

Abstract
Synaptic scaling stabilizes neuronal firing through the homeostatic regulation of postsynaptic strength, but the mechanisms by which chronic changes in activity lead to bidirectional adjustments in synaptic AMPA receptor (AMPAR) abundance are incompletely understood. Furthermore, it remains unclear to what extent scaling up and scaling down use distinct molecular machinery. PSD-95 is a scaffold protein proposed to serve as a binding “slot” that determines synaptic AMPAR content, and synaptic PSD-95 abundance is regulated by activity, raising the possibility that activity-dependent changes in the synaptic abundance of PSD-95 or other membrane-associated guanylate kinases (MAGUKs) drives the bidirectional changes in AMPAR accumulation during synaptic scaling. We found that synaptic PSD-95 and SAP102 (but not PSD-93) abundance were bidirectionally regulated by activity, but these changes were not sufficient to drive homeostatic changes in synaptic strength. Although not sufficient, the PSD-95 MAGUKs were necessary for synaptic scaling, but scaling up and down were differentially dependent on PSD-95 and PSD-93. Scaling down was completely blocked by reduced or enhanced PSD-95, through a mechanism that depended on the PDZ1/2 domains. In contrast, scaling up could be supported by either PSD-95 or PSD-93 in a manner that depended on neuronal age and was unaffected by a superabundance of PSD-95. Together, our data suggest that scaling up and down of quantal amplitude is not driven by changes in synaptic abundance of PSD-95 MAGUKs, but rather that the PSD-95 MAGUKs serve as critical synaptic organizers that use distinct protein–protein interactions to mediate homeostatic accumulation and loss of synaptic AMPAR.
Introduction
Synaptic scaling has emerged as an important form of synaptic plasticity that contributes to the homeostatic regulation of neuronal activity both in vitro and in vivo (Turrigiano and Nelson, 2004; Turrigiano, 2008; Pozo and Goda, 2010). During synaptic scaling, miniature EPSC (mEPSC) amplitude is bidirectionally regulated to compensate for chronic changes in neuronal firing, and these homeostatic adjustments in synaptic strength can mostly be accounted for by changes in synaptic AMPA receptor (AMPAR) (Turrigiano, 2008). Although the molecular underpinnings of synaptic scaling have recently come under intense scrutiny (Shepherd et al., 2006; Stellwagen and Malenka, 2006; Cingolani et al., 2008; Ibata et al., 2008; Seeburg et al., 2008; Gainey et al., 2009; Goold and Nicoll, 2010), the mechanisms by which chronic changes in activity lead to homeostatic adjustments in the abundance of synaptic AMPAR are incompletely understood, and it remains unclear to what extent scaling up and down use distinct molecular mechanisms.
AMPARs diffuse into and out of synaptic sites in which they interact with and are stabilized by scaffold proteins (Malinow and Malenka, 2002; Newpher and Ehlers, 2008). PSD-95 is a member of the membrane associated guanylate kinase (MAGUK) family that includes SAP102 and PSD-93. PSD-95 binds indirectly to AMPAR, regulates synaptic strength, and has been proposed to serve as a “slot” protein that determines synaptic AMPAR content (Schnell et al., 2002; Colledge et al., 2003; Ehrlich and Malinow, 2004; Elias and Nicoll, 2007; Keith and El-Husseini, 2008). PSD-95 accumulates at synapses and is necessary for the enhanced AMPAR accumulation induced by chronic activity blockade in hippocampal neurons (Noritake et al., 2009) and physically interacts with SPAR (Pak et al., 2001), a postsynaptic protein important for scaling down (Seeburg et al., 2008). These observations raise the possibility that activity-dependent changes in the abundance of synaptic PSD-95 or other MAGUKs could drive the bidirectional changes in AMPAR accumulation that underlie synaptic scaling; alternatively PSD-95 might function as a signaling scaffold to mediate synaptic scaling.
To distinguish between these possibilities, we performed gain- and loss-of-function experiments to probe the role of the PSD-95 MAGUKs in regulating bidirectional synaptic scaling in neocortical neurons. We found that, although synaptic PSD-95 abundance was bidirectionally regulated by activity, this was not sufficient to drive homeostatic changes in AMPAR accumulation. Although not sufficient, PSD-95 was necessary for synaptic scaling, in a manner that depended on the direction of scaling and neuronal age. In young neurons, scaling down required PSD-95, whereas scaling up could be supported by either PSD-95 or PSD-93. PSD-95 expression increased with age, and, in older neurons, scaling up as well as down became critically dependent on PSD-95. Finally, PSD-95 overexpression blocked scaling down through a mechanism that required the PDZ1/2 domains but had no effect on scaling up at any age. Together, these data demonstrate that PSD-95 plays distinct and developmentally regulated roles in scaling up and down. Our data suggest that, in neocortical pyramidal neurons, synaptic PSD-95 abundance is not a direct determinant of quantal amplitude but rather that the PSD-95 MAGUKs serve as critical synaptic organizers that mediate the homeostatic accumulation and loss of synaptic AMPAR.
Materials and Methods
Neuronal cultures and transfection.
Neuronal cultures were prepared as described previously (Wierenga et al., 2005). Cultures were made from dissociated visual cortex of postnatal day 1–3 Long–Evans rats and were plated on a confluent layer of astrocytes. All experiments were performed on day in vitro (DIV) 7–9 for young cultures and DIV 14–16 for older cultures. All experimental conditions were compared with age-matched controls from sister cultures. Electrophysiology and immunocytochemistry were performed on pyramidal neurons, which can be identified by their characteristic morphology, including a thick, long apical-like dendrite and a teardrop-shaped soma. Neurons were transfected 2–4 d before experimentation using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. All experiments were performed 2–4 d after transfection unless otherwise noted. TTX (2 μm), DNQX (20 μm), bicuculline (50 μm), and picrotoxin (PTX) (100 μm) were added to cultures for 1 d and in some experiments were refreshed 4–6 h before the experiment.
DNA constructs.
The following DNA constructs were used in the experiments as described previously: PSD-95–enhanced green fluorescent protein (EGFP) (Pratt et al., 2008), short hairpin RNAs (shRNAs) against PSD-95, PSD-93, and SAP102 (gifts from Dr. Nicoll, University of California, San Francisco, San Francisco, CA, and Dr. Fukata, National Institute for Physiological Sciences, Okazaki, Japan) (Elias et al., 2006; Noritake et al., 2009). The PSD-95 deletion mutants (ΔGK, ΔPDZ1/2, and PDZ1/2) were gifts from Dr. Nicoll and Dr. Sabatini (Harvard Medical School, Boston, MA) (Schnell et al., 2002; Sturgill et al., 2009). Transmembrane AMPA receptor regulatory protein (TARP) γ3 was a gift from Dr. Nicoll (Shi et al., 2009). Transfections with hairpins were performed for 2–4 d before 24 h of drug treatment. In control experiments, we compared the degree of knockdown (KD) at 3 and 6 d and found no difference: the degree of PSD-95 KD after 3 d transfection (n = 15 neurons) was 97 ± 11% of that after 6 d (n = 11 neurons, p = 0.45).
Immunostaining.
The following primary antibodies were used: PSD-95 (1:500; MA1-046; Affinity BioReagents), PSD-93 (1:500; AB5168, Millipore), PSD-93 (1:2000; ab2930; Abcam), SAP102 (1:500; 75-058; NeuroMab), VGluT-1 (1:500; 135 304; Synaptic Systems), and pan-TARPs (1:500; 07-577; Millipore). Intracellular staining of PSD-95, PSD-93, SAP102, and pan-TARPs were performed as described previously (Wierenga et al., 2005; Ibata et al., 2008). Alexa Flour 488, Texas Red, and Cascade Blue (1:200; Invitrogen) were used as secondary antibodies. All images were acquired on an Olympus IX-70 or IX-81 microscopes under an oil-immersion 60× objective using an Orca ER camera. A 6% neutral density filter was used to avoid photobleaching during image acquisition. Images were obtained and analyzed using Openlab (Improvision) or MetaMorph (Molecular Devices) software. The total fluorescence intensity of puncta was measured using the “granularity” function in MetaMorph. To allow pooling of data across imaging experiments, the data generated from each experiment were normalized to the mean control value for the respective experiment. To generate cumulative distributions of puncta fluorescence intensity (see Fig. 1B), 30 puncta from each neuron were randomly selected.
Figure 1.

Selective regulation of PSD-95 MAGUK accumulation during synaptic scaling. A, Sample images showing the differential regulation of PSD-95 MAGUK proteins by chronic activity blockade or activity elevation for 1 d. Scale bar, 10 μm. B, Cumulative distributions of puncta fluorescence intensity for PSD-95, PSD-93, and SAP102, for control, activity blockade, and activity elevation. a.u., Arbitrary units. C, Summary of the changes in puncta fluorescence of PSD-95 MAGUK proteins during synaptic scaling protocols. Values were normalized to control. D, Summary of the changes in puncta length density of PSD-95 MAGUK proteins. n = 21–50 for each condition. Different from control, **p < 0.01, ***p < 0.001.
Culture electrophysiology.
Whole-cell recordings were performed as described previously (Turrigiano et al., 1998; Wierenga et al., 2005). Briefly, after 2–4 d of transfection and 24 ± 2 h of activity blockade (TTX or DNQX) or activity elevation (PTX or bicuculline), cultured neurons were perfused at 25°C with artificial CSF (ACSF) containing the following (in mm): 126 NaCl, 5 KCl, 1 NaHPO4, 25 NaHCO3, 2 CaCl2, and 14 dextrose, pH 7.4 (osmolarity 325–330; oxygenated with 95% O2/5% CO2 throughout the recording). All recordings for mEPSCs contain TTX (0.1 μm), AP-5 (50 μm), and picrotoxin (25 μm) to isolate AMPA mEPSCs. Internal recording solution contains the following (in mm): 120 KMeSO4, 10 KCl, 2 MgSO4, 10 K-HEPES, 0.5 EGTA, 3 K2ATP, 0.3 NaGTP, 10 Na2phosphocreatine. mESPC recordings were performed on pyramidal neurons voltage clamped to −70 mV using an Axopatch 200B amplifier. Neurons in which Vm > −50 mV, series resistance > 20 MΩ, or any of these parameters changed by >20% during the recording were excluded from analysis. To generate cumulative distributions of mEPSC amplitude under each condition, 25 mEPSC events were randomly selected from each neuron. Neurons with fewer than 25 mEPSC events were excluded from analysis. However, mEPSC frequency was reduced markedly in SAP102 knockdown and PSD-95/SAP102 double knockdown conditions, and so to evaluate changes in mEPSC frequency for these experiments, we included in our frequency analysis all recordings with at least one mEPSC event. For current-clamp recording, the cells were perfused at 30°C with normal ACSF. The baseline firing rate in PSD-95 KD neurons was not significantly different from control (PSD-95 KD, 90.8 ± 40.7% of control; control, n = 20 neurons; PSD-95 KD, n = 10 neurons; p = 0.97). Furthermore, acute PTX treatment induced strong bursting activity under all conditions similar to that reported previously (data not shown) (Turrigiano et al., 1998). All physiology data were analyzed with in-house Igor Pro software.
Statistics.
All the data were expressed as mean ± SEM for the number of neurons indicated, unless otherwise noted. Statistical analyses were performed with unpaired Student's t tests or for multiple comparisons ANOVA followed by corrected t tests. For comparisons of cumulative distributions, a Kolmogorov–Smirnov (K–S) test was used. A p value equal to or smaller than 0.05 was considered statistically significant.
Results
Activity-dependent synaptic accumulation of PSD-95 MAGUKs
The mechanisms underlying altered receptor accumulation during synaptic scaling remain incompletely understood. It has been suggested that the number of synaptic AMPARs is determined by the synaptic levels of scaffolding proteins such as the PSD-95 family of MAGUKs, which included PSD-95, PSD-93, and SAP102 (El-Husseini et al., 2000; Schnell et al., 2002; Colledge et al., 2003; Stein et al., 2003; Ehrlich and Malinow, 2004; Elias et al., 2006). We began by examining the possibility that bidirectional changes in the accumulation of MAGUKs underlie the bidirectional changes in AMPAR accumulation during synaptic scaling. As a first step toward testing this, we examined the synaptic accumulation of PSD-95, PSD-93, and SAP102 by using antibodies against the endogenous proteins and quantifying the intensity of staining at punctate dendritic accumulations (Fig. 1); the majority (∼70%) of these puncta colocalize with synaptic markers (Pratt et al., 2003, 2008; Wierenga et al., 2005, 2006) and so represent putative synaptic sites. Activity blockade [using TTX or DNQX (Turrigiano et al., 1998)] or activity elevation [using bicuculline or picrotoxin (Turrigiano et al., 1998; Seeburg et al., 2008)] for 24 h bidirectionally regulated the intensity of PSD-95 and SAP102 puncta. In contrast, synaptic levels of PSD-93, a closely related MAGUK, were not significantly affected by either manipulation (Fig. 1), indicating that MAGUKs are differentially regulated by chronic changes in activity. The puncta densities of the MAGUKs were not significantly affected by synaptic scaling protocols (Fig. 1D) (ANOVA; PSD-95, p = 0.08; PSD-93, p = 0.16; SAP102, p = 0.14).
Changes in synaptic PSD-95 are not sufficient to drive homeostatic changes in AMPAR accumulation
PSD-95 is known to regulate the amplitude of evoked AMPA currents in hippocampal pyramidal neurons (El-Husseini et al., 2000; Schnell et al., 2002; Stein et al., 2003; Ehrlich and Malinow, 2004), suggesting that the bidirectional changes in synaptic accumulation of PSD-95 described above might drive the bidirectional changes in AMPAR accumulation during synaptic scaling. However, almost nothing is known about the role of the PSD-95 MAGUKs in regulating basal AMPA-mediated transmission in neocortical pyramidal neurons, and only one previous study has examined the effects of PSD-95 overexpression on neocortical AMPA minis and found no effect (Béïque and Andrade 2003). If increased synaptic PSD-95 drives scaling up in neocortical neurons, then directly increasing synaptic PSD-95 through overexpression should mimic the effects of activity blockade. To test this, we transfected neurons at low efficiency with PSD-95–EGFP and recorded mEPSCs 2–4 d after transfection. Despite a fourfold to fivefold increase in synaptic PSD-95 protein levels (Fig. 2A,B), there was no significant change in mean mEPSC amplitude (Fig. 2C–E). An inspection of the cumulative distribution of mEPSC amplitudes revealed that PSD-95 overexpression had a small effect toward the high end of the amplitude distribution (Fig. 2D) (p = 0.027, K–S test); this is in marked contrast to activity blockade, which scales up the entire amplitude distribution uniformly (see Figs. 4A, 6C). Other studies have also found only modest or no effects of acute PSD-95 overexpression on mEPSC amplitude (Béïque and Andrade 2003; Stein et al., 2003; Ehrlich and Malinow, 2004; Kelsch et al., 2008). Consistent with previous reports (Béïque and Andrade 2003; Stein et al., 2003; Ehrlich and Malinow, 2004), PSD-95 overexpression significantly increased mEPSC frequency (Fig. 2E) and also increased the length density of PSD-95 puncta (Fig. 2B), suggesting that PSD-95 overexpression might increase the number of functional synaptic sites.
Figure 2.

Overexpression of PSD-95 increases mEPSC frequency but not amplitude. A, Example images showing PSD-95 protein levels (red) in control dendrite and in dendrite transfected with PSD-95–EGFP (green) for 2–3 d. Scale bar, 5 μm. B, Quantification of fluorescent intensity and length density of PSD-95 puncta in control (untrans) and PSD-95-overexpressing neurons (PSD-95). ***p < 0.001. C, Example traces of mEPSC recordings from each condition. D, Cumulative distributions of mEPSC amplitude for control neurons or neurons overexpressing PSD-95. p = 0.027, K–S test. E, Mean mEPSC amplitude and frequency. Different from control, *p < 0.05. Number of neurons in each condition indicated on bars.
Figure 4.

Functional compensation between PSD-95 and PSD-93 during scaling up in young cortical neurons. A, Cumulative distributions of mEPSC amplitudes for control and activity blockade for nontransfected (top left), PSD-93 KD (top right), PSD-95 KD (middle left), PSD-95/SAP102 double KD (middle right), PSD-93/PSD-95 double KD (bottom left), and PSD-93/PSD-95/SAP102 triple KD (bottom right) conditions. Insets, Average mEPSC waveforms for each condition. B, Summary of mean mEPSC amplitude for each condition. Different from control, *p < 0.05, **p < 0.01, ***p < 0.001. n.s., Not significant. Number of neurons in each condition indicated on bars.
Figure 6.
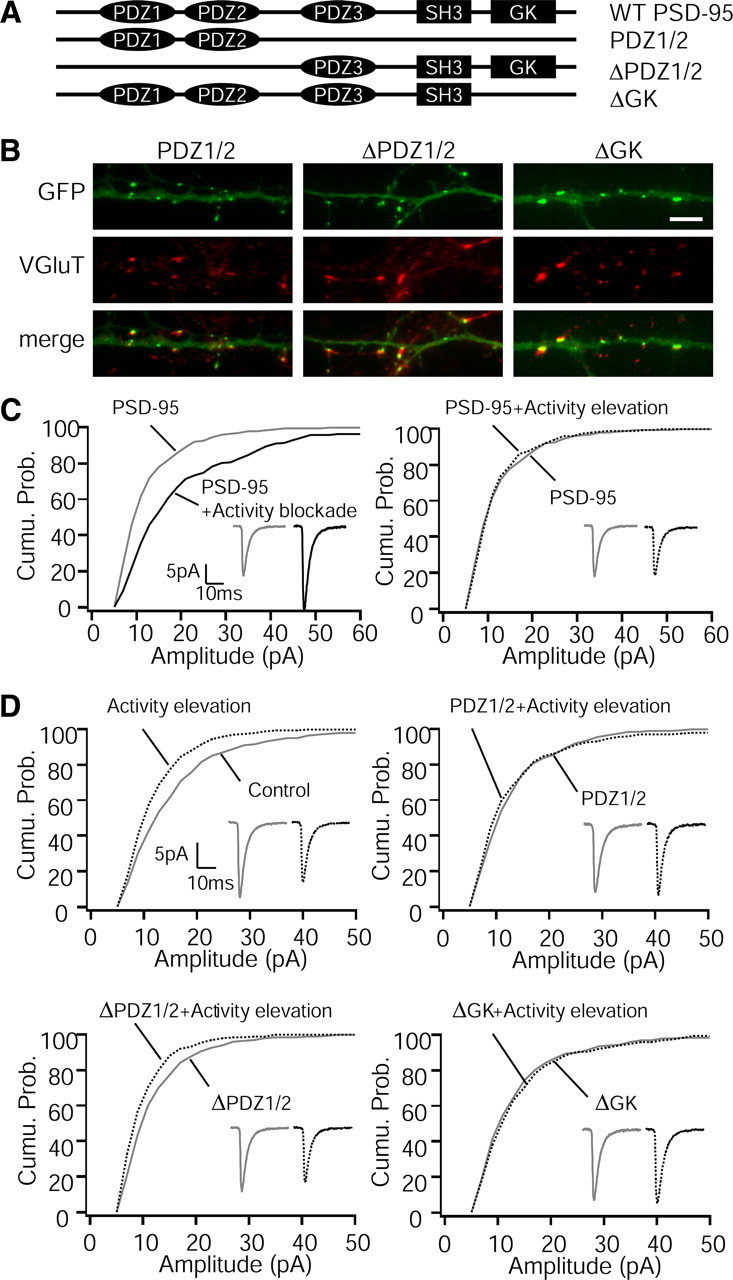
Scaling down is mediated through PSD-95 PDZ1/2 domain interactions in young cortical neurons. A, Diagram illustrates the domain structure of wild-type (WT) PSD-95 and the various PSD-95 mutant constructs tested here. B, Example images showing punctate expression patterns of three PSD-95 mutants tagged with GFP (green), VGluT (red), and merge. Scale bar, 5 μm. C, Cumulative distributions of mEPSC amplitudes in neurons overexpressing PSD-95. Left: n = 18 for control, 9 for activity elevation, p < 0.001, K–S test. Right: n = 18 for control, 10 for activity elevation, p = 0.39, K–S test. Insets, Average mEPSC waveforms for each condition. D, Cumulative distributions of mEPSC amplitudes for control and activity elevation, for nontransfected neurons (top left; n = 46 for control, 44 for activity elevation; p < 0.001, K–S test) or neurons transfected with the PDZ1/2 construct (top right; n = 12 for control, 7 for activity elevation; p = 0.14, K–S test), the ΔPDZ1/2 construct (bottom left; n = 20 for control, 18 for activity elevation; p < 0.001, K–S test), or the ΔGK construct (bottom right; n = 12 for control, 10 for activity elevation; p = 0.7, K–S test).
To examine the role of endogenous PSD-95 in regulating mEPSC amplitude, we used shRNAs directed against PSD-95 to knock down the endogenous protein; this shRNA and those used below for SAP102 and PSD-93 have been extensively characterized and are specific for their targets with no apparent off-target effects (Elias et al., 2006, 2008). Neurons were transfected at low efficiency with the PSD-95 hairpin and GFP, and 3 d after transfection, we examined PSD-95 protein levels in transfected neurons by immunostaining (Fig. 3A). The hairpin decreased the fluorescent intensity and density of PSD-95 puncta to 34.4 and 47.3% of control, respectively, giving an overall reduction in synaptic PSD-95 levels of ∼84% (Fig. 3B), whereas transfected with a scrambled hairpin had no effect on PSD-95 expression (intensity: scrambled, 111.5 ± 11.2% of control, p = 0.34; density: scrambled, 88.5 ± 7.6% of control, p = 0.32; n = 11 and 21 neurons, respectively). Knockdown of PSD-95 had no significant effect on baseline mEPSC amplitude (Fig. 3C–E). A similar analysis for PSD-93 and SAP102 revealed that no single knockdown had significant effects on mini amplitude (Figs. 3E, 4B). Furthermore, various combinations of double or triple knockdowns also failed to significantly affect baseline mEPSC amplitude (Figs. 3E, 4B). mEPSC frequency was unaffected by KD of PSD-95 (Fig. 3E) or PSD-93 alone (data not shown) but was significantly reduced by SAP102 KD and PSD-95/SAP102 double KD (Fig. 3E). Together, our data show that synaptic PSD-95 MAGUK levels have only subtle effects on baseline mEPSC amplitude in young neocortical neurons, and activity-dependent changes in synaptic MAGUK abundance are not sufficient to account for synaptic scaling of quantal amplitude.
Figure 3.

Knockdown of PSD-95 and SAP102 does not affect mEPSC amplitude. A, Example images showing PSD-95 protein levels (red) in control (untrans) dendrite and in dendrite transfected with shRNA directed against PSD-95 (green) for 3 d. Scale bar, 10 μm. B, Quantification of fluorescent intensity and length density of PSD-95 puncta in control and KD neurons. ***p < 0.001. C, Example raw traces of mEPSC recording from each condition. D, Cumulative distributions of mEPSC amplitude from control, PSD-95 KD, SAP102 KD, and PSD-95/SAP102 double KD neurons. E, Mean mEPSC amplitude and frequency. Different from control, **p < 0.01. Number of neurons in each condition indicated on bars.
Functional compensation among PSD-95 MAGUKs during scaling up in young cortical neurons
MAGUKs are thought to play both scaffolding and signaling roles within the PSD (Elias and Nicoll, 2007; Steiner et al., 2008; Xu et al., 2008). Although changes in synaptic MAGUK levels are not sufficient to drive synaptic scaling, one or more of the PSD-95 family of MAGUKS might be necessary for the regulated enhancement or reduction in mEPSC amplitude induced by synaptic scaling protocols. To test the role of PSD-95 MAGUKs in synaptic scaling in young neocortical neurons (DIV 7–9), we first examined the effects of acute KD on scaling up. Activity blockade significantly scaled up mEPSC amplitude in control, PSD-95 KD, or PSD-93 KD neurons to a similar extent (Fig. 4), suggesting that neither PSD-95 nor PSD-93 is essential for the expression of synaptic scaling up in young neurons. However, PSD-95 MAGUKs are known to compensate for each other under some conditions (Elias et al., 2006, 2007). To address the possible functional redundancy of PSD-95 MAGUKs during scaling up, we examined scaling up after double or triple knockdown of PSD-95 MAGUKs. Scaling up was intact in PSD-95/SAP102 double KD neurons (Fig. 4A), indicating that SAP102 is not required for scaling up. In contrast, double KD of PSD-95/PSD-93 or triple KD of PSD-95/PSD-93/SAP102 completely blocked scaling up (Fig. 4A,B). Together, these results show that either PSD-95 or PSD-93 are sufficient to support scaling up, suggesting that these two MAGUKs play redundant roles in scaling up and thus can compensate for each other. Consistent with this ability to compensate for each other, PSD-95 and PSD-93 puncta were highly colocalized (64.2 ± 3.8%, n = 11 neurons), supporting their ability to compensate for each other. Interestingly, even on a PSD-95/SAP102 double KD background, chronic activity blockade did not affect the synaptic accumulation of PSD-93 (for fluorescence intensity, activity blockade was 114.8 ± 9.1% of control, p = 0.16; for length density, activity blockade was 105.5 ± 6.4% of control; n = 17 and 20 neurons, respectively), underscoring the point that the role of PSD-93 in mediating scaling up was independent of changes in synaptic abundance.
PSD-95 is necessary for scaling down
It is unclear whether scaling up and scaling down of mEPSC amplitude is driven by similar or distinct signaling pathways (Ibata et al., 2008; Turrigiano, 2008; Goold and Nicoll, 2010), so next we tested the necessity of PSD-95 MAGUKs for scaling down. As reported previously (Turrigiano et al., 1998; Seeburg et al., 2008), elevating firing for 24 h (using picrotoxin or bicuculline) significantly reduced mEPSC amplitude in nontransfected neurons (Fig. 5). KD of SAP102 or PSD-93 (Fig. 5) did not block scaling down. Strikingly, KD of PSD-95 alone completely blocked scaling down; in fact, activity enhancement increased mEPSCs slightly in KD neurons (Fig. 5), and the double KD of PSD-95/SAP102 looked similar to KD of PSD-95 alone (Fig. 5). Thus, PSD-95 plays an essential role in scaling down, and, in contrast to scaling up, PSD-93 is unable to compensate for loss of PSD-95.
Figure 5.

PSD-95 is necessary for scaling down in young cortical neurons. Cumulative distributions of mEPSC amplitudes for control and activity elevation for nontransfected (top left), PSD-93 KD (top right), SAP102 KD (middle left), PSD-95 KD (middle right), and PSD-95/SAP102 double KD conditions (bottom left). Insets, Average mEPSC waveforms for each condition. Bottom right, Summary of mean mEPSC amplitude; activity elevation expressed as percentage of control for each condition. Different from control, *p < 0.05, **p < 0.01. Number of neurons in each condition indicated on bars.
PDZ1/2 domains of PSD-95 are required for scaling down
Our data suggest that interactions between PSD-95 and other proteins are necessary for the regulated reduction in quantal amplitude that underlies synaptic scaling down. This raised the possibility that a superabundance of PSD-95 might have dominant-negative effects on scaling down, by interfering with these protein–protein interactions. Consistent with our knockdown data, scaling down was completely blocked by PSD-95 overexpression, whereas scaling up was unaffected (Fig. 6C). To ask how critically dependent scaling down is on PSD-95 levels, we reduced the concentration of PSD-95 cDNA and found that elevating synaptic PSD-95 by as little as 1.5- to 2.5-fold was still able to block scaling down (scaling down condition was 96 ± 5% of control; n = 13 and 17 neurons, respectively; p = 0.66). Thus, unlike scaling up, scaling down is highly sensitive to the amount of synaptic PSD-95 and can be disrupted by either too much or too little of this MAGUK.
Distinct domains in PSD-95 bind to a variety of synaptic proteins and serve unique functional and structural roles (Fig. 6A). PSD-95 has three PDZ (PSD-95/Discs large/zona occludens-1) domains, the first two of which are important in binding to TARPs [and thus indirectly to AMPAR (Chen et al., 2000; Nicoll et al., 2006)], and Src homology 3 (SH3) and guanylate kinase (GK) domains, which are critical for long-term depression (LTD) induction and bind to proteins such as the A-kinase anchor protein AKAP and the spine-associated RapGAP protein SPAR (Pak et al., 2001; Xu et al., 2008; Bhattacharyya et al., 2009). Even mild PSD-95 overexpression blocks scaling down, so we were not able to use a KD-and-replace strategy to examine the dependence of scaling down on particular PSD-95 domains. As an alternative strategy, we tested the ability of different PSD-95 deletion mutants tagged with GFP to act as dominant negatives during scaling down (Fig. 6A). All three of these mutants exhibited punctate expression patterns and were colocalized with the presynaptic marker vesicular glutamate transporter (VGluT) (Fig. 6B). We found that truncated PSD-95 proteins missing only the GK domain (ΔGK) or the GK, SH3, and PDZ3 domains (PDZ1/2) were still able to block scaling down (Fig. 6D). In contrast, the reciprocal mutation missing only the first two PDZ domains (ΔPDZ1/2) was unable to block scaling down (Fig. 6D). These results indicate that the PDZ1/2 domains are necessary for the dominant-negative effects of PSD-95, suggesting that PSD-95 regulates scaling down through PDZ1/2-mediated protein–protein interactions. This differentiates scaling down from other forms of synaptic plasticity that also reduce AMPAR number, such as LTD, that rely on the SH3 and GK domains for their effects (Xu et al., 2008; Bhattacharyya et al., 2009); interestingly, a truncated form of PSD-95 that contains the SH3–GK domains can act as a dominant negative for LTD, suggesting that scaling down and LTD rely on distinct protein–protein interactions between PSD-95 and downstream signaling molecules.
PSD-95 binds directly to TARP family members through its PDZ1/2 domains (Chen et al., 2000; Nicoll et al., 2006). If this interaction is important for scaling down, then overexpression of a TARP might also disrupt scaling down. To test this, we overexpressed γ3 [a TARP highly enriched in cortical neurons (Sager et al., 2009)], which significantly increased TARP expression both synaptically and extrasynaptically (Fig. 7B). Overexpression of γ3 significantly reduced baseline mEPSC amplitude and completely blocked (or occluded) scaling down (Fig. 7A,C). Although the effect of γ3 overexpression on AMPA transmission has not previously been described, overexpression of γ8 similarly reduces mEPSC amplitude without affecting the total number of surface AMPAR (Milstein and Nicoll, 2009). The reduction in baseline mEPSC amplitude suggests that excess γ3 binds to synaptic MAGUKs and thus reduces binding of endogenous AMPAR/TARP complexes and could disrupt scaling down by providing an excess of TARP/MAGUK complexes that are not bound to AMPAR. Although the precise mechanism by which γ3 overexpression disrupts scaling down remains unclear, these data support the interpretation that PDZ1/2 interactions are critical for the regulated reduction in quantal amplitude that underlies scaling down.
Figure 7.

Overexpression of γ3 TARP reduces baseline mEPSC amplitude and blocks scaling down in young cortical neurons. A, Cumulative distributions of mEPSC amplitudes for control and activity elevation, for nontransfected neurons (left) and neurons transfected with γ3 (right). B, Example immunostaining images showing the expression of TARPs for nontransfected (untrans) and γ3 overexpressing (OE) neurons. Scale bar, 5 μm. C, Summary of mean mEPSC amplitude for each condition. Different from control, *p < 0.05, **p < 0.01. n.s., Not significant. Number of neurons in each condition indicated on bars.
The requirement of synaptic scaling up for PSD-95 depends on neuronal age
PSD-95 expression progressively increases during development in vivo (Sans et al., 2000) and is important for regulating mEPSC frequency in hippocampal neurons from mature, but not young, animals (Béïque et al., 2006; Elias et al., 2006, 2008), suggesting that the synaptic function(s) of PSD-95 may be developmentally regulated. In contrast to our finding that PSD-95 is not essential for scaling up in young (DIV 7–9) cortical neurons, a recent study using older hippocampal neurons (DIV 18–21) found that PSD-95 KD blocked the enhancement of AMPAR accumulation during activity blockade (Noritake et al., 2009). To determine whether the role of PSD-95 in regulating baseline transmission and synaptic scaling changes with time, we first examined the expression profile of PSD-95 in young and older cortical neurons. Both the density and intensity of PSD-95 puncta colocalized with VGluT increased significantly between DIV 7–9 and DIV 14–16 (Fig. 8A). In keeping with the increase in puncta density, we observed a substantial increase in mEPSC frequency in older cultures (young, 0.80 ± 0.14 Hz, n = 26; older, 3.36 ± 0.42 Hz; n = 25; p < 0.001). Together, these data suggest that the number of PSD-95-containing excitatory synapses is progressively increased with time in vitro, as is the amount of PSD-95 at individual synaptic puncta.
Figure 8.

PSD-95 is necessary for both scaling up and down in older cultures. A, Left, Sample images showing changes of PSD-95 expression at different neuronal age in cortical neurons. Right, Quantification of fluorescent intensity and density of PSD-95 puncta during DIV 7–9 and DIV 14–16. Different from DIV 7–9, *p < 0.05, ***p < 0.001. Scale bar, 5 μm. B, Mean mEPSC frequency for nontransfected, PSD-95 overexpression, and PSD-95 KD. Different from nontransfected, **p < 0.01, ***p < 0.001. C, Quantification of fluorescent intensity of PSD-95 puncta for control, activity blockade, and activity elevation in older cultures (DIV14–16). *p < 0.05, ***p < 0.001. D, Mean mEPSC amplitude for each condition. Different from control, *p < 0.05, **p < 0.01, ***p < 0.001. Number of neurons in each condition indicated on bars.
Synaptic PSD-95 levels in older neurons are bidirectionally regulated by synaptic scaling protocols (Fig. 8C), as they are in younger neurons (Fig. 1). Next, we asked whether the dependence of baseline mEPSC amplitude and synaptic scaling on PSD-95 is age dependent. In old as in young neurons, overexpression of PSD-95 had no significant affect on mEPSC amplitude (Fig. 8D). In contrast, KD induced a small but significant (10.4%) reduction in mEPSC amplitude in old (Fig. 8D) (p < 0.05) but not in young neurons (Figs. 3E, 4B). The degree of KD in older neurons (89%) was comparable with that achieved in younger neurons (84%). Also in contrast to young neurons in which KD of PSD-95 did not reduce mEPSC frequency (Fig. 3E), in old neurons, KD and overexpression of PSD-95 bidirectionally regulated mEPSC frequency (Fig. 8B). Thus, PSD-95 plays a more important role in regulating baseline mEPSC properties in older than in younger neurons, but modulation of synaptic PSD-95 levels was not sufficient to reproduce synaptic scaling at either age.
Next we asked whether PSD-95 is critical for synaptic scaling in older neurons. In older neurons as for young neurons (Fig. 6C), overexpression of PSD-95 preferentially blocked scaling down and had no effect on scaling up (Fig. 8D). Surprisingly, in marked contrast to young neurons, both scaling up and scaling down were completely blocked by KD of PSD-95 (Fig. 8D), indicating that PSD-95 is essential for bidirectional expression of synaptic scaling in older neurons. Because KD but not overexpression blocks scaling up in older neurons, we were able to perform a rescue experiment to control for off-target effects of the hairpin; PSD-95 overexpression was able to rescue scaling up in older PSD-95 KD neurons (Fig. 8D). These data indicate that the molecular mechanisms of synaptic scaling up change during development to become exclusively dependent on PSD-95. The differential sensitivity of scaling up and down to overexpression of PSD-95, however, suggests that even in older neurons PSD-95 plays distinct roles in scaling up and down.
Discussion
The molecular changes within the postsynaptic density that lead to altered AMPAR abundance during synaptic scaling remain essentially unknown. Here we investigated the possibility that activity-dependent changes in the abundance of the PSD-95 MAGUK family of scaffold proteins drive bidirectional changes in AMPAR accumulation during synaptic scaling. We found that, although synaptic PSD-95 and SAP102 (but not PSD-93) abundance was bidirectionally regulated by activity, these changes were not sufficient to drive synaptic scaling. Although not sufficient, the PSD-95 MAGUKs were necessary for the expression of synaptic scaling, but scaling up and down were differentially dependent on PSD-95 and PSD-93. Scaling down was completely blocked by reduced or enhanced PSD-95, through a mechanism that depended on the PDZ1/2 domains. In contrast, scaling up could be supported by either PSD-95 or PSD-93 in a manner that depended on neuronal age and was unaffected by a superabundance of PSD-95. Together, our data suggest that scaling up and down of quantal amplitude is not driven by changes in the synaptic abundance of PDS-95 MAGUKs but rather that the PSD-95 MAGUKs serve as critical synaptic organizers that mediate the regulated accumulation and loss of synaptic AMPAR during homeostatic plasticity.
The factors that regulate the number of AMPAR at synapses under basal conditions and during various forms of synaptic plasticity are poorly understood. One proposal is that the number of slots, or AMPAR binding sites, at synapses directly determines the number of AMPAR (Malinow and Malenka, 2002). MAGUKs are putative slot proteins widely believed to play a pivotal role in determining the number of synaptic AMPAR (Schnell et al., 2002; Colledge et al., 2003; Nicoll et al., 2006; Elias and Nicoll, 2007) and have been implicated in mediating several forms of synaptic plasticity (Migaud et al., 1998; Ehrlich and Malinow, 2004; Carlisle et al., 2008; Xu et al., 2008; Noritake et al., 2009). We found that MAGUKs are differentially regulated by chronic activity manipulations. Synaptic PSD-95 accumulation was increased by activity blockade and reduced by activity elevation, consistent with previous studies (Kim et al., 2007; Noritake et al., 2009) but at odds with another, which found bidirectional regulation but in the opposite direction (Ehlers, 2003); the reason for this discrepancy is not clear. SAP102 was also bidirectionally regulated by chronic changes in activity, whereas synaptic PSD-93 levels were unaffected. Thus, synaptic scaling protocols produce complex changes in the composition of the postsynaptic density.
Manipulations of PSD-95 can dramatically alter evoked synaptic transmission (El-Husseini et al., 2000; Béïque and Andrade, 2003; Stein et al., 2003; Ehrlich and Malinow, 2004; Elias et al., 2006; Schlüter et al., 2006; Xu et al., 2008), but we found that PSD-95 overexpression or KD had only moderate effects on quantal amplitude, in keeping with previous reports (El-Husseini et al., 2000; Stein et al., 2003; Ehrlich and Malinow, 2004; Béïque and Andrade, 2003; Gray et al., 2006; Kelsch et al., 2008). Acute knockdown of various combinations of PSD-95/PSD-93/SAP102 also had little or modest effects on mEPSC amplitude, suggesting that basal mEPSC amplitude is tightly regulated and is not critically dependent on variations in MAGUK protein levels. In contrast to the modest effects of PSD-95 MAGUK KD on quantal amplitude, mEPSC frequency was strongly influenced by manipulations of PSD-95 and SAP102, as reported previously for PSD-95 (Sans et al., 2000; Béïque et al., 2006; Elias et al., 2006, 2008). We found that SAP102 KD reduced mEPSC frequency in young neurons, whereas PSD-95 KD became important in regulating mEPSC frequency in more mature neocortical neurons, when synaptic PSD-95 levels were higher. Our data on quantal amplitude and frequency are thus consistent with the model that synaptic PSD-95/SAP102 abundance modulates the number of functional AMPA-containing synapses rather than directly determining the number of receptors at individual synaptic sites (Béïque and Andrade, 2003); this would account for why PSD-95 has profound effects on evoked transmission but modest effects on quantal amplitude. Together, our data show that activity-dependent changes in synaptic PSD-95 MAGUK abundance are not sufficient to mediate the changes in mEPSC amplitude that characterize synaptic scaling.
Although changes in synaptic PSD-95 abundance do not drive synaptic scaling, our data demonstrate that PSD-95 MAGUKs play an essential and complex scaffolding/signaling role in homeostatic plasticity. Little is currently known about the degree to which scaling up and down use the same or distinct signaling pathways. Both occur through cell-autonomous changes in somatic calcium and involve changes in calcium/calmodulin-dependent kinase kinase and calcium/calmodulin-dependent kinase IV activation and transcriptional regulation (Ibata et al., 2008; Goold and Nicoll, 2010). In contrast, some other signaling or AMPAR trafficking elements such as Arc and TNFα appear to be critical only for scaling up (Shepherd et al., 2006; Stellwagen and Malenka, 2006). Our data demonstrate that PSD-95 MAGUKs play central but distinct roles in scaling up and down. In young neurons, PSD-95 is essential for activity-dependent scaling down of mEPSCs, whereas scaling up can be supported by either PSD-95 or PSD-93. PSD-95 and PSD-93 have ∼71% sequence homology and the same domain structure but do differ in the structure of the PDZ1 domain and so could interact preferentially with different binding partners (Fiorentini et al., 2009); our data suggest that scaling down relies on protein–protein interactions unique to PSD-95, whereas in young neurons scaling up relies on protein–protein interactions common to both MAGUKs. Adding to this complexity, in older neurons, PSD-93 can no longer compensate for loss of PSD-95 during scaling up, indicating for the first time that the dependence of synaptic scaling on particular signaling elements changes with neuronal age. Because we are taking a KD rather than knockout approach, we cannot completely exclude the possibility that the differential dependence of scaling up and down on PSD-95 in young neurons is attributable to a difference in the sensitivity of these two pathways to the small amount of PSD-95 that remains after KD, but the observations that (1) the remaining PSD-95 cannot support scaling up when PSD-93 is also knocked down, (2) in older neurons a similar degree of PSD-95 KD is able to block both scaling up and down, and (3) that a superabundance of PSD-95 blocks scaling down but not scaling up suggest that this explanation is unlikely. Rather, we propose that the pathways that lead to upregulation and downregulation of synaptic AMPAR abundance during synaptic scaling rely on distinct PSD-95 (and/or PSD-93)-mediated protein–protein interactions.
Interestingly, both KD and overexpression of PSD-95 can prevent scaling down, whereas scaling up is unaffected by overexpression in both young and older neurons. Scaling down thus appears to depend on maintenance of a critical level of synaptic PSD-95, presumably because too much PSD-95 interferes with downstream signaling elements necessary for the expression of scaling down. PSD-95 can interact through its GK domain with SPAR, a synaptic scaffolding protein known to be critical for scaling down (Pak et al., 2001; Seeburg and Sheng, 2008; Seeburg et al., 2008), and LTD is dependent on the SH3–GK domains of PSD-95, which can act as a dominant negative for LTD (Xu et al., 2008). In contrast, we found that the SH3–GK domains are not essential for the dominant-negative effects of PSD-95, indicating that PSD-95 acts to regulate scaling down through a pathway that is independent of SPAR and distinct from that mediating LTD. Instead, scaling down was critically dependent on the PDZ1/2 domains of PSD-95, which are known to physically interact with numerous scaffolding and signaling proteins, including the TARPs, which link PSD-95 to AMPAR (Kim and Sheng, 2004; Nicoll et al., 2006). Overexpression of γ3 [a TARP highly expressed in neocortex (Sager et al., 2009)] also blocked scaling down, indicating that a superabundance of either the PSD-95 PDZ1/2 domains or a complementary binding partner disrupt scaling down. This suggests that PSD-95 mediates scaling down by targeting the PSD-95–TARP interaction to reduce synaptic AMPAR stability.
Although PSD-95 is emerging as a critical player in several important forms of synaptic plasticity, different forms of plasticity appear to rely on different domains of PSD-95. Whereas LTD depends on the SH3–GK domains, scaling down depends on the PDZ1/2 domains. Whereas scaling down is critically dependent on PSD-95, scaling up can be supported by either PSD-95 or PSD-93 and is unaffected by PSD-95 overexpression. Similarly, enhancement of synaptic strength during long-term potentiation (LTP) and scaling up are differentially dependent on PSD-95; KD does not block LTP (Ehrlich et al., 2007) but does block scaling up in older neurons, whereas overexpression occludes LTP (Stein et al., 2003; Ehrlich and Malinow, 2004) but does not affect scaling up. Together with these studies on Hebbian forms of plasticity, our data show that PSD-95 is a centrally placed scaffold/signaling molecule that, by virtue of its many interaction domains, can act as a platform to simultaneously mediate multiple mechanistically distinct forms of Hebbian and homeostatic plasticity.
Footnotes
This work was supported by National Institutes of Health Grant NS 36853. We thank L. Wang and B. Baxter for technical assistance and R. Nicoll, B. Sabatini, M. Fukata, D. Bredt, and W. Xu for kindly providing DNA constructs.
References
- Béïque JC, Andrade R. PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J Physiol. 2003;546:859–867. doi: 10.1113/jphysiol.2002.031369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béïque JC, Lin DT, Kang MG, Aizawa H, Takamiya K, Huganir RL. Synapse-specific regulation of AMPA receptor function by PSD-95. Proc Natl Acad Sci U S A. 2006;103:19535–19540. doi: 10.1073/pnas.0608492103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Biou V, Xu W, Schlüter O, Malenka RC. A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat Neurosci. 2009;12:172–181. doi: 10.1038/nn.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlisle HJ, Fink AE, Grant SG, O'Dell TJ. Opposing effects of PSD-93 and PSD-95 on long-term potentiation and spike-timing dependent plasticity. J Physiol. 2008;586:5885–5900. doi: 10.1113/jphysiol.2008.163469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- Cingolani LA, Thalhammer A, Yu LM, Catalano M, Ramos T, Colicos MA, Goda Y. Activity-dependent regulation of synaptic AMPA receptor composition and abundance by beta3 integrins. Neuron. 2008;58:749–762. doi: 10.1016/j.neuron.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Klein M, Rumpel S, Malinow R. PSD-95 is required for activity-driven synapse stabilization. Proc Natl Acad Sci U S A. 2007;104:4176–4181. doi: 10.1073/pnas.0609307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- Elias GM, Nicoll RA. Synaptic trafficking of glutamate receptors by MAGUK scaffolding proteins. Trends Cell Biol. 2007;17:343–352. doi: 10.1016/j.tcb.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52:307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Elias GM, Elias LA, Apostolides PF, Kriegstein AR, Nicoll RA. Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD-95 underlies synapse development. Proc Natl Acad Sci U S A. 2008;105:20953–20958. doi: 10.1073/pnas.0811025106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentini M, Nielsen AK, Kristensen O, Kastrup JS, Gajhede M. Structure of the first PDZ domain of human PSD-93. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2009;65:1254–1257. doi: 10.1107/S1744309109043267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainey MA, Hurvitz-Wolff JR, Lambo ME, Turrigiano GG. Synaptic scaling requires the GluR2 subunit of the AMPA receptor. J Neurosci. 2009;29:6479–6489. doi: 10.1523/JNEUROSCI.3753-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold CP, Nicoll RA. Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron. 2010;68:512–528. doi: 10.1016/j.neuron.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray NW, Weimer RM, Bureau I, Svoboda K. Rapid redistribution of synaptic PSD-95 in the neocortex in vivo. PLoS Biol. 2006;4:e370. doi: 10.1371/journal.pbio.0040370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Keith D, El-Husseini A. Excitation control: balancing PSD-95 function at the synapse. Front Mol Neurosci. 2008;1:4. doi: 10.3389/neuro.02.004.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsch W, Lin CW, Lois C. Sequential development of synapses in dendritic domains during adult neurogenesis. Proc Natl Acad Sci U S A. 2008;105:16803–16808. doi: 10.1073/pnas.0807970105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Futai K, Jo J, Hayashi Y, Cho K, Sheng M. Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95. Neuron. 2007;56:488–502. doi: 10.1016/j.neuron.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Milstein AD, Nicoll RA. TARP modulation of synaptic AMPA receptor trafficking and gating depends on multiple intracellular domains. Proc Natl Acad Sci U S A. 2009;106:11348–11351. doi: 10.1073/pnas.0905570106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newpher TM, Ehlers MD. Glutamate receptor dynamics in dendritic microdomains. Neuron. 2008;58:472–497. doi: 10.1016/j.neuron.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Tomita S, Bredt DS. Auxiliary subunits assist AMPA-type glutamate receptors. Science. 2006;311:1253–1256. doi: 10.1126/science.1123339. [DOI] [PubMed] [Google Scholar]
- Noritake J, Fukata Y, Iwanaga T, Hosomi N, Tsutsumi R, Matsuda N, Tani H, Iwanari H, Mochizuki Y, Kodama T, Matsuura Y, Bredt DS, Hamakubo T, Fukata M. Mobile DHHC palmitoylating enzyme mediates activity-sensitive synaptic targeting of PSD-95. J Cell Biol. 2009;186:147–160. doi: 10.1083/jcb.200903101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak DT, Yang S, Rudolph-Correia S, Kim E, Sheng M. Regulation of dendritic spine morphology by SPAR, a PSD-95-associated RapGAP. Neuron. 2001;31:289–303. doi: 10.1016/s0896-6273(01)00355-5. [DOI] [PubMed] [Google Scholar]
- Pozo K, Goda Y. Unraveling mechanisms of homeostatic synaptic plasticity. Neuron. 2010;66:337–351. doi: 10.1016/j.neuron.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt KG, Watt AJ, Griffith LC, Nelson SB, Turrigiano GG. Activity-dependent remodeling of presynaptic inputs by postsynaptic expression of activated CaMKII. Neuron. 2003;39:269–281. doi: 10.1016/s0896-6273(03)00422-7. [DOI] [PubMed] [Google Scholar]
- Pratt KG, Taft CE, Burbea M, Turrigiano GG. Dynamics underlying synaptic gain between pairs of cortical pyramidal neurons. Dev Neurobiol. 2008;68:143–151. doi: 10.1002/dneu.20577. [DOI] [PubMed] [Google Scholar]
- Sager C, Tapken D, Kott S, Hollmann M. Functional modulation of AMPA receptors by transmembrane AMPA receptor regulatory proteins. Neuroscience. 2009;158:45–54. doi: 10.1016/j.neuroscience.2007.12.046. [DOI] [PubMed] [Google Scholar]
- Sans N, Petralia RS, Wang YX, Blahos J, 2nd, Hell JW, Wenthold RJ. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci. 2000;20:1260–1271. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter OM, Xu W, Malenka RC. Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron. 2006;51:99–111. doi: 10.1016/j.neuron.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci U S A. 2002;99:13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg DP, Sheng M. Activity-induced Polo-like kinase 2 is required for homeostatic plasticity of hippocampal neurons during epileptiform activity. J Neurosci. 2008;28:6583–6591. doi: 10.1523/JNEUROSCI.1853-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg DP, Feliu-Mojer M, Gaiottino J, Pak DT, Sheng M. Critical role of CDK5 and Polo-like kinase 2 in homeostatic synaptic plasticity during elevated activity. Neuron. 2008;58:571–583. doi: 10.1016/j.neuron.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lu W, Milstein AD, Nicoll RA. The stoichiometry of AMPA receptors and TARPs varies by neuronal cell type. Neuron. 2009;62:633–640. doi: 10.1016/j.neuron.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein V, House DR, Bredt DS, Nicoll RA. Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J Neurosci. 2003;23:5503–5506. doi: 10.1523/JNEUROSCI.23-13-05503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner P, Higley MJ, Xu W, Czervionke BL, Malenka RC, Sabatini BL. Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron. 2008;60:788–802. doi: 10.1016/j.neuron.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Sturgill JF, Steiner P, Czervionke BL, Sabatini BL. Distinct domains within PSD-95 mediate synaptic incorporation, stabilization, and activity-dependent trafficking. J Neurosci. 2009;29:12845–12854. doi: 10.1523/JNEUROSCI.1841-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Wierenga CJ, Ibata K, Turrigiano GG. Postsynaptic expression of homeostatic plasticity at neocortical synapses. J Neurosci. 2005;25:2895–2905. doi: 10.1523/JNEUROSCI.5217-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga CJ, Walsh MF, Turrigiano GG. Temporal regulation of the expression locus of homeostatic plasticity. J Neurophysiol. 2006;96:2127–2133. doi: 10.1152/jn.00107.2006. [DOI] [PubMed] [Google Scholar]
- Xu W, Schlüter OM, Steiner P, Czervionke BL, Sabatini B, Malenka RC. Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron. 2008;57:248–262. doi: 10.1016/j.neuron.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]