


Abstract
Transfer RNA (tRNA) contains a number of complex ‘hypermodified’ nucleosides that are essential for a number of genetic processes. Intermediate forms of these nucleosides are rarely found in tRNA despite the fact that modification is not generally a complete process. We propose that the modification machinery is tuned into an efficient ‘assembly line’ that performs the modification steps at similar, or sequentially increasing, rates to avoid build-up of possibly deleterious intermediates. To investigate this concept, we measured steady-state kinetics for the final two steps of the biosynthesis of the mnm5s2U nucleoside in Escherichia coli tRNAGlu, which are both catalysed by the bifunctional MnmC enzyme. High-performance liquid chromatography-based assays using selectively under-modified tRNA substrates gave a Km value of 600 nM and kcat 0.34 s−1 for the first step, and Km 70 nM and kcat 0.31 s−1 for the second step. These values show that the second reaction occurs faster than the first reaction, or at a similar rate at very high substrate concentrations. This result indicates that the enzyme is kinetically tuned to produce fully modified mnm5(s2)U while avoiding build-up of the nm5(s2)U intermediate. The assay method developed here represents a general approach for the comparative analysis of tRNA-modifying enzymes.
INTRODUCTION
tRNA molecules are heavily modified with many non-canonical nucleosides, which perform a number of biological functions including structural stabilization (1) and optimization of codon binding (2). These modifications range from simple methylations to complex ‘hypermodified’ residues involving multistep biosyntheses. The hypermodified nucleosides are particularly interesting, as the cellular machinery allocates a relatively large amount of energy to the biosyntheses of these modifications, indicating a high importance to cellular processes. These nucleosides are most often located in or adjacent to the anticodon (3), and are involved in key translation processes anticodon–stem loop stabilization (2), codon binding and wobble base pairing (4,5). Recent studies on the quantification of modified nucleosides show that tRNA is not always fully modified, instead containing a significant number of unmodified positions [(6,7) and unpublished data]. However, partially modified precursors of hypermodified nucleosides are generally not observed, with the exception of those present in tRNAs that always contain the partially modified form (3,8–10). Together, these results suggest that biosynthetic pathways are organized to give complete hypermodified nucleosides, avoiding partially modified intermediate nucleosides. Biosynthetic tuning of this type would require a coordinated ‘assembly line’ process in which sequential modifications are performed at similar, or increasing, rates in order to efficiently construct the final products without a build-up of intermediates. We propose that this process is likely to be predominantly controlled by tuning of the activities and abundances of the tRNA-modifying proteins. Alternatively, biosynthetic tuning could involve selective degradation of partially modified tRNAs (11) or control of enzyme compartmentalization (12) in eukaryotes.
The 5-methylaminomethyl(-2-thio)uridine (mnm5(s2)U) residue, which is present in position 34 of the anticodon of Escherichia coli tRNAs Glu, Lys, Arg and, probably, Gln (3), is particularly interesting due to its involvement in wobble base pairing (4,5,13,14) and its complex four-step biosynthetic pathway (Scheme 1). The s2 group is inserted by the MnmA enzyme (15,16), while the mnm5 group is independently formed in a three-step sequence. Initially, (s2)U is converted to 5-carboxymethylaminomethyl(-2-thio)uridine (cmnm5(s2)U) by a two-enzyme complex involving addition of glycine to a reactive intermediate (17–19). The cmnm5(s2)U base is then converted to mnm5s2U in two steps by the bifunctional enzyme MnmC (20–22), which initially carries out a flavin adenine dinucleotide (FAD)-mediated demodification to 5-aminomethyl(-2-thio)uridine (nm5(s2)U), followed by methylation to mnm5(s2)U using S-adenosylmethionine (SAM) as cofactor. To test our proposal that the modification steps are coordinated by tuning of the kinetics and abundance of biosynthesis enzymes, we investigated the activities of the two modification steps performed by MnmC. As these two reaction steps are both performed by a single enzyme, differences in enzyme abundance for the individual steps are excluded. Therefore, control of enzyme kinetics would be necessary for our proposed assembly-line type process. To our knowledge, the partial modification nm5(s2)U has not been reported in normal cellular tRNA, indicating that this intermediate is avoided by the biosynthetic machinery. The presence or absence of under-modified cmnm5(s2)U is not clear due to the natural occurrence of this nucleoside at certain tRNA positions.
Scheme 1.

Biosynthesis of mnm5(s2)U in tRNA. X = O (in U) or S (in s2U), R = cmnm5, nm5 or mnm5. Modified positions are coloured in red.
Kinetic assay of complex tRNA-modifying enzymes is generally problematic due to the difficulty of obtaining defined under-modified tRNA substrates and the measurement of reaction progress. Previous cmnm5(s2)U → mnm5(s2)U assays have used either total tRNA from cells lacking MnmC (20–22) or substantially under-modified tRNA from in vitro transcription (18). The most accurate assay results to date indicate that the first step is rate limiting (20), but could only measure the methylation reaction and did not have the defined substrates necessary for full kinetic analysis. In order to obtain such substrates for reaction with MnmC, we overexpressed tRNAGlu in an E. coli expression strain lacking MnmC, then isolated the selectively under-modified cmnm5s2U-containing tRNA species using anion-exchange high-performance liquid chromatography (HPLC). The second substrate (containing nm5s2U) was formed by reaction of the cmnm5s2U-containing tRNA with MnmC in the absence of the SAM cofactor. Reaction of each substrate with MnmC was assayed using anion-exhange HPLC to monitor reaction progress. To further assess the importance of the MnmC-mediated modifications, we additionally measured growth curves for an MnmC knockout E. coli strain and its corresponding wild-type strain.
MATERIALS AND METHODS
Preparation of recombinant MnmC protein
His-tagged MnmC was cloned and expressed as previously reported (21) in E. coli BL21 cells, and purified as follows. All steps were performed at 0–4°C. Harvested cells were suspended in buffer A (50 mM Tris–HCl, pH 8.0, 200 mM KCl, 10 mM MgCl2, 3 mM β-mercaptoethanol, 10% glycerol) with the addition of lysozyme (0.2 mg/ml), then lysed using a French press. The lysate was cleared by centrifugation (24 000g for 30 min) and filtration (0.45 µM) and was applied to a HiScreen IMAC FF column (GE Healthcare) charged with Ni2+, using an ÄKTApurifier system (GE Healthcare). The column was washed with buffer A supplemented with 5 mM imidazole then MnmC was eluted with buffer A containing 0.5 M imidazole. The protein obtained was concentrated and exchanged into buffer B (buffer A lacking KCl) using an Amicon Ultra 30 000 MWCO centrifugal filter (Millipore) then applied to a 1 ml Mono Q column (GE Healthcare) equilibrated with the same buffer. The enzyme was eluted with a linear gradient of buffer C (buffer A containing 500 mM KCl), concentrated then applied to a Superdex 200 GL10/300 size-exclusion column and eluted with buffer C. The purified protein was concentrated to 10 mg/ml in buffer D (buffer A containing only 50 mM KCl) and stored at −80°C. A yield of ∼10 mg of purified protein was obtained from 4 l of expression culture. Protein concentration was calculated by Bradford assay.
MnmC knockout from an E. coli expression strain
The MnmC gene was removed from T7 express E. coli (NEB) by replacement with a chloramphenicol resistance cassette. The cassette was made by polymerase chain reaction (PCR) amplification of the cmr gene from the pDEST17 plasmid (Invitrogen) using Phusion polymerase (Finnzymes) and the following primers, each incorporating 50 bases of the MnmC gene: 5′-TGAAACACTACTC CATACAACCTGCCAACCTCGAATTTAATGCTGAGGGTGCGGCCGCATTAGGCACCCC and 5′-TACCCCGCCTTAACCGCTTTACCCTTCAACAATTTCCGCACCCATAACC GTTACGCCCCGCCCTGCCACT. Chemically competent T7 express cells were transformed with the pSIM6 plasmid (23), kindly supplied by Donald L. Court, then made electrocompetent and transformed with the PCR-amplified Cam cassette. After pregrowth in LB medium then selection on Cam plates, several colonies were chosen and confirmed by PCR to contain the Cam gene and no MnmC gene (primers: (5′-CTGAACCATATGAAACA CTACTCCATACAACCTGCC, 5′-CTGAACAGATCTTTACCCCGCCTTAACCGCTTTA CCCTTCAAC). The cells were made electrocompetent then stored at −80°C in 10% glycerol.
Cloning, expression and purification of under-modified tRNAGlu
The tRNAGlu gene (corresponding to RNA sequence GUCCCCUUCGUCUAGAGGCCCA GGACACCGCCCUUUCACGGCGGUAACAGGGGUUCGAAUCCCCUAGGGGACGCCA) was amplified by PCR from NEB5α E. coli (NEB) using Phusion polymerase and the following primers to give an EcoRI cleavage site and the T7 promoter 5′ of the tRNA sequence, and BsaI and HindIII cleavage sites 3′ of the tRNA: 5′-GAATTCTAATACGACT CACTATAGTCCCCTTCGTCTAGAGGCCCAGGA and 5′-AAGCTTGGTCTCATGGCGT CCCCTAGGGGATTCGAACC. The PCR product was cleaved with the EcoRI and HindIII restriction enzymes, and ligated into the pSGAT2 plasmid. The presence of the tRNAGlu gene was confirmed by sequencing. The expression plasmid was then used to transform electrocompetent ΔMnmC T7 express cells (see above). Transformed cells were grown in LB-medium supplemented with carbenicillin (100 µg/ml) to an OD600 of 0.5, then induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for 6 h and harvested. The cell pellets were phenol extracted as reported previously (6) to give total cell tRNA (∼20 mg per litre of E. coli culture), which was dissolved at ∼10 mg/ml in buffer E (100 mM Tris, pH 8, 50 mM MgCl2) after ethanol precipitation.
The crude tRNA was then separated by two sequential anion exchange HPLC purifications using a DNAPac PA100 22 × 250 mm column (Dionex) and a Merck Hitachi Lachrom system. The first purification was done in buffer F (100 mM NaOAc, pH 5.0, 50 mM MgCl2) with a gradient of 205–210 mM NaCl, followed by concentration of the tRNA using an Amicon Ultra 10 000 MWCO centrifugal filter (Millipore) and a further purification in buffer E using a gradient of 205–215 mM NaCl The purified tRNA was exchanged into buffer E and stored at −20°C. To obtain nm5s2U-containing tRNA, the cmnm5s2U-containing tRNA after pH 5 purification (400 µg) was incubated with MnmC (40 µg) in 1 ml of buffer G (50 mM Tris, 20 mM NH4Cl) at 37°C for 1 h before the pH 8 purification. For mnm5s2U-containing tRNA, the same procedure was carried out using 100 µg cmnm5s2U-tRNA and 10 µg MnmC with the addition of SAM (final conc. 800 µM).
Characterization of tRNA
The purified tRNA was analysed by digestion with RNases A or T1, followed by matrix-assisted laser desorption/ionization-mass spectrometry (MALDI-MS) (24). For identification of pseudouridine bases, tRNA (2 µM) and acrylonitrile (1.8 M) were initially dissolved in TEA-AcOH buffer (0.98 M), ethanol (36%) at a total volume of 34 µl, incubated at 70°C for 2 h, then lyophilized, redissolved in water and exchanged into pure water using a centrifugal filter.
RNase digests were performed by incubation of tRNA (0.2 µg) with RNase A (0.1 µg, Fermentas) or RNase T1 (1 U, Ambion) in 3 mg/ml 3-HPA, 1% acetonitrile at a total volume of 3 µl at 37°C for 2 h (for RNase A) or 7 h (for RNase T1). The hydrolysed tRNA was desalted three times using a 0.025 µM drop dialysis filter (Millipore) then 0.5 µl of the solution was mixed with 0.5 µl of matrix (0.6 M 3-HPA, 32 mM picolinic acid, 18 mM diammonium citrate, 10% acetonitrile) on a MALDI sample plate and allowed to dry. MALDI-TOF MS was performed using a Bruker Autoflex II in negative mode with 19 kV and 16 kV ionization source voltages, 8.55 kV lens voltage, 20 kV reflector voltage and 200 ns pulsed ion extraction. Masses were externally calibrated using oligonucleotides of known sequence, or internally calibrated using invariant tRNA fragments from the sample (e.g. for accurate analysis of the anticodon–stem loop fragment).
Assay of MnmC
For assay of the FAD-dependent MnmC activity, the following final reaction mixture was used: cmnm5s2U-containing tRNA (0.13–2.0 µM), MnmC (1.7 nM), Tris (60 mM, pH 8.0), NH4Cl (20 mM) KCl (2.5 mM), MgCl2 (0.65 mM), β-mercaptoethanol (0.15 mM), glycerol (0.5%), bovine serum albumin (BSA; 5 µg/ml) [note: KCl, β-mercaptoethanol, glycerol and BSA were only present as they were used in the enzyme storage buffer]. All components except the enzyme were assembled on ice, then preincubated at 37°C in a water bath for 3 min. The enzyme (in buffer D plus 0.1 mg/ml BSA) was then added, and aliquots of at least 20 ng tRNA were removed within the first 5 min of reaction, diluted at least 2 × with buffer F then injected onto a DNAPac PA100 4 × 250 mm column (Dionex) and analysed in buffer E with a gradient of 175–180 mM NaCl. Reaction volumes were typically 40 µl. Each assay was carried out in triplicate.
For assay of the SAM-dependent MnmC activity, the following final reaction mixture was used: nm5s2U containing tRNA (25–400 nM), MnmC (42 pM), SAM (500 µM), Tris (60 mM, pH 8.0), NH4Cl (20 mM) KCl (2.5 mM), MgCl2 (0.65 mM) β-mercaptoethanol (0.15 mM), glycerol (0.5%), ethanol (0.6%), BSA (5 µg/ml) [note: KCl, β-mercaptoethanol, glycerol and BSA and ethanol were only present as they were used in the enzyme or SAM storage buffers]. The reaction was performed as described for the FAD-dependent activity above, using an HPLC gradient of 160–165 mM NaCl and typical reaction volumes of 160 µl.
Reaction progress was calculated from the ratio of the areas of the product and starting material UV absorbance peaks (260 nm) in the HPLC chromatograms. In the case of the SAM-dependent reaction, the product and starting material peaks overlapped, and were deconvoluted using the eXPFit14 add-in (www.chem.qmul.ac.uk/software/eXPFit.htm) for Microsoft Excel. Reaction progress was then plotted versus time, and the linear slope of each plot measured as the initial reaction rate at the corresponding substrate concentration. Michaelis–Menten curves were fitted to the initial rate versus [S] data using the EZ-Fit5 program (Perrella Scientific).
To ensure that the UV absorbance data gave a linear response for the product and substrate peak areas, calibration injections were performed at a range of concentrations of cmnm5s2U, nm5s2U and mnm5s2U containing tRNA. To ensure that the SAM concentration of 500 µM was saturating, preliminary nm5s2U to mnm5s2U assays were carried out with a range of SAM concentrations. SAM concentrations from 100 to 1000 µM were found to give similar reaction rates, indicating that the KM for SAM is much <500 µM.
Bacterial growth rate experiments
Bacterial strains were obtained from the Keio collection of E. coli knockouts (obtained from the NBRP-E.coli at NIG). Growth rates of the wild-type (BW25113) and ΔMnmC (JW5380) strains were measured at 37°C in rich (LB) medium by recording optical densities at 600 nm (OD600) at various time points. Pre-cultures were initially grown exponentially for several generations before dilution into fresh medium for data collection. Each strain was grown in triplicate under identical conditions. Growth rates were calculated by fitting an exponential curve of the type y = Aekt.
RESULTS
Preparation of selectively under-modified tRNAGlu substrates for MnmC
In order to prepare selectively under-modified tRNAGlu, we assembled a tRNA overexpression system (25) using an E. coli strain that lacks the MnmC gene. As the expression construct, the E. coli tRNAGlu gene was cloned into a protein expression plasmid (pSGAT2) containing a T7 promoter for inducible expression. An additional T7 promoter was added directly 5′ to the tRNA gene in order to expedite tRNA processing (as the RNA transcripts from this promoter should begin at the first position of the tRNA and therefore not require enzymatic 5′-cleavage). To obtain an expression strain lacking MnmC, we used the recombineering method (23) to replace the MnmC gene in T7 expression cells with an antibiotic resistance cassette. The tRNA expression plasmid was then used to transform ΔMnmC cells, from which tRNAGlu was expressed and isolated by phenol extraction (6). The major tRNA ‘modivariants’ (10) obtained were then separated by anion-exchange HPLC (Figure 1a and b) then partially sequenced by MALDI-MS analysis of RNase digests (Figure 2). All major peaks in the HPLC chromatogram were found to be tRNAGlu based on this MALDI sequencing (Figure 2a–c and Supplementary Figure S1), showing that a very high level of expression was sustained by the cells. A single modivariant (tRNA 1, Figure 1a and c) was found to contain the desired cmnm5s2U modification (Figure 2c) as well as the other expected mass-detectable tRNAGlu modifications (T and m2A, Figure 2a–c). The presence of the two expected pseudouridine (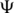 ) nucleotides in this tRNA was then confirmed by reaction with acrylonitrile followed by RNase/MS analysis (Supplementary Figure S2) (24). The other modivariants obtained lacked anticodon–stem loop modifications (Supplementary Figure S1), as labelled in Figure 1c.
) nucleotides in this tRNA was then confirmed by reaction with acrylonitrile followed by RNase/MS analysis (Supplementary Figure S2) (24). The other modivariants obtained lacked anticodon–stem loop modifications (Supplementary Figure S1), as labelled in Figure 1c.
Figure 1.

Overexpressed tRNAGlu from a ΔMnmC E. coli strain. (a) HPLC of total extracted tRNA. HPLC buffers: 100 mM Tris pH 8, 50 → 150 mM MgCl2. The four major modivariants are labelled 1–4. Each was isolated by HPLC for subsequent MS analysis of modified nucleosides. (b) Representative purified tRNA (tRNA 5, see below) after anion-exchange purifications at pH 5 and 8. HPLC buffers: 100 mM Tris pH 8, 50 mM MgCl2, 0 → 500 mM NaCl. [Note: The MgCl2 and NaCl gradients in (a) and (b) were used interchangeably for analytical purposes] (c) Diagrams showing the modified nucleosides present in tRNAs 1–5 and those present in tRNAs 5 and 6, the products of reaction of tRNA 1 with MnmC (see below). Nucleosides labelled in red (or orange or yellow) were identified by MS (Figure 2 and Supplementary Figure S1). The presence of 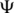 nucleosides labelled in grey was not confirmed. tRNA 1 contains cmnm5s2U34 and all other expected modifications, while tRNA 2–4 are less fully modified at anticodon–stem loop postions as shown. The nm5s2U and mnm5s2U modifications obtained by reaction with MnmC are shown in orange and yellow, respectively, to differentiate from cmnm5s2U.
nucleosides labelled in grey was not confirmed. tRNA 1 contains cmnm5s2U34 and all other expected modifications, while tRNA 2–4 are less fully modified at anticodon–stem loop postions as shown. The nm5s2U and mnm5s2U modifications obtained by reaction with MnmC are shown in orange and yellow, respectively, to differentiate from cmnm5s2U.
Figure 2.

MALDI-MS spectra of RNase digests of tRNA 1, 5 and 6. (a) RNase A digest of tRNA 1 (cmnm5s2U-tRNAGlu). (b) RNase T1 digest of tRNA 1. (c) Expanded section of (b) showing the anticodon–stem loop fragment of tRNA 1. Extra peaks are assigned as follows. a: 3165.5, Fragment A [AAUCCCCUAGcp (cp = cyclic phosphate)]; b: 3182.9, Fragment B (AAUCCCCUAGp); c: 3203.7, Fragment A – H + K and Fragment B – H + Na; d: 3221.3, Fragment B – H + K; e: 3235.6, Fragment C (CCCUcmnm5s2UUCm2ACGcp); f: 3242.4, Fragment A – 2H + 2K; g: 3258.4, Fragment B – 2H + 2K; h: 3274.4, Fragment C – H + K and Fragment D (CCCUcmnm5s2UUCm2ACGp) – H + Na; i: 3290.9, Fragment D – H + K; j: 3296.0, Fragment B – 3H + 3K. (d) Section of a RNase T1 digest of tRNA 5 (nm5s2U-tRNAGlu) showing the nm5s2U-containing fragment. Extra peaks are assigned as follows. a: 3165.3, Fragment A (AAUCCCCUAGcp); b: 3177.8, Fragment E (CCCUnm5s2UUCm2ACGcp), c: 3182.9, Fragment B (AAUCCCCUAGp); d: 3204.2, Fragment B – H + Na; e: 3215.8, Fragment E – H + K; f: 3221.2, Fragment B – H + K; g: 3233.5, Fragment F (CCCUnm5s2UUCm2ACGp) – H + K; h: 3243.3, Fragment B – 2H + Na + K; i: 3258.5, Fragment B – 2H + 2K; j: 3270.1, Fragment F – 2H + 2K; k: 3296.1, Fragment B – 3H + 3K; l: 3307.8, Fragment F – 3H + 3K. (e) Section of an RNase T1 digest of tRNA 6 (mnm5s2U-tRNAGlu) showing the mnm5s2U-containing fragment. Extra peaks are assigned as follows. a: 3165.4, Fragment A (AAUCCCCUAGcp); b: 3182.9, Fragment B (AAUCCCCUAGp); c: 3191.0, Fragment G (CCCUmnm5s2UUCm2ACGcp); d: 3204.9, Fragment B – H + Na; e: 3221.0, Fragment B – H + K; f: 3229.3, Fragment G – H + K; g: 3246.4, Fragment H (CCCUmnm5s2UUCm2ACGp) – H + K; h: 3258.9, Fragment B – 2H + 2K; i: 3267.3, Fragment G – 2H + 2K; j: 3283.9, Fragment H – 2H + 2K; k: 3296.5, Fragment B – 3H + 3K; l: 3306.7, Fragment A – 3H + 3K; m: 3322.0, Fragment H – 3H + 3K; n: Fragment B – 4H + 4K. The tRNA fragments analysed in (a) and (b) are labelled with mass and sequence on each spectrum, and coloured in red on each corresponding tRNA diagram. The data in (c)–(e) are calibrated to the constant fragment B (AAUCCCCUAGp, 3182.9) to allow accurate determination of the variable anticodon fragment.
The purified cmnm5s2U-containing tRNA 1 was reacted with purified recombinant MnmC (21) in the absence or presence of the SAM cofactor to give nm5s2U or mnm5s2U-containing tRNA, respectively (tRNA 5 and 6, Figure 1c). These tRNA products were further purified by anion-exchange HPLC and analysed by MALDI-MS of RNase T1 digests of the tRNA products. Mass peaks corresponding to the anticodon–stem loop fragments confirmed the identity of each tRNA (Figure 2d and e). High purity of the tRNAs 1 and 5 was determined from the lack of contaminating under-modified fragments in MALDI-MS spectra, the single major HPLC peak detected for each tRNA, and the observed complete reaction with MnmC as measured by HPLC.
Development of an HPLC-based assay for both activities of MnmC
Anion-exchange HPLC of the cmnm5s2U- mnm5s2U- and nm5s2U-containing tRNAs showed that each elutes at a different retention time (Figure 3a and b), allowing an HPLC-based assay to be used to measure both activities of the enzyme. As full separation of tRNA 5 and 6 was not obtained, a computational peak deconvolution was employed to calculate the area of each peak (Figure 3b). Preliminary studies confirmed the linear response of the areas of the product and substrate peaks, and the approximate saturating concentration of the SAM cofactor (∼100 µM). Subsequently, steady-state kinetic assays for each reaction step were carried out. The conditions for each assay were chosen to be similar in order to compare kinetic constants, and were based on conditions known to optimize enzyme activity (20) with the addition of MgCl2 to stabilize tRNA (26). Rate versus substrate concentration curves are shown in Figure 3c and d. Fitting a rectangular hyperbola to these curves gives the Michaelis–Menten constants shown in Table 1. The Km for the second reaction is substantially lower (∼9-fold) than that of first reaction, and the kcat constant is not significantly different. This shows that the enzyme binds the second substrate (tRNA 5) tighter than the first (tRNA 1). This clearly will result in a higher rate of reaction for the second step, or similar rates at very high substrate concentrations where the kcat constant becomes dominant. The kcat/Km value, a general indicator of activity, is correspondingly eight times higher for the second reaction step, showing substantially higher activity at intermediate substrate concentrations.
Figure 3.

HPLCs and Michaelis–Menten plots for each MnmC catalysed reaction. (a) Representative HPLC showing complete separation of tRNA 1 from tRNA 5. HPLC gradient: 100 mM Tris, 50 mM MgCl2, 175 → 180 mM NaCl over 1 → 20 min. (b) Representative HPLC showing partial separation of tRNA 5 from tRNA 6, and calculated peaks for each tRNA. HPLC gradient: 100 mM Tris, 50 mM MgCl2, 160 → 165 mM NaCl over 1 → 30 min. (c) Michaelis–Menten plot for the FAD-dependent cmnm5s2U → nm5s2U demodification. (d) Michaelis–Menten plot for the SAM-dependent nm5s2U → mnm5s2U methylation. Rates in (c) and (d) represent the amount of substrate formed in a 40 µl reaction per minute per milligram enzyme.
Table 1.
Michaelis–Menten constants for each MnmC catalysed reaction
| Reaction | Km (nM) | kcat (s−1) | kcat/Km (µM−1 s−1) |
|---|---|---|---|
| cmnm5s2U → nm5s2U | 600 ± 200 | 0.34 ± 0.04 | 0.56 |
| nm5s2U → mnm5s2U | 70 ± 40 | 0.31 ± 0.05 | 4.5 |
To further support our idea that partially hypermodified nucleosides negatively affect translation processes, we measured the growth rate of a well-defined MnmC knockout strain from the Keio collection of E. coli knockouts (Figure 4). Exponential fits to the growth curves of the ΔMnmC and wild-type strains reveal a larger growth constant, k, for the wild-type strain. As the knockout strain is expected to contain the cmnm5 modification in place of mnm5, the result shows that this cmnm5 under-modification is detrimental to cell growth.
Figure 4.

Growth curves showing the impact of an MnmC gene knockout on E. coli growth rate. Strains are from the Keio Collection: BW25113 (wild type) and JW5380 (ΔMnmC). For each strain, the geometrical symbols on the graph (triangle, square, diamond) each represent the data points for one independent growth experiment.
DISCUSSION
The MnmC enzyme catalyses the final two steps of the biosynthesis of the mnm5(s2)U34 nucleoside in tRNA. The mnm5 biosynthesis pathway seems to be tuned to give only the fully modified residue, in particular avoiding nm5 under-modification [cmnm5s2U has been detected in vivo (8); however, it is not clear whether this is due to under-modification or the natural presence of cmnm5 in certain tRNAs]. The biosynthetic machinery could be regulated in a number of ways, for example by selective degradation of partially modified tRNA (11), controlled expression [or compartmentalization, in eukaryotes (12)] of particular modifying enzymes or by optimization of enzyme activities and specificities. This study reveals that the two transformations catalysed by MnmC are regulated by enzyme kinetics, as the second substrate is bound tighter than the first. Even at relatively high substrate concentrations (∼3 µM), the two reactions would be performed at similar rates, resulting in a relatively low concentration of the partially modified intermediate. Degradation of the under-modified tRNA cannot be ruled out as an additional mechanism; however, this seems unlikely given the existing kinetic control and the inefficiency associated with degradation of a highly modified tRNA.
In terms of the applicability of our in vitro study to in vivo enzyme activity, several aspects need to be considered. It is possible that the modification reactions are affected in vivo by other cellular components. Importantly, Mg2+ and 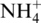 concentrations were reported previously to affect enzyme activity. However, this is unlikely to substantially change the results reported here, as both activities of MnmC were reported to be similarly affected by changes in the concentrations of these ions (20). Intracellular SAM concentrations are reported to be similar to those used in our assay (27), so our result is likely to be relevant in this respect. Other modifying enzymes have been found to be dependent on the state of tRNA processing, such as TilS, which modifies tRNAIle2 at a precursor stage (28). None of the cmnm5- or mnm5-containing tRNA obtained in the tRNAGlu overexpressions here and in a previous study (10) was found to lack other modifications, implying that MnmC acts on tRNA at a late stage of processing. Therefore, it is likely that our substrates represent those normally modified by MnmC.
concentrations were reported previously to affect enzyme activity. However, this is unlikely to substantially change the results reported here, as both activities of MnmC were reported to be similarly affected by changes in the concentrations of these ions (20). Intracellular SAM concentrations are reported to be similar to those used in our assay (27), so our result is likely to be relevant in this respect. Other modifying enzymes have been found to be dependent on the state of tRNA processing, such as TilS, which modifies tRNAIle2 at a precursor stage (28). None of the cmnm5- or mnm5-containing tRNA obtained in the tRNAGlu overexpressions here and in a previous study (10) was found to lack other modifications, implying that MnmC acts on tRNA at a late stage of processing. Therefore, it is likely that our substrates represent those normally modified by MnmC.
The fact that the mnm5(s2)U pathway is tuned to give full modification suggests that the partially modified nucleoside (particularly nm5(s2)U) is particularly detrimental to translation. Our result that ΔMnmC E. coli, which contain cmnm5(s2)U instead of mnm5(s2)U, grows significantly slower than wild-type E. coli, supports this proposal. It is likely that the observed slow growth of the MnmC knockout is a result of disruption of the reported fine tuning of A and G wobble pairing with mnm5s2U (14). Interestingly, some species completely lack an MnmC-type modification and instead use cmnm5s2U in the 34 position of tRNA e.g. Bacillus subtilis (3) and Saccharomyces cerevisiae (29). It is unclear at this stage as to why other organisms expend energy to further modify cmnm5s2U to mnm5s2U.
The assay method reported here is a general approach that should be applicable to comparative assay of most tRNA modifications. Although a number of very effective radioactivity-based assays have been reported (20,30), these require specific radioactive substrates and are not applicable to modifications for which no radioactive incorporation can be measured (e.g. the cmnm5s2U → nm5s2U demodification). Analysis of reaction progress by reverse-phase HPLC of full RNase digests is also possible (18) but is complicated by the extra digestion step and the requirement for quantification standards. Our analysis of intact tRNA using HPLC also allows verification of tRNA stability during reaction, which is often not taken into account (31). We believe our general approach will allow better comparison of the activities of tRNA-modifying enzymes in future studies.
In summary, we reveal that the full modification of the mnm5s2U tRNA modification is regulated by kinetic tuning of the activities of the MnmC enzyme. Our method represents a general approach to the comparative analysis of tRNA-modifying enzymes, and should be applicable to the study of other modification pathways. With the methods for selective preparation and assay of modified tRNA developed in this article, future studies will be carried out to further elucidate the regulation of tRNA modification and the impact of partial hypermodification on genetic processes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Alexander von Humboldt Foundation [postdoctoral fellowship to D.P.]; the Deutsche Forschungsgemeinschaft [grant numbers CA275/8-4, SFB 749]; Excellence Cluster CIPSM. Funding for open access charge: Deutsche Forschungsgemeinschaft [SFB 749].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Markus Müller for training in microbiological techniques.
REFERENCES
- 1.Motorin Y, Helm M. tRNA stabilization by modified nucleotides. Biochemistry. 2010;49:4934–4944. doi: 10.1021/bi100408z. [DOI] [PubMed] [Google Scholar]
- 2.Agris PF. Bringing order to translation: the contributions of transfer RNA anticodon-domain modifications. EMBO Rep. 2008;9:629–635. doi: 10.1038/embor.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jühling F, Mörl M, Hartmann RK, Sprinzl M, Stadler PF, Pütz J. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–D162. doi: 10.1093/nar/gkn772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agris PF, Vendeix FAP, Graham WD. tRNA’s wobble decoding of the genome: 40 years of modification. J. Mol. Biol. 2007;366:1–13. doi: 10.1016/j.jmb.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 5.Takai K, Yokoyama S. Roles of 5-substituents of tRNA wobble uridines in the recognition of purine-ending codons. Nucleic Acids Res. 2003;31:6383–6391. doi: 10.1093/nar/gkg839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brückl T, Globisch D, Wagner M, Müller M, Carell T. Parallel isotope-based quantification of modified tRNA nucleosides. Angew. Chem. Int. Ed. 2009;48:7932–7934. doi: 10.1002/anie.200902740. [DOI] [PubMed] [Google Scholar]
- 7.Saikia M, Fu Y, Pavon-Eternod M, He C, Pan T. Genome-wide analysis of N1-methyl-adenosine modification in human tRNAs. RNA. 2010;16:1317–1327. doi: 10.1261/rna.2057810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buck M, Connick M, Ames BN. Complete analysis of tRNA modified nucleosides by high-performance liquid chromatography: The 29 modified nucleosides of Salmonella typhimurium and Escherichia coli tRNA. Anal. Biochem. 1983;129:1–13. doi: 10.1016/0003-2697(83)90044-1. [DOI] [PubMed] [Google Scholar]
- 9.McCloskey JA, Graham DE, Zhou S, Crain PF, Ibba M, Konisky J, Söll D, Olsen GJ. Post-transcriptional modification in archaeal tRNAs: identities and phylogenetic relations of nucleotides from mesophilic and hyperthermophilic Methanococcales. Nucleic Acids Res. 2001;29:4699–4706. doi: 10.1093/nar/29.22.4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Madore E, Florentz C, Giegé R, Sekine S-i, Yokoyama S, Lapointe J. Effect of modified nucleotides on Escherichia coli tRNAGlu structure and on its aminoacylation by glutamyl-tRNA synthetase. Eur. J. Biochem. 1999;266:1128–1135. doi: 10.1046/j.1432-1327.1999.00965.x. [DOI] [PubMed] [Google Scholar]
- 11.Phizicky EM, Alfonzo JD. Do all modifications benefit all tRNAs? FEBS Lett. 2010;584:265–271. doi: 10.1016/j.febslet.2009.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hopper AK, Pai DA, Engelke DR. Cellular dynamics of tRNAs and their genes. FEBS Lett. 2010;584:310–317. doi: 10.1016/j.febslet.2009.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy FV, IV, Ramakrishnan V, Malkiewicz A, Agris PF. The role of modifications in codon discrimination by tRNALysUUU. Nat. Struct. Mol. Biol. 2004;11:1186–1191. doi: 10.1038/nsmb861. [DOI] [PubMed] [Google Scholar]
- 14.Krüger MK, Pedersen S, Hagervall TG, Sørensen MA. The modification of the wobble base of tRNAGlu modulates the translation rate of glutamic acid codons in vivo. J. Mol. Biol. 1998;284:621–631. doi: 10.1006/jmbi.1998.2196. [DOI] [PubMed] [Google Scholar]
- 15.Kambampati R, Lauhon CT. MnmA and IscS are required for in vitro 2-thiouridine biosynthesis in Escherichia coli. Biochemistry. 2003;42:1109–1117. doi: 10.1021/bi026536+. [DOI] [PubMed] [Google Scholar]
- 16.Numata T, Ikeuchi Y, Fukai S, Suzuki T, Nureki O. Snapshots of tRNA sulphuration via an adenylated intermediate. Nature. 2006;442:419–424. doi: 10.1038/nature04896. [DOI] [PubMed] [Google Scholar]
- 17.Yim L, Moukadiri I, Björk GR, Armengod M-E. Further insights into the tRNA modification process controlled by proteins MnmE and GidA of Escherichia coli. Nucleic Acids Res. 2006;34:5892–5905. doi: 10.1093/nar/gkl752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moukadiri I, Prado S, Piera J, Velázquez-Campoy A, Björk GR, Armengod ME. Evolutionarily conserved proteins MnmE and GidA catalyze the formation of two methyluridine derivatives at tRNA wobble positions. Nucleic Acids Res. 2009;37:7177–7193. doi: 10.1093/nar/gkp762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi R, Villarroya M, Ruiz-Partida R, Li Y, Proteau A, Prado S, Moukadiri I, Benítez-Páez A, Lomas R, Wagner J, et al. Structure-function analysis of E. coli MnmG (GidA), a highly-conserved tRNA-modifying enzyme. J. Bacteriol. 2009;191:7614–7619. doi: 10.1128/JB.00650-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hagervall TG, Edmonds CG, McCloskey JA, Björk GR. Transfer RNA(5-methylaminomethyl-2-thiouridine)-methyltransferase from Escherichia coli K-12 has two enzymatic activities. J. Biol. Chem. 1987;262:8488–8495. [PubMed] [Google Scholar]
- 21.Bujnicki JM, Oudjama Y, Roovers M, Owczarek S, Caillet J, Droogmans L. Identification of a bifunctional enzyme MnmC involved in the biosynthesis of a hypermodified uridine in the wobble position of tRNA. RNA. 2004;10:1236–1242. doi: 10.1261/rna.7470904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roovers M, Oudjama Y, Kaminska KH, Purta E, Caillet Jl, Droogmans L, Bujnicki JM. Sequence–structure–function analysis of the bifunctional enzyme MnmC that catalyses the last two steps in the biosynthesis of hypermodified nucleoside mnm5s2U in tRNA. Proteins. 2008;71:2076–2085. doi: 10.1002/prot.21918. [DOI] [PubMed] [Google Scholar]
- 23.Sharan SK, Thomason LC, Kuznetsov SG, Court DL. Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc. 2009;4:206–223. doi: 10.1038/nprot.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mengel-Jørgensen J, Kirpekar F. Detection of pseudouridine and other modifications in tRNA by cyanoethylation and MALDI mass spectrometry. Nucleic Acids Res. 2002;30:e135. doi: 10.1093/nar/gnf135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sylvers LA, Rogers KC, Shimizu M, Ohtsuka E, Söll D. A 2-thiouridine derivative in tRNAGlu is a positive determinant for aminoacylation by Escherichia coli glutamyl-tRNA synthetase. Biochemistry. 1993;32:3836–3841. doi: 10.1021/bi00066a002. [DOI] [PubMed] [Google Scholar]
- 26.Madore E, Florentz C, Giegé R, Lapointe J. Magnesium-dependent alternative foldings of active and inactive Escherichia coli tRNA(Glu) revealed by chemical probing. Nucleic Acids Res. 1999;27:3583–3588. doi: 10.1093/nar/27.17.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hallidaya NM, Hardiea KR, Williamsa P, Winzera K, Barrett DA. Quantitative liquid chromatography–tandem mass spectrometry profiling of activated methyl cycle metabolites involved in LuxS-dependent quorum sensing in Escherichia coli. Anal. Biochem. 2010;403:20–29. doi: 10.1016/j.ab.2010.04.021. [DOI] [PubMed] [Google Scholar]
- 28.Nakanishi K, Bonnefond L, Kimura S, Suzuki T, Ishitani R, Nureki O. Structural basis for translational fidelity ensured by transfer RNA lysidine synthetase. Nature. 2009;461:1144–1148. doi: 10.1038/nature08474. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Yan Q, Guan M-X. Combination of the loss of cmnm5U34 with the lack of s2U34 modifications of tRNALys, tRNAGlu, and tRNAGln altered mitochondrial biogenesis and respiration. J. Mol. Biol. 2010;395:1038–1048. doi: 10.1016/j.jmb.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Lanen SG, Kinzie SD, Matthieu S, Link T, Culp J, Iwata-Reuyl D. tRNA modification by S-adenosylmethionine:tRNA ribosyltransferase-isomerase. J. Biol. Chem. 2003;278:10491–10499. doi: 10.1074/jbc.M207727200. [DOI] [PubMed] [Google Scholar]
- 31.Uhlenbeck OC. Keeping RNA happy. RNA. 1995;1:4–6. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.