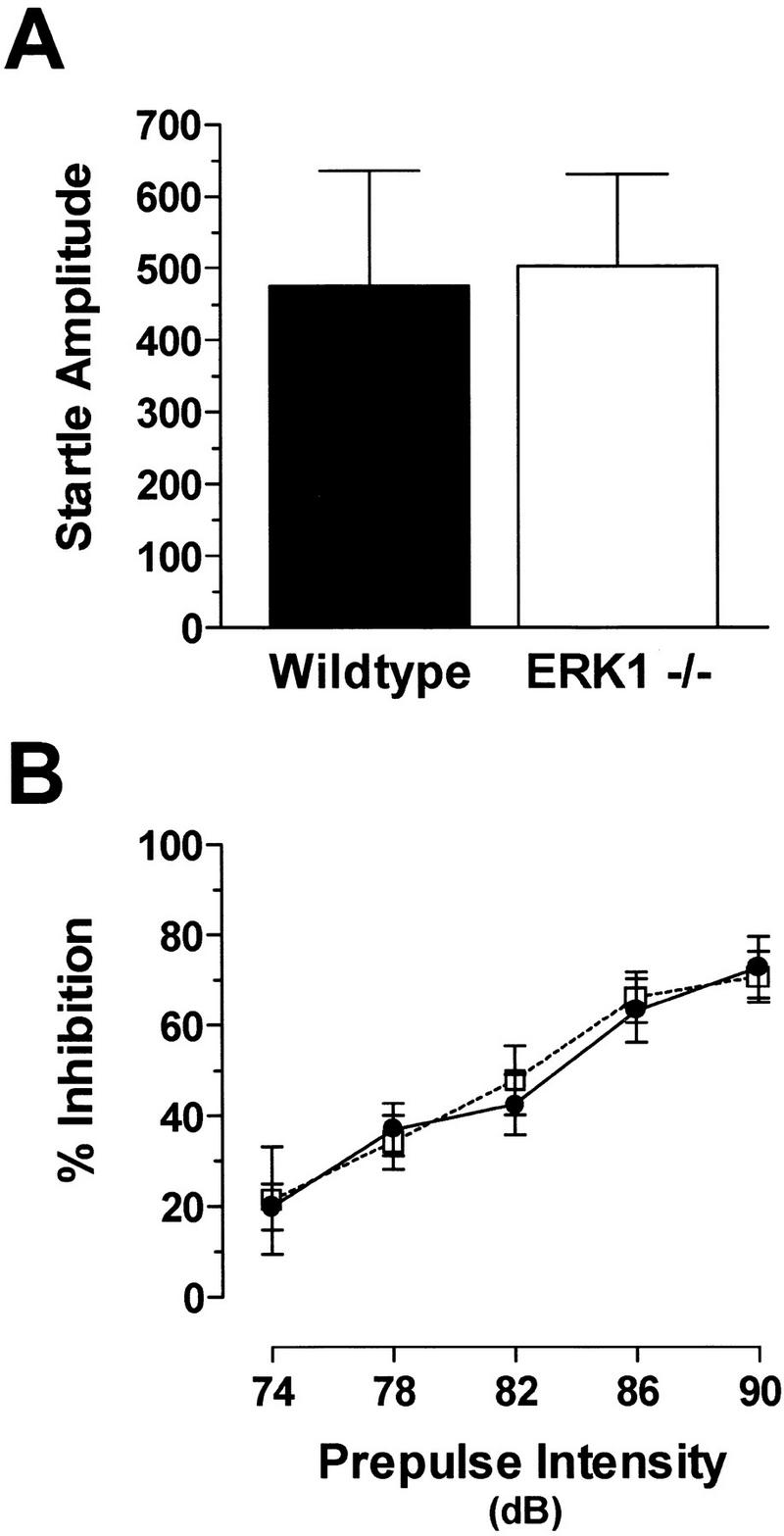
No difference in startle amplitude or prepulse inhibition between ERK1−/− and wild-type mice. (A) Acoustic startle amplitude to the 120-dB stimulus for wild-type and ERK1 knockout mice. (B) Inhibition of the startle response by prepulses of varying intensities for ERK1−/− (open box; n = 14) and wild-type (filled circle; n = 12) mice.
Emotional Learning is Unimpaired in ERK1−/− Mice
Previous studies have demonstrated a pronounced dependence of fear conditioning on normal ERK activation in the central nervous system. To assay emotional learning in our ERK1-deficient mice, we used a standard fear-conditioning paradigm. In fear conditioning, animals learn to associate neutral stimuli with an aversive one. The mice were placed in a novel environment or context and were exposed to two pairings of a tone and mild footshock. Fear learning was assessed 24 h later by measuring freezing behavior in response to re-presentation of either the context (the training cage) or the auditory cue within a completely different context. To test long-term retention of these associative memories, contextual and cue tests were also conducted 2 wk after training.
ERK1-deficient and wild-type mice displayed similar levels of conditioned fear (Fig. 5). There was no difference in the levels of freezing during the training phase, suggesting that acquisition of conditioned fear is identical in the two sets of mice (Fig. 5A). Compared to controls, freezing behavior in response to re-presentation of the context (Fig. 5B) and to the delivery of the cue within a new context (Fig. 5C) was not significantly different for ERK1 knockout mice. Longer-term fear retention in the two groups also appeared similar; fear associated with both the context (Fig. 5D) and the cue (Fig. 5E) were intact when tested 2 wk after training. Once again, there were no significant differences between mutant and wild-type mice detected in these retention tests.
Figure 5.

ERK1 is not required for normal fear conditioning. (A) Freezing responses during the training phase are shown. Within a novel environment, an acoustic stimulus (solid bar) was paired twice with a footshock (arrow) during training. Baseline behavior (before presentation of the tone) and shock response (after the footshock) were similar for both ERK1−/− ( open box; n = 10) and wild-type (filled circle; n = 8) groups. (B) Compared to controls, ERK1-deficient mice demonstrated normal freezing to the context 24 h after receiving two pairings of tone and shock. (C) Freezing in response to re-presentation of the auditory CS (solid bar) in a different context 24 h after training also revealed unimpaired cue learning in ERK1 knockout mice. (D) Contextual fear conditioning was still intact when tested 14 d after training in both ERK1−/− and wild-type mice. (E) Re-presentation of the auditory CS (solid bar) 14 d after fear conditioning yielded freezing levels in ERK1−/− mice similar to wild-type controls. These results suggest unimpaired long-term retention of fear learning.
We assessed another emotional learning task in the ERK1 knockout mice: passive avoidance. In this test, animals learn to suppress their natural tendency to seek out dark areas over well-lit areas after their entry into a dark compartment is paired with a mild footshock. There was no significant difference between ERK1 knockout mice and their littermate controls in step-through latency, the measure of passive avoidance (Fig. 6). In both sets of mice, time obviously had a significant effect, as subjects showed a substantial increase in step-through latencies following training on day 1 [F(2,45) = 17.36, P < 0.0001]. Both mutant mice and controls successfully suppressed their natural preference for dark areas by avoiding the compartment within which they were shocked.
Figure 6.

ERK1-deficient mice are unimpaired in passive avoidance learning. Time to enter the dark half of the chamber, or step-through latency, was similar on both testing days for ERK1−/− mice ( open box; n = 10) and littermate wildtype controls (filled circle; n = 7).
Normal Hippocampal Physiology in ERK1−/−
Hippocampal synaptic plasticity is critically dependent on the activity of the ERKs (English and Sweatt 1996, 1997). Induction of LTP leads to activation of both ERK1 and ERK2; however, ERK2 is the predominant isoform activated following synaptic activation (e.g., see Fig. 1; English and Sweatt 1996, 1997). Loss of the ERK1 isoform of MAPK had no deleterious effect on baseline synaptic transmission at Schaffer collateral synapses. Input–output functions generated by stimulation of area CA3 and field recordings of pyramidal neurons in area CA1 were similar for mutant and wild-type hippocampal slices (Fig. 7A). Paired-pulse facilitation (PPF) was also assessed in these slices. This is a form of short-term plasticity believed to result from residual calcium in the presynaptic cell that enhances neurotransmitter release. PPF in ERK1 knockout mice was virtually identical to wild-type controls at all interstimulus intervals tested (Fig. 7B). These results suggest that ERK1 is not necessary for normal hippocampal synaptic transmission and is not an integral component of the machinery underlying short-term plasticity.
Figure 7.

Normal synaptic transmission and synaptic plasticity in ERK1 knockouts. (A) Baseline synaptic transmission shown with input-output curves for ERK1−/− (open box; n = 9 slices) and littermate wildtype controls (filled circle; n = 10). (B) Paired-pulse facilitation in hippocampal slices from ERK1-deficient and wild-type mice. (C) LTP induced with a pair of 100-Hz tetani in area CA1 is similar for ERK1−/− and wild-type hippocampal slices. Inset, representative traces from control and mutant slices before (gray) and after (black) tetanic stimulation. Scale bars are 0.2 mV by 4 msec. The arrow indicates the delivery of two trains of 100-Hz, 1-sec stimulation.
On the basis of the necessity for ERK activation in the induction of LTP in the rat, we were especially intrigued to analyze hippocampal LTP in mice specifically lacking ERK1. In these experiments, LTP was induced with two 1-sec, 100-Hz tetani separated by 20 sec. We observed no significant differences between ERK1 knockout mice and littermate controls in either posttetanic potentiation (PTP) or LTP (Fig. 7C). This lack of phenotype does not appear to be caused by any compensatory change in ERK2 protein levels or enhanced ERK2 activation in the ERK1 knockout mice (Fig. 1). Overall, these data indicate that area CA1 hippocampal physiology, in terms of baseline synaptic transmission, PPF, and tetanus-induced long-term plasticity, is not dependent on the activity of the ERK1 isoform of MAPK.
DISCUSSION
The MAPK Cascade in LTP and Learning
An extraordinary number of recent studies have implicated the MAPK cascade in synaptic plasticity and learning in a wide variety of systems. Activation of the ERK isoforms of MAPK have been demonstrated to be necessary for the induction of NMDA receptor-dependent LTP in area CA1 of the rat hippocampus (English and Sweatt 1996, 1997; Atkins et al. 1998; Impey et al. 1998a; Winder et al. 1999; Wu et al. 1999; but also see Liu et al. 1999), NMDA receptor-independent LTP in area CA1 (Kanterewicz et al. 2000), LTP in the dentate gyrus (Coogan et al. 1999), LTP in vivo (Davis et al. 2000; Rosenblum et al. 2000), and LTP of the amygdalar inputs into the insular cortex (Jones et al. 1999). ERK activation has also been shown to be necessary in long-term facilitation of the sensory-motor synapse in Aplysia (Martin et al. 1997) and in an in vitro Pavlovian conditioning paradigm in Hermissenda (Crow et al. 1998). In addition, long-term depression (LTD) in the rat hippocampus is associated with long-lasting decreases in ERK immunoreactivity (Norman et al. 2000).
Behavioral studies have also demonstrated a significant role for MAPK in learning and memory. Our laboratory previously reported a necessity for ERK activation in fear conditioning in both the rat (Atkins et al. 1998) and the mouse (Selcher et al. 1999). Contextual and cued fear learning in both of these species was blocked by application of the MEK inhibitor SL327. ERK activation was shown to be necessary for another form of associative learning, conditioned taste aversion. Microinjection of a MEK inhibitor into the insular cortex shortly before pairing of a novel taste to a malaise-inducing agent attenuated long-term taste-aversion memory (Berman et al. 1998). MEK inhibitors also impair spatial learning in the Morris water maze in both of these rodent species, pointing to a necessary role for ERK in this form of learning (Blum et al. 1999; Selcher et al. 1999).
Taken together, these findings build a convincing argument for a requirement of the ERK isoforms of MAPK in the molecular events that underlie information storage at both the synaptic and behavioral levels. The goal of this study was to address the specific contribution of one of these ERK isoforms. To this end, ERK1 knockout mice were tested in various behavioral and physiological paradigms to elucidate the role of this particular protein in sensory, motor, and learning systems.
One important caveat to this study, as with other studies involving knockout animals, involves developmental effects of the mutation. The fact that the gene and its product were missing throughout development means that the knockout affects every ERK1-dependent function during development. Because the MAPK pathway is so important developmentally, the potential for these types of effects was especially worrisome. In addition, we considered whether the absence of ERK1 during development allowed compensatory mechanisms to emerge that could potentially mask the normal role of ERK1. The ERK1-null mice, however, appeared to show normal anatomical and functional development. In fact, the only apparent derangement in the knockout mice in terms of sensory or motor functioning was a modest increase in activity measured in the open field test. Although hyperactivity can often impact other behavioral tests (e.g., fear conditioning), no other phenotype was uncovered in testing, suggesting that the enhanced activity in these mice was not a confounding factor for subsequent tests. We also saw no obvious compensatory changes in the basal levels of ERK2 or in stimulated levels of phosphorylated ERK2 in the hippocampi of knockout mice. Therefore, behavioral and physiological characterization of these animals should provide an accurate assessment of the role of ERK1 in mouse learning.
No Role for ERK1 in LTP and Learning
The results of this study clearly demonstrate that the ERK1 isoform of MAPK is not necessary for associative learning in the mouse. No differences were observed in the acquisition or retention (24 h and 2 wk after training) of contextual or cued fear conditioning between the two groups of mice; there were also no differences in their baseline freezing rates or in their shock responses. In addition, no learning phenotype was observed in the passive avoidance task. In this test, animals were shocked on entering the dark half of a two-chambered shuttle box. When tested 24 h later, there was no significant difference between the ERK1 knockout mice and wild-type controls in their latencies to enter the darkened chamber.
The lack of a behavioral phenotype in contextual fear conditioning and passive avoidance, two hippocampus-dependent tasks (Stubley-Weatherly et al. 1996; Logue et al. 1997; Impey et al. 1998b), taken together with the absence of an overt physiological phenotype, suggest that hippocampal function is essentially intact in the ERK1 knockout mice. We hypothesize that other hippocampus-dependent behaviors, such as spatial learning, should be undisturbed in these mutant mice. Pilot experiments testing the ERK1 knockout mice in the hidden-platform version of the Morris water maze demonstrated that the ERK1-deficient mice exhibited no apparent differences in escape latencies during training trials (data not shown). In addition, probe trials demonstrated that both sets of mice used a selective search strategy by spending significantly more time searching in the quadrant of the pool in which the platform was located during training. On the basis of these findings, it seems rather unlikely that ERK1 is playing a critical role in mammalian spatial learning.
A Selective Role for ERK2?
What do these findings reveal about the role of ERK2 in synaptic plasticity and learning? One explanation for the lack of a functional effect would suggest that ERK1 and ERK2 play redundant roles. In this scenario, ERK2 can compensate for the loss of ERK1 in these knockout mice, thereby preventing the detection of a learning impairment. A second explanation would allow for the possibility that ERK2 plays a predominant role in the plastic changes accompanying learning, and its selective activation is necessary for learning to occur.
Consistent with the first idea is the fact that in most in vitro systems, the two ERK isoforms are coordinately regulated. Both are activated solely by MEK1 and MEK2, share very similar substrate profiles, and display a high degree of sequence homology. The absence of an overt phenotype and the finding that the animals are behaviorally similar to wild-type littermates argue that the ERK isoforms have significant functional redundancy.
Nevertheless, we favor the second hypothesis, as a number of recent findings suggest that ERK2 may be more selectively involved in mammalian learning. Relative to ERK1, for example, the ERK2 isoform of MAPK shows higher basal levels of activation in the hippocampus as assessed using phospho-selective antibodies. Hippocampal ERK2 also displays a high degree of responsiveness to a variety of signal transduction pathways critical to synaptic plasticity and learning (Roberson et al. 1999; Schmid et al. 1999; Norman et al. 2000). More interesting, perhaps, is that our laboratory has demonstrated previously selective activation of ERK2 following induction of a physiological model for learning. In these experiments, phospho-ERK2 but not phospho-ERK1 levels were significantly increased in area CA1 of the rat hippocampus 1 h after delivery of LTP-inducing tetanic stimulation (English and Sweatt 1996). The preferential activation of the ERK2 isoform of MAPK in the same region of the rat hippocampus was also observed in experimental animals 1 h after exposure to contextual fear conditioning (Atkins et al. 1998). Hopefully, future investigations using ERK2 deficient mice will allow the direct testing of these two alternative possibilities.
MATERIALS AND METHODS
Subjects
ERK1 knockout and littermate wild-type mice were generated on a mixed 129/SvImJ × CD1 genetic background. The genetic background of the subjects used in these experiments was 98% 129/SvImJ following multiple (roughly six or seven) backcrossings. ERK1-null mice and littermate wild-types were shipped from Case Western Reserve School of Medicine and were held at Baylor College of Medicine in quarantine for ∼4 wk. Behavioral testing was begun 1 wk following release from quarantine. To characterize the phenotype of these ERK1 null mice, subjects were put through a battery of behavioral tasks adapted from Paylor et al. (1998). Animals were tested in the open field test, rotarod test, acoustic startle, prepulse inhibition, cued and contextual fear conditioning, and passive avoidance learning. Animals were housed on a 12-h light/dark schedule. All experiments were performed in accordance with the Baylor College of Medicine Institutional Animal Care and Use Committee and with national regulations and policies.
Western Blot Analysis
For Western Blot samples, mouse brains were immediately removed and perfused with ice-cold saline (125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 25 mM D-glucose, 2 mM CaCl2, 1 mM MgCl2, saturated with 95% O2/5% CO2 at pH 7.4). Regions of the brain were dissected, then homogenized in 2 mL of buffer (20 mM Tris-HCl pH 7.5, 1 mM EGTA, 1 mM EDTA, 25 μg/mL aprotonin, 25 μg/mL leupeptin, 1 mM Na4P2O7, 500 μM phenylmethylsulfonyl fluoride, 4 mM nitrophenylphosphate, 1 mM sodium orthovanadate). Laemmli sample buffer was immediately added to the homogenate, and the samples were boiled at 95°C for 10 min. Samples were electrophoresed on a 10% SDS-polyacrylamide gel, blotted electrophoretically to Immobilon-P or PVDF, and washed in TTBS buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.05% Tween-20). Membranes were blocked in TTBS with 3% bovine serum albumin (BSA). The following primary antibodies were used: activated MAPK (1:1000; New England Biolabs) and panERK (1:3000; UBI). All blots were incubated with a secondary antibody conjugated to horseradish peroxidase (1:30,000) and developed using the enhanced chemiluminescence method (Amersham). To assess changes in the activation of MAPK, phosphorylation levels were normalized relative to the total MAPK level. Protein concentration was determined using the BCA assay.
Histological Analysis
ERK1−/− mice and control littermates were anesthetized with 0.04 cc/g avertin and perfused with 4% paraformaldehyde in 0.1M phosphate buffer for 10 min. The dissected tissue was fixed in 4% paraformaldehyde for an additional hour at room temperature. Samples were rinsed with PBS and infiltrated with 30% sucrose overnight at 4°C. The tissues were embedded in paraplast (Oxford) and 10–μm sections were cut on a Leitz 1512 rotary microtome. The tissues were stained with hematoxylin and eosin and counterstained with luxol Blue.
Open Field Test
Each subject was placed in the center of a clear arena (40 × 40 × 30 cm) in standard room-lighting conditions. The open field was divided into 256 equally sized squares by 16 photoreceptor beams on each side of the arena. Locomotor activity was quantitated using a Digiscan optical animal activity system (RXYZCM [16], Accuscan Electronics). Activity measures represent the number of photobeam interruptions in both the horizontal and vertical planes collected in 2-min intervals over a 30-min testing period. Additional activity data include the total distance traveled in the horizontal plane (in cm) and the ratio of the distance traveled in the 16 most centrally located squares compared to the total distance traveled (center distance/total distance ratio).
Rotarod Test
Two days after open field testing, motor coordination, balance, and motor learning were examined with the accelerating rotarod task. In this test, the animal was required to maintain its balance upon placement on a rotating bar. Over the course of each 5-min trial, the speed of the rotarod accelerated from 4 to 40 rpm. The amount of time before the subject fell from the rod was measured. Mice underwent four rotarod trials per day for two consecutive days with an intertrial interval of 1 h.
Acoustic Startle and Prepulse Inhibition
Three days later, acoustic startle and prepulse inhibition of startle were assayed as described previously (Paylor and Crawley 1997). Briefly, mice were placed in a Plexiglas cylinder within a testing chamber (70-dB background noise level) and left undisturbed for 5 min. Acoustic startle was then measured by the SR-Lab System (San Diego Instruments) in response to a 40-msec, 120-dB soundburst. Prepulse inhibition, a phenomenon whereby the acoustic startle response is reduced upon presentation of a sound (or prepulse) just prior to the startle stimulus, was also assessed in the two sets of mice. Test sessions consisted of seven different types of trials. In addition to the single acoustic startle stimulus trials, there were also five different prepulse-plus-acoustic-startle stimulus trials. Prepulse sounds of 74, 78, 82, 86, and 90 dB were presented 100 msec before the startle stimulus and were 20 msec in duration. We also employed a trial in which no stimulus was presented, to gauge baseline movement. Testing consisted of six blocks of seven trials with an intertrial interval averaging 15 sec. Startle responses were recorded for 65 msec starting with the onset of the startle stimulus. The peak startle amplitude was used.
Fear Conditioning
One week after prepulse inhibition testing, conditioned fear was assessed as described in Selcher et al. (1999), except that a 2x-pairing protocol was employed. For cue and contextual fear conditioning, animals were placed in the fear conditioning apparatus for 3 min, then a 30-sec acoustic conditioned stimulus (CS; white noise, 80 dB) was delivered. During the last second of the CS, a 1-sec shock unconditioned stimulus (US; 0.5 mA) was applied to the grid floor. CS-US pairings were given twice with a 90-sec interval between the two pairings. To assess contextual learning, the animals were returned to the training context 24 h post-training, and freezing behavior was scored for 5 min. To assess cue learning, the animals were placed in a different context (novel odor, lighting, cage floor, and visual cues) following contextual testing. Baseline behavior was measured for 3 min in the novel context, and then the tone was presented for 3 min. Fourteen days after training, fear retention was assessed. Two-week retention testing was identical to the contextual and cue tests conducted 24 h after training. Freezing behavior was assessed using a time sampling procedure whereby the animal was observed for approximately 1 sec every 5 sec. The experimenter was blind to genotype.
Passive Avoidance
Emotional learning was also tested in a step-through passive avoidance task. Animals were placed in a conditioning chamber separated into two compartments, one illuminated and one dark, by a partition containing a trap door. On the training day, subjects were placed in the illuminated section and allowed to move freely in the chamber. The time necessary for the mouse to move into the dark compartment, or step-through latency, was measured for each animal. When the mouse had completely entered the dark half of the chamber, the trap door was shut and a mild footshock (1 sec, 0.5 mA) was delivered. After 10 sec in the dark section, the animal was removed and returned to its home cage. Testing was conducted 24 h later and consisted of placing the animal back into the lit compartment. Step-through latencies were once again measured. Mice displaying normal passive avoidance learning avoided the dark section of the chamber and thus had much higher latencies following training.
Electrophysiological Characterization
Hippocampal slice preparation and electrophysiology were performed as described (English and Sweatt 1996). Transverse hippocampal slices (400 μM) from ERK1−/− and wild-type littermate control mice (4–8 wk old) were prepared with a Vibratome and perfused (1 mL/min) with a standard saline solution (see saline recipe above) and allowed to equilibrate for 60–90 min in a Fine Science Tools interface chamber at 30°C. For extracellular field recordings, glass recording electrodes (1–3 MΩ) filled with ACSF were placed in the stratum radiatum of area CA1, while a bipolar platinum stimulating electrode was also placed in stratum radiatum of area CA3 for stimulation of Schaffer collateral afferents. Test stimulus intensity was adjusted to elicit a field excitatory postsynaptic potential (fEPSP) that was ∼30–40% of the maximum response. Responses (obtained at 0.05 Hz) were monitored for at least 20 min to ensure a stable baseline. The high-frequency stimulation paradigm employed in these studies consisted of two trains of 1-sec, 100-Hz stimulation at the test stimulus intensity with an intertrain interval of 20 sec.
Acknowledgments
We thank M.-A. Pendergast for help with histology. This work was supported by the National Institutes of Health (grant no. MH 57014 to J.D.S., and grant no. DA 14127 to J.C.S.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jsweatt@bcm.tmc.edu; FAX (713) 798-3946.
Article and publication are at www.learnmem.org/cgi/doi/10.1101/lm.37001.
REFERENCES
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- Abeliovich A, Paylor R, Chen C, Kim JJ, Wehner JM, Tonegawa S. PKCg mutant mice exhibit mild deficits in spatial and contextual learning. Cell. 1993;75:1263–1271. doi: 10.1016/0092-8674(93)90614-v. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Berman DE, Hazvi S, Rosenblum K, Seger R, Dudai Y. Specific and differential activation of mitogen-activated protein kinase cascades by unfamiliar taste in the insular cortex of the behaving rat. J Neurosci. 1998;18:10037–10044. doi: 10.1523/JNEUROSCI.18-23-10037.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum S, Moore AN, Adams F, Dash PK. A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J Neurosci. 1999;19:3535–3544. doi: 10.1523/JNEUROSCI.19-09-03535.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coogan AN, O'Leary DM, O'Connor JJ. P42/44 MAP kinase inhibitor PD98059 attenuates multiple forms of synaptic plasticity in rat dentate gyrus In vitro. J Neurophysiol. 1999;81:103–110. doi: 10.1152/jn.1999.81.1.103. [DOI] [PubMed] [Google Scholar]
- Crow T, Xue-Bian J-J, Siddiqi V, Kang T, Neary JT. Phosphorylation of mitogen-activated protein kinase by one-trial and multi-trial classical conditioning. J Neurosci. 1998;18:3480–3487. doi: 10.1523/JNEUROSCI.18-09-03480.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Vanhoutte P, Pages C, Caboche J, Laroche S. The MAPK/ERK cascade targets both Elk-1 and cAMP response element- binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J Neurosci. 2000;20:4563–4572. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English J, Pearson G, Wilsbacher J, Swantek J, Karandikar M, Xu S, Cobb MH. New insights into the control of MAP kinase pathways. Exp Cell Res. 1999;253:255–270. doi: 10.1006/excr.1999.4687. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- ————— A requirement for the mitogen-activated protein kinase cascade in hippocampal long-term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB- dependent transcription and ERK nuclear translocation. Neuron. 1998a;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR. Stimulation of the cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci. 1998b;1:595–601. doi: 10.1038/2830. [DOI] [PubMed] [Google Scholar]
- Jones MW, French PJ, Bliss TV, Rosenblum K. Molecular mechanisms of long-term potentiation in the insular cortex In vivo. J Neurosci. 1999;19:RC36. doi: 10.1523/JNEUROSCI.19-21-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanterewicz BI, Urban NN, McMahon DB, Norman ED, Giffen LJ, Favata MF, Scherle PA, Trzskos JM, Barrionuevo G, Klann E. The extracellular signal-regulated kinase cascade is required for NMDA receptor-independent LTP in area CA1 but not area CA3 of the hippocampus. J Neurosci. 2000;20:3057–3066. doi: 10.1523/JNEUROSCI.20-09-03057.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornhauser JM, Greenberg ME. A kinase to remember: Dual roles for MAP kinase in long-term memory. Neuron. 1997;18:839–842. doi: 10.1016/s0896-6273(00)80322-0. [DOI] [PubMed] [Google Scholar]
- Liu J, Fukunaga K, Yamamoto H, Nishi K, Miyamoto E. Differential roles of Ca(2+)/calmodulin-dependent protein kinase II and mitogen-activated protein kinase activation in hippocampal long-term potentiation. J Neurosci. 1999;19:8292–8299. doi: 10.1523/JNEUROSCI.19-19-08292.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue SF, Paylor R, Wehner JM. Hippocampal lesions cause learning deficits in inbred mice in the Morris water maze and conditioned-fear task. Behav Neurosci. 1997;111:104–113. doi: 10.1037//0735-7044.111.1.104. [DOI] [PubMed] [Google Scholar]
- Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER. MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron. 1997;18:899–912. doi: 10.1016/s0896-6273(00)80330-x. [DOI] [PubMed] [Google Scholar]
- Norman ED, Thiels E, Barrionuevo G, Klann E. Long-term depression in the hippocampus in vivo is associated with protein phosphatase-dependent alterations in extracellular signal- regulated kinase. J Neurochem. 2000;74:192–198. doi: 10.1046/j.1471-4159.2000.0740192.x. [DOI] [PubMed] [Google Scholar]
- Paylor R, Crawley JN. Inbred strain differences in prepulse inhibition of the mouse startle response. Psychopharmacology (Berl) 1997;132:169–180. doi: 10.1007/s002130050333. [DOI] [PubMed] [Google Scholar]
- Paylor R, Nguyen M, Crawley JN, Patrick J, Beaudet A, Orr-Urtreger A. Alpha 7 nicotinic subunits are not necessary for hippocampal-dependent learning or sensorimotor gating: A behavioral characteristic of Acra7-deficient mice. Learn Mem. 1998;5:302–316. [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum K, Futter M, Jones M, Hulme EC, Bliss TV. ERKI/II regulation by the muscarinic acetylcholine receptors in neurons. J Neurosci. 2000;20:977–985. doi: 10.1523/JNEUROSCI.20-03-00977.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Nadel NV, Sullivan GM, Harris A, LeDoux JE. Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn Mem. 1999;6:97–110. [PMC free article] [PubMed] [Google Scholar]
- Schmid RS, Graff RD, Schaller MD, Chen S, Schachner M, Hemperly JJ, Maness PF. NCAM stimulates the Ras-MAPK pathway and CREB phosphorylation in neuronal cells. J Neurobiol. 1999;38:542–558. [PubMed] [Google Scholar]
- Selcher JC, Atkins CM, Trzaskos JM, Paylor R, Sweatt JD. A necessity for MAP kinase activation in mammalian spatial learning. Learn Mem. 1999;6:478–490. doi: 10.1101/lm.6.5.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S. Impaired spatial learning in a-calcium-calmodulin kinase II mutant mice. Science. 1992;257:206–211. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- Stubley-Weatherly L, Harding JW, Wright JW. Effects of discrete kainic acid-induced hippocampal lesions on spatial and contextual learning and memory in rats. Brain Res. 1996;716:29–38. doi: 10.1016/0006-8993(95)01589-2. [DOI] [PubMed] [Google Scholar]
- Walz R, Roesler R, Quevedo J, Rockenbach IC, Amaral OB, Vianna MR, Lenz G, Medina JH, Izquierdo I. Dose-dependent impairment of inhibitory avoidance retention in rats by immediate post-training infusion of a mitogen-activated protein kinase kinase inhibitor into cortical structures. Behav Brain Res. 1999;105:219–223. doi: 10.1016/s0166-4328(99)00077-7. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Atkins CM, Selcher JC, Varga AW, Mirnikjoo B, Paylor R, Leitges M, Sweatt JD. A Role for the beta Isoform of Protein Kinase C in Fear Conditioning. J Neurosci. 2000;20:5906–5914. doi: 10.1523/JNEUROSCI.20-16-05906.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER. ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron. 1999;24:715–726. doi: 10.1016/s0896-6273(00)81124-1. [DOI] [PubMed] [Google Scholar]
- Wu SP, Lu KT, Chang WC, Gean PW. Involvement of mitogen-activated protein kinase in hippocampal long- term potentiation. J Biomed Sci. 1999;6:409–417. doi: 10.1007/BF02253672. [DOI] [PubMed] [Google Scholar]