

Abstract
Inflammation of the lower urinary tract occurs frequently in people. The causes remain obscure, with the exception of urinary tract infection. Animal models have proven useful for investigating and assessing mechanisms underlying symptoms associated with lower urinary tract inflammation and options for suppressing these symptoms. This review will discuss various animal models of lower urinary tract inflammation, including feline spontaneous (interstitial) cystitis, neurogenic cystitis, autoimmune cystitis, cystitis induced by intravesical instillation of chemicals or bacterial products (particularly lipopolysaccharide or LPS), and prostatic inflammation initiated by transurethral instillation of bacteria. Animal models will continue to be of significant value in identifying mechanisms resulting in bladder inflammation, but the relevance of some of these models to the causes underlying clinical disease is unclear. This is primarily because of the lack of understanding of causes of these disorders in people. Comparative and translational studies are required if the full potential of findings obtained with animal models to improve prevention and treatment of lower urinary tract inflammation in people is to be realized.
Inflammation of the lower urinary tract is relatively common and is most often the result of urinary tract infection. Urinary tract infection is the second most common infectious disorder of humans and results in 8.3 million doctor visits annually (1). All but about 5% of these patients exhibit symptoms of inflammation, including pain, urgency, and increased frequency (2). Painful Bladder Syndrome/Interstitial Cystitis (PBS/IC) is a poorly characterized syndrome of unknown cause(s) characterized by pain, increased urgency, and increased frequency. A recent review of this disorder found studies estimating a prevalence ranging from less than 1% to 11% of all women above the age of 19 (3), and it is thought that PBS/IC affects more than one million patients in the US alone (4). The diagnosis of PBS/IC is typically made by exclusion of other causes of symptoms (including infection), and inflammation of the bladder is confirmed by biopsy in many patients diagnosed with PBS/IC (5,6). Pain characteristically accompanies inflammation of the lower urinary tract. Inflammation is also commonly identified in prostate biopsy samples obtained from patients with benign prostatic hyperplasia (BPH) (7-9). Although pain is not consistently correlated with the presence of BPH, a subset of these patients complains of pain associated with the lower urinary tract (10,11). While a variety of neoplastic or parasitic disorders may occasionally cause inflammation of the lower urinary tract, these disorders occur far less frequently.
Clearly, it would be unethical and immoral to perform mechanistic studies using human subjects, but investigators should keep in mind the influence genetics and environment may have on results. Animal models (that typically use inbred stains of rodents) of clinical disorders resulting in lower urinary tract inflammation suffer from many of the same limitations associated with clinical research focused on human (outbred) subjects. Bacterial cystitis can be induced in rodents, but the response to infection varies widely dependent on the strain of the host and the particular bacteria used to induce infection (12,13). When using animal models to investigate mechanisms resulting in lower urinary tract inflammation, one must be aware of the impact of genetic differences among strains and species on results obtained. Although it is perfectly reasonable to assume that the biological response of the lower urinary tract of animals to a particular infectious or irritating stimulus is consistent and reproducible and accurately reflects the physiology of that species, genetic differences within and between species may render the direct relevance of the findings to specific human patients suspect. The use of animal models to investigate lower urinary tract inflammation is also complicated by the nearly complete lack of understanding of the underlying causes and pathogenesis of PBS/IC. In the absence of a clear understanding of the factors resulting in the onset of this disorder, it is difficult to state with absolute certainty that results of studies using animal models recapitulate events resulting in PBS/IC.
Thus, results of experiments using animal models of lower urinary tract inflammation should be viewed as having greatest relevance to mechanisms of pathophysiology leading to inflammation of the lower urinary tract. This does not mean that animal models of lower urinary tract inflammation are without value. Results of experiments performed with animal models of lower urinary tract inflammation have clearly provided the basis for development of therapies that relieve symptoms in some patients. Some examples of this include intravesical instillation of resiniferatoxin or botulinin toxin A and development of various antimicrobial agents. Resiniferatoxin is a potent analogue of capsaicin that desensitizes the transient receptor potential vanilloid type 1 (TRPV1), a cation channel that is thought to play a key role in transmitting nociceptive sensation from the bladder (14,15). Intravesical resiniferatoxin decreases spinal c-fos expression and increases bladder filling volume prior to stimulation of reflex micturition in rats with chronic inflamed urinary bladders (16). Injection of botulinum toxin type A into the bladder walls of rats (17) or intravesical instillation of this compound into the bladders of rats (18) helped to normalize bladder function in the presence of chemical cystitis or partial outflow obstruction, respectively. However, the fact that not all patients consistently respond to these treatments simply reinforces the complexity of the pathogenesis of inflammation of the lower urinary tract.
The purpose of this review is to describe animal models of lower urinary tract inflammation and to discuss the positive aspects and limitations of these models. Table 1 lists animal models of lower urinary tract inflammation and the references cited for each in this review.Models that will be discussed include feline spontaneous (interstitial) cystitis, neurogenic cystitis, autoimmune cystitis, and cystitis induced by intravesical instillation of chemicals or bacterial products (particularly lipopolysaccharide or LPS). A recently-described model of prostatic inflammation initiated by transurethral instillation of bacteria will also be described.
Table 1.
Animal Models of Lower Urinary Tract (LUT) Inflammation
| Cause of LUT Inflammation |
Species | Advantages | Disadvantages | References |
|---|---|---|---|---|
| Spontaneous Inflammation |
Cats | Develops without external intervention |
Etiology remains unknown | 19 – 28, 35, 36, 39 |
| Neurogenic Inflammation |
Mice, Rats, Guinea Pigs |
Viral model results in inflammation without external manipulation of the bladder |
Difficult to be certain that inflammation solely arises from activation of nerves |
41 – 50, 54 – 59, 65, 71, 73 – 75, 78 80, 82 - 88 |
| Autoimmunity | Mice, Rats, Guinea Pigs |
IC/PBS patients have a high co- morbidity of immune-related disease |
Target(s) of immune response in bladder remain uncertain |
94, 98 - 103 |
| Intravesical Instillation or Urinary Excretions of Irritants |
Mice, Rats | Can control severity and duration of inflammation |
Causes indiscriminate damage to glycosaminoglycan layer, urothelium, and wall of bladder by multiple mechanisms |
16, 17, 104, 105, 119 – 136, |
| Intravesical Instillation of Bacterial Products |
Mice, Rats | Bacterial infection occurs naturally in humans |
Difficult to recapitulate genetic and environmental factors influencing response of bladder |
12, 13, 106, 107 – 115, 118, |
| Transurethral Instillation of Bacteria into the Prostate |
Mice | Consistently produces acute or chronic inflammation |
Role of infection remains unclear in BPH, prostatic pain, or prostatic inflammation in patients |
150 |
Feline Spontaneous (Interstitial) Cystitis
It has long been recognized that some domestic cats develop bladder disease that is characterized by increased urinary frequency, urgency, and hematuria (19). Early studies suggested that this disorder may be due to infection of the bladder by calicivirus or herpesvirus (20-22). However, despite the ability of these investigators to experimentally replicate the disorder by intravesical viral instillation, other investigators have been unable to duplicate these findings, and most authorities do not believe that there is an underlying viral cause of this syndrome in cats (reviewed in 23). Similarly, although bacteria have been identified in the urine from some of these cats, careful study demonstrated that bacteria could be cultured from the urine in a very small percentage (possibly less than 1%) of these cats (24-26).
In 1993, Buffington et al. reported that, similar to patients with IC/PBS, these cats excrete less glycosaminoglycan in the urine and further suggested that this disorder in cats was analogous to IC/PBS in humans (27). Subsequent studies using cats with this disorder demonstrated numerous similarities to patients with IC/PBS, including increased permeability of the bladder wall (28). There is significant interest in communication between the urothelium (epithelial cells lining the bladder) and afferent innervation of the bladder, and it has been demonstrated that urothelial cells express receptors for many neurotransmitters, as well as having the capacity to produce compounds that mediate pain and inflammation (reviewed in 29). Substance P is a neurotransmitter that plays a key role in transmission of painful sensations (30), and nerve growth factor is a neurotrophin that also stimulates nociception in peripheral tissues (31). It has been reported that urothelial cells from the bladders of cats with spontaneous (or interstitial) cystitis express increased amounts of substance P and nerve growth factor (32), as has been observed in bladder biopsies from humans with IC/PBS (33,34). Similarly, alterations in the profile of expression of purinergic receptors and release of ATP by urothelial cells has been described in both cats with spontaneous cystitis (35,26) and humans with IC/PBS (37,38). Both of these substances have the capacity to stimulate nociceptive input and contribute to inflammation. A more comprehensive analysis of similarities between feline spontaneous (interstitial) cystitis and IC/PBS in humans has been published (39).
Feline spontaneous (interstitial) cystitis has the distinct advantage of occurring in the absence of administration of exogenous substances that injure the bladder. While many similarities between cats with this disorder and humans with IC/PBS have been identified, it is still unclear whether or not this disorder in cats is analogous to IC/PBS in humans, if for no other reason than the fact that the underlying causes of, and mechanisms resulting in, IC/PBS are have not been definitively identified. Thus, as with many other animal models of human disease, studying cats with spontaneous cystitis of unknown cause(s) can provide clues to the processes that result in, or cause sequela to, IC/PBS in humans, but it remains difficult to accept the concept that this model replicates all aspects of IC/PBS in humans.
Neurogenic Cystitis
Neurogenic Cystitis
Neurogenic inflammation has classically been considered the result of antidromic stimulation of afferent sensory nerves (thought to be primarily unmyelinated C fibers or lightly myelinated Aδ fibers in the bladder) resulting in release of neurotransmitters that cause pain and inflammation (40,41). This phenomenon has been reported to affect several organs, including the bladder, skin, gut, lungs, airways, eyes, and joints (42-45). Neuropeptides that mediate neurogenic inflammation include substance P, neurokinin A, and calcitonin gene-related peptide (CGRP), and these substances are released by nerve fibers that express transient receptor vanilloid 1 (TRPV1) channel (46). Interestingly, it has also been reported that neurons expressing both α1-adrenoceptors and TRPV1 are present in lumbar and sacral dorsal root ganglia and that exposure of these neurons to phenylephrine stimulated release of substance P, providing strong evidence that the bladder also receives afferent innervation from the sympathetic nervous system (47). Other investigators have reported that sympathetic nerves have the capacity to synthesize and release substance P, neurokinin A, and CGRP, and it is widely accepted that the sympathetic nervous system participates in neurogenic inflammation (48-50).
The TRPV1 channel is activated by capsaicin or resiniferatoxin, resulting in death or desensitization of the afferent fiber, and desensitization of afferent fibers is thought to be the mechanism underlying pain relief provided by intravesicular instillation of these compounds in patients with IC/PBS (14,15,51). Unfortunately, initial contact of nerve fibers with these substances is accompanied by intense pain, and nerve fibers regenerate or regain sensitivity over time, limiting efficacy in some patients and duration of relief of pain subsequent to these treatments in most patients (52,53).
Release of substance P, neurokinin A, and CGRP has been associated with smooth muscle contraction, vasodilation, increased vascular permeability, and facilitated neurotransmitter release from intramural nerves in the bladder wall (54,55). Some of these effects are a direct result of interaction with specific receptors for these neuropeptides on the vasculature or smooth muscle, but many are the result of stimulation of release of prostaglandins, bradykinin, and cytokines from leukocytes and other cells. There is a reciprocal release of mediators of pain and inflammation in response to neuropeptides entailing subsequent further release of neuropeptides in response to these noxious substances, suggesting a circular, self-perpetuating process that leads to persistence of pain and inflammation after the initial insult has resolved (56-59). It is assumed that, under normal circumstances, homeostatic mechanisms act to limit this cyclical process, but it is possible that suppression or loss of homeostatic mechanisms could result in chronic pain and inflammation of the bladder or other organs.
Mast cells appear to play a particularly crucial role in neurogenic inflammation. Mast cells have received particular attention in studies of IC/PBS in humans because they are present in increased numbers within the bladder wall in at least a subset of these patients (60-62). Mast cells have also been observed in close contact with sensory nerves within the bladder wall (63-66). Mast cells synthesize and release a number of vasoactive and chemotactic factors, including histamine, prostaglandins, leukotrienes, serotonin, bradykinin, tumor necrosis factor-α, and platelet activating factor (67-69). Degranulation resulting in release of pro-inflammatory factors by mast cells typically occurs subsequent to crosslinking of antigen with immunoglobulin bound to the surface of the mast cell, and this is referred to as immediate or Type I hypersensitivity (70). However, non-immunologic stimuli such as substance P, bradykinin, tumor necrosis factor-α, and nerve growth factor also have the capacity to stimulate release of mast cell contents (68,69). We have previously reported that experimental cystitis induced in mice by intravesical instillation of substance P or E. coli lipopolysaccharide was far less severe in mice that were genetically deficient in mast cells compared to that observed in congenic wild-type mice (71).
Initiating factors for neurogenic inflammation vary widely and include antigens, cold, heat, bacterial or viral infection, or direct stimulation of nerves (43-45). Models of antigen- and viral-induced cystitis will be discussed in this section.
Antigen-Induced Cystitis
Exposure of the airways of sensitized guinea pigs to antigen has long been used as a model to study airway disease and asthma (72). Experimental cystitis induced by systemic sensitization of guinea pigs to ovalbumin and subsequent intravesical instillation of this antigen was described in 1991 (73). Intravesical instillation of ovalbumin into the bladders of naïve guinea pigs has no effect. Since this model is dependent on a Type I hypersensitivity response, mast cells clearly play a crucial role in development of cystitis. This model was adapted for use in mice, and it was demonstrated that intravesical instillation of antigen consistently caused cystitis in wild-type control mice, but not in mice rendered genetically deficient of mast cells (74).
Multiple studies have demonstrated an integral role for neurogenic-mediated processes in models of antigen-induced cystitis. Exposure of bladder tissue from sensitized guinea pigs to antigen resulted in release of neuropeptides, whereas treatment of bladder tissue from naïve guinea pigs with antigen failed to stimulate neuropeptide release (56). The neurokinin-1 receptor (NK-1) is the primary receptor mediating the direct effects of substance P in neurogenic inflammation (75). Stimulation of NK-1 located on postcapillary venules has been demonstrated to increase vascular permeability, resulting in edema (42,76,77). Significantly, absence of functional NK-1 receptors prevented antigen-induced cystitis in mice, suggesting that activation of NK-1 by substance P is an essential component of antigen-induced cystitis (78). Incubation of bladder tissue from sensitized guinea pigs with antigen also stimulates release of prostaglandins, leukotrienes, histamine, and bradykinin (59,79,80). Absence of antigen-induced cystitis in mice lacking functional NK-1 receptors provides strong evidence that neurogenic inflammation plays a key role in this model of cystitis (78).
Advantages of antigen-induce cystitis primarily relate to avoiding the need to instill infectious or noxious substances systemically or intravesically and the fact that the mechanisms underlying development of bladder inflammation entail intrinsic immune and neurological responses. It has also been noted that patients with IC/PBS have a relatively higher incidence of allergies and asthma than the general population (81). However, disadvantages of this model arise primarily from the lack of clear evidence for a similar pathogenesis in patients with IC/PBS.
Viral-Induced Cystitis
It was observed in 1997 that injection of modified pseudorabies virus into the abductor caudae dorsalis tail muscles of the rat caused hemorrhagic cystitis that was prevented by denervation of the bladder (82). The importance of the role of the CNS in this process was further confirmed by results of experiments by this group demonstrating that lesions of areas of the spinal cord (bilateral dorsolateral or ventrolateral funiculectomy) or brainstem (lesions of Barrington’s nucleus/locus coeruleus area) associated with innervation of the bladder prevented cystitis subsequent to injection of virus into the tail muscles (83). Further studies using this model in mice demonstrated that release of pro-inflammatory and nociceptive substances by mast cells in the lamina propria of the bladder resulted in pain, inflammation, and loss of barrier function of the urothelium (84-87). Referred somatic pain induced by viral cystitis in mice was suppressed by antagonists of the NK-1 or histamine-2 receptors (84). These investigators further reported that the cytokine tumor necrosis factor-α may play a key role in migration of mast cells into the bladder wall (87).
Subsequent to injection into the tail muscles, the virus was only cultured from the spinal cord, not the bladder or urine (86). Histologically, virus was observed in the superficial layers of the dorsal horn of the thoracolumbar and sacral spinal cord, as well as the brainstem (83,88). Combined, these results strongly suggest that the effects of injection of virus into the tail muscles is dependent upon irritation of the relevant areas of the CNS resulting in release of neuropeptides and ingress of mast cells within the bladder wall. While this model demonstrates that innervation of the bladder has the capacity, when stimulated appropriately, to cause cystitis, its utility, as with most other models of bladder disorders in humans is limited to confirming the capacity of potential mechanistic pathways to participate in the onset and persistence of bladder disorders.
Autoimmune Cystitis
Around 40 years ago, it was suggested that self-antibodies directed against the bladder could be the cause of IC/PBS (89), and other investigators have subsequently reported findings suggesting that chronic autoimmune disorders may be the underlying cause of some bladder disorders in humans (90-93). These observations are particularly intriguing in light of the higher incidence of autoimmune disorders (e.g., rheumatoid arthritis, Sjogren’s Syndrome, systemic lupus erythematosis, and others) in patients diagnosed with IC/PBS than that observed in the general population (94-97).
Experimental cystitis has been induced in mice (98-100) and rats (101) by injection of homogenized bladder tissue obtained from syngeneic rodents combined with adjuvant. Cystitis in these animals is characterized by increased urinary frequency and decreased voiding volume. Histologically, bladders in sensitized animals typically exhibited increased vascularity, increased numbers of leukocytes within the bladder wall, increased permeability to 14C-urea, and occasional mucosal ulceration (98-101). Interestingly, cystitis was induced by adoptive transfer by intraperitoneal injection of naïve mice with suspensions of homogenates of spleen and lymph nodes obtained from sensitized mice (98) and rats (101). The successful induction of cystitis by adoptive transfer demonstrates that capacity of primed immune cells to recognize and respond to normal bladder tissue.
Autoimmune cystitis was also induced by generation of transgenic mice in which the urothelium expressed ovalbumin (OVA) as a result of insertion of the gene sequence for OVA driven by the uroplakin II gene promoter (102,103). The transgenic mice (Tg-OVA) did not exhibit spontaneous cystitis, but intravenous injection of CD8+ T cells obtained from Tg-OVA mice that had been incubated in vitro with an OVA peptide consistently induced cystitis. Creation of double-transgenic Tg-OVA mice that expressed CD8+ T cells that were responsive to OVA resulted in mice that spontaneously developed cystitis.
These studies, in combination with clinical observations, strongly support the possibility that autoimmunity plays a significant role in IC/PBS. Unfortunately, direct support for this in the form of identification of one or more antigens that specifically stimulate cystitis in patients is still lacking. The role of autoimmunity in the pathogenesis of IC/PBS is further confounded by a lack of reports that imunosuppressive therapy prevents or diminishes symptoms of IC/PBS. However, it is still possible that an autoimmune response may trigger a series of events that results in persistent symptoms of IC/PBS in the absence of an active autoimmune reaction.
Cytitis Induced by Irritants
Experimental cystitis has been induced by intravesical instillation of a variety of irritants, including bacterial lipopolysaccharide (LPS), acid, turpentine, mustard oil, croton oil, and acrolein. Systemic treatment of rodents with cyclophosphamide is one of the most common methods used to initiate cystitis. Cyclophosphamide is an antineoplastic agent, and hemorrhagic cystitis is a common complication observed in patients treated with this drug. Cyclophosphamide is metabolized by the liver to acrolein, and the presence of acrolein within the bladder is thought to be the cause of cystitis in patients or animals that receive cyclophosphamide (104,105). Chemical irritants, including cyclophosphamide or acrolein, directly damage the urothelium and other cells of the bladder, resulting in varying degrees of erosion of the mucosa, edema, hemorrhage, and leukocytic infiltration of the bladder wall.
Exogenous irritants have been used to induce inflammation of the bladder in rats and mice to create cystitis to investigate mechanisms underlying pain and inflammation associated with cystitis. The advantage of these models is that they allow control of the timing, duration, and severity of inflammation. It is also possible to investigate various strategies intended to diminish the severity of pain and inflammation in these controlled models. Unfortunately, studies that investigate treatment options are typically designed to administer the particular intervention being evaluated prior to initiation of bladder inflammation. While this may imply efficacy in patients that have intermittent cystitis, these results may or may not be applicable to patients who have established inflammation at the time of examination and treatment. Other disadvantages of these models primarily relate to whether or not the mechanisms that underlie the response of the bladder and nervous system to these compounds are relevant to those that result in inflammation and associated pain in patients with cystitis.
E. coli LPS is the bacterial product most commonly instilled into the bladder to initiate cystitis. Instillation of E. coli LPS alone into the bladders of mice stimulates cystitis characterized by edema, hemorrhage, and infiltration of neutrophils into the bladder wall (106), and there is evidence that LPS may cross the bladder wall, enter lymphatics or blood vessels, and ultimately be deposited in other organs, including the lungs and rectum (107). In rats, protamine sulfate is typically instilled into the bladder to destroy the glycosaminoglycan layer prior to infusion of LPS to induce cystitis (108-111). Interestingly, systemic administration of E. coli LPS by intraperitoneal or intravenous injection also stimulates inflammation of the bladder in rats and mice characterized primarily by edema within the bladder wall (71,108,112). The toll-like receptor 4 (TLR4) is thought to be the transmembrane receptor responsible for cellular response to LPS (113,114), and instillation of uropathogenic E. coli into the bladders of mice that spontaneously express dominant negative TLR4 (C3H/HeJ mice) resulted in bacterial colonization of the bladder wall in the absence of inflammation (115).
Bacillus Calmette-Gueirin (BCG), prepared from an attenuated strain of Mycobacterium bovis, was initially used as a vaccine against tuberculosis (116). BGG is now commonly instilled into the bladder to treat superficial, noninvasive cancer (117). It has recently been reported that instillation of BCG into the bladders of mice stimulates profound inflammation (118).
A survey of relevant literature suggests that the vast majority of experimental studies of the onset and sequela of cystitis utilize exposure of the bladder to irritant chemicals. It was reported over 20 years ago that intravesical instillation of turpentine, mustard oil, or croton oil stimulated cystitis characterized by edema, hemorrhage and leukocytic infiltration of the bladder wall (119). Bladder inflammation has also been induced by intravesical administration of hydrochloric (120,121) or acetic (122,123) acid in rats and mice. These models consistently produce cystitis and damage to the urothelium that is accompanied by activation of a variety of signaling pathways too numerous to summarize in this review (29,124-129) .
Hemorrhagic cystitis has been reported as a complication of treatment of patients with cyclophosphamide since the mid-1960’s (130). Systemic treatment of rodents with cyclophosphamide as a model to induce cystitis has been used for at least 40 years (131) and is one of the most common experimental models of bladder inflammation. Cyclophosphamide has not been reported to stimulate inflammation of other organs of the urinary tract (132), and, as mentioned previously, its irritant effects on the bladder have been attributed to contact of the bladder surface with acrolein.
Direct instillation of acrolein into the bladder has been shown to induce cystitis (133,134). This model has the potential advantage relative to systemic administration of cyclophosphamide of not requiring hepatic metabolism of cyclophosphamide to acrolein, and at least one study reported differential rates of metabolism of cyclophosphamide and subsequent excretion of acrolein into the urine by two strains of mice (135). We investigated cystitis induced by intravesical instillation of acrolein in mice to determine whether or not the severity of cystitis could be more tightly controlled and also whether or not differences in response to acrolein could be detected among strains of mice (Figure 1) (136). We found that the intensity of inflammation could be varied in direct proportion to the concentration of acrolein instilled, and we also observed that, when identical volumes and concentrations were administered intravesically, acrolein-induced cystitis was more severe in C57bl/6n and C3H/OuJ mice than in C3H/HeJ mice. Part of the motivation for performing this study was the concern that the severity of cystitis induced by some models could be so severe that subtle differences in signaling or response to potential therapeutic interventions could be overwhelmed by massive tissue damage and inflammation. The results of this study indicate that strains of rats and mice, as well as the intensity of inflammation induced, should be considered when selecting models of cystitis to investigate mechanisms underlying bladder inflammation or response to various interventions.
Figure 1.


Four (A) and 24 hours (B) after instillation of acrolein (6, 10, or 100 μg; 15 μl total volume) or 15 μl phosphate buffered saline (PBS; control) into the bladders of female C57BL6N mice, it was observed that the severity of inflammation as indicated by intramural edema and hemorrhage correlated with increasing concentrations of acrolein. (L, lumen; D, detrusor); 40x original; scale bar = 200 μm. (With permission from: Bjorling DE, Elkahwaji JE, Bushman W, Janda LM, Boldon K, Hopkins WJ, Wang ZY. Acute acrolein-induced cystitis in mice. BJU Int 2007;99:1523-1529.)
Prostatic inflammation
Benign prostatic hyperplasia (BPH) affects more than 50 percent of men past the age of 50, and as many as one-third of all men develop significant BPH-related lower urinary tract symptoms (LUTS) that require treatment (137). LUTS include increased urinary frequency, urgency, nocturia, weak urinary stream, straining to void, and a sense of incomplete emptying, and the incidence of LUTS increases with age in men (138). Historically, LUTS in men with BPH have been attributed to physical obstruction of the bladder outflow tract by prostatic encroachment on the urethral lumen (139-142). However, in a recent study of men with LUTS, half the study population (42/84) had no evidence of bladder outlet obstruction (143), and it has been proposed that LUTS associated with benign prostatic hyperplasia (BPH) are actually the result of 3 components: prostatic enlargement, alpha-adrenergic receptor-mediated narrowing of the urethra, and prostatic inflammation (144). In a retrospective study of 3,942 prostatic biopsies from BPH patients, inflammation (primarily chronic inflammation) was observed in 1,700 (43.1%) (8). Inflammation was observed in all histological specimens obtained from 80 men without symptoms of prostatic inflammation who underwent transurethral prostatectomy for treatment of BPH, suggesting that inflammation is extremely common in patients with symptoms of BPH who have no other symptoms of prostatic inflammation (9).
A role for bacterial colonization or infection in the etiology of BPH is plausible but remains controversial. Analysis of material obtained by biopsy of the prostate using the polymerase chain reaction (PCR) demonstrated the presence of bacterial 16S ribosomal RNA, suggesting the presence of bacteria in prostates with histological evidence of inflammation (145,146). Bacteria were also cultured from 38% of tissue samples obtained by transurethral prostatectomy from patients who had negative urine cultures prior to surgery (147). It also appears that urinary reflux into the prostatic ducts is a common occurrence (148) and bacterial colonization/infection in surgical specimens of BPH may be more common than previously assumed.
It has also been suggested that prior infection or the presence of normal commensal organisms may sensitize afferent innervation of the prostate, contributing to prostate-associated pain (149). One of the co-authors (WB) has previously described a model of prostatic inflammation that was induced by a single transurethral inoculation with E. coli 1677 (150). This strain of E. coli was isolated from a patient with a severe urinary tract infection (151). Twelve week old male C3H/HeOuJ (OuJ) mice were anesthetized, a urethral catheter was inserted, and bacteria (2×106 CFU) in a 20 μl volume were instilled. Mice were sacrificed 5 days and 12 weeks after instillation of bacteria. Histology of the prostate 5 days after instillation of bacteria demonstrated edema, shedding of epithelial cells, and infiltration of neutrophils into the stroma and ducts (Figure 2). Prostate glands removed from mice sacrificed 12 weeks after infection had varying degrees of atypical hyperplasia, dysplasia, epithelial proliferation, lymphocytic infiltration, and evidence of oxidative DNA damage. Instillation of phosphate-buffered saline had no effect on prostate histology.
Figure 2.
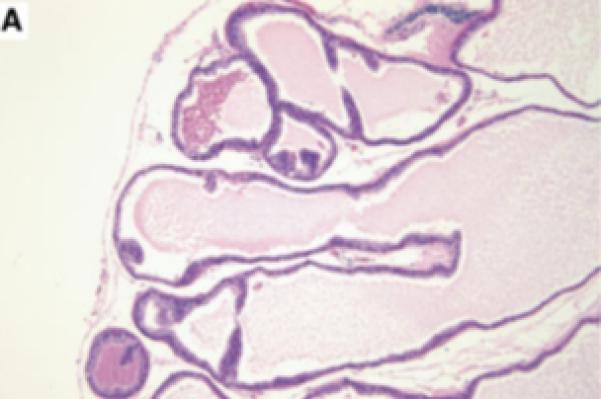
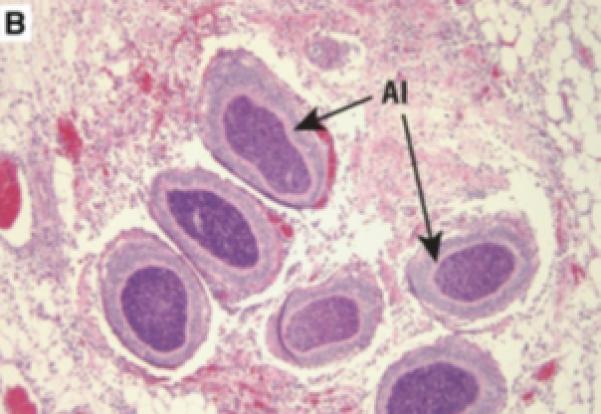
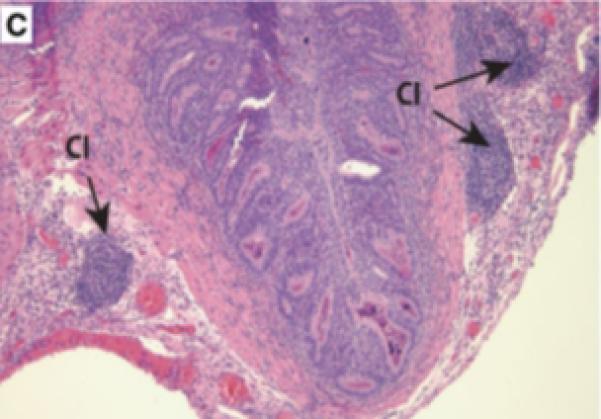
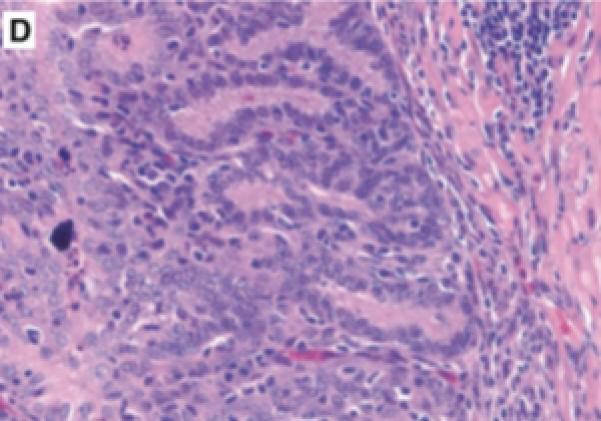
Prostatic inflammation was induced by a single transurethral instillation of E. coli 1677 in C3H/OuJ mice as previously described (141). A. Instillation of saline failed to cause any histological response when tissues from mice sacrificed 5 days after infusion were stained with hematoxylin and eosin. B. Five days after instillation of E. coli 1677, evidence of acute inflammation (AI) characterized by edema, inflammatory cell infiltrate, and exfoliation of epithelial cells into the ducts was observed. C. and D. Chronic inflammation (CI) consisting of dense accumulation of lymphocytes was present in the coagulating gland 12 weeks after instillation of bacteria, as well as evidence of early dysplastic changes in the ductal wall (D). 10x original. (With permission from: Elkahwaji JE, Zhong W, Hopkins WJ, Bushman W.Chronic bacterial infection and inflammation incite reactive hyperplasia in a mouse model of chronic prostatitis. Prostate 2007;67:14-21.)
We have since modified the protocol to include treatment of mice with nitrofurantoin (3.4 mg/kg, sc, twice daily) for 1 day prior and daily for 3 days after instillation of bacteria to prevent bacterial cystitis. Mechanical sensitivity of the hind paws was assessed in these mice using von Frey monofilaments to investigate the effects of prostatic inflammation on referred mechanical hypersensitivity. Our preliminary data indicate that a single infusion of E. coli resulted in localized prostatic infection and inflammation and increased sensitivity to application of mechanical stimuli to the hind paws (unpublished observations).
These results indicate that this may be a useful model of prostatic inflammation. They further suggest that prostatic inflammation has the capacity to sensitize the afferent nervous system, providing support for the notion that LUTS associated with prostatic inflammation could be due to neural plasticity induced by inflammation of the prostate.
Summary
A wide variety of animal models of inflammation of the lower urinary tract has been used over an extended period of time. The principal benefits of this research relative to lower urinary tract inflammation in humans relate to potential mechanisms underlying the onset and persistence of pain and inflammation. With the exception of disorders arising from bacterial causes, initiating causes of inflammatory diseases in humans remain obscure. Unfortunately, no currently-available animal model, including infection models, perfectly mimics disease that occurs in humans. These models have therefore have mainly been of benefit in identifying potential mechanisms underlying clinical disease. Further comparative and translational studies are required to better characterize causes of clinical disease in humans and the utility of animal models in studying these causes.
Acknowledgments
Funding: NIH R01 DK066349 (DEB) NIH R01 DK0757 (WB)
References
- 1.Ambulatory Care Visits to Physician Offices, Hospital Outpatient Departments, and Emergency Departments: United States, 1999–2000. National Center for Health Statistics, Centers for Disease Control and Prevention, U.S. Dept. of Health and Human Services; Hyattsville, MD: (Series 13).Vital and Health Statistics. 2004 September;(No. 157) [PubMed]
- 2.Nicolle LE, Bradley S, Colgan R, Rice JC, Schaeffer A, Hooton TM. Infectious Diseases Society of America guidelines for the diagnosis and treatment of asymptomatic bacteriuria in adults. Clin Infect Dis. 2005;40:643–654. doi: 10.1086/427507. [DOI] [PubMed] [Google Scholar]
- 3.Berry SH, Bogart LM, Pham C, Liu K, Nyberg L, Stoto M, Suttorp M, Clemens JQ. Development, validation and testing of an epidemiological case definition of interstitial cystitis/painful bladder syndrome. J Urol. 2010;183:1848–1852. doi: 10.1016/j.juro.2009.12.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clemens JQ, Joyce GF, Wise M, Payne CK. Litwin MS, Saigal CS, editors. Interstitial cystitis and painful bladder syndrome. US Department of Health and Human Services, National Institutes of Health, US Government Printing Office; Washington, DC: Urologic Diseases in America. 2007:123–154. NIH publication 07–5112.
- 5.Wyndaele JJ, Van Dyck J, Toussaint N. Cystoscopy and bladder biopsies in patients with bladder pain syndrome carried out following ESSIC guidelines. Scand J Urol Nephrol. 2009;43:471–475. doi: 10.3109/00365590903199007. [DOI] [PubMed] [Google Scholar]
- 6.Erickson DR, Tomaszewski JE, Kunselman AR, Stetter CM, Peters KM, Rovner ES, Demers LM, Wheeler MA, Keay SK. Urine markers do not predict biopsy findings or presence of bladder ulcers in interstitial cystitis/painful bladder syndrome. J Urol. 2008;179:1850–1856. doi: 10.1016/j.juro.2008.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delongchamps NB, de la Roza G, Chandan V, Jones R, Sunheimer R, Threatte G, Jumbelic M, Haas GP. Evaluation of prostatitis in autopsied prostates—is chronic inflammation more associated with benign prostatic hyperplasia or cancer? J Urol. 2008;179:1736–1740. doi: 10.1016/j.juro.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Silverio F, Gentile V, De Matteis A, Mariotti G, Giuseppe V, Luigi PA, Sciarra A. Distribution of inflammation, pre-malignant lesions, incidental carcinoma in histologically confirmed benign prostatic hyperplasia: a retrospective analysis. Eur Urol. 2003;43:164–175. doi: 10.1016/s0302-2838(02)00548-1. [DOI] [PubMed] [Google Scholar]
- 9.Nickel JC, Downey J, Young I, Boag S. Asymptomatic inflammation and/or infection in benign prostatic hyperplasia. BJU Inter. 1999;84:976–981. doi: 10.1046/j.1464-410x.1999.00352.x. [DOI] [PubMed] [Google Scholar]
- 10.Nickel JC, Elhilali M, Vallancien G, ALF-ONE Study Group Benign prostatic hyperplasia (BPH) and prostatitis: prevalence of painful ejaculation in men with clinical BPH. BJU Inter. 2005;95:571–574. doi: 10.1111/j.1464-410X.2005.05341.x. [DOI] [PubMed] [Google Scholar]
- 11.O’Sullivan M, Murphy C, Deasy C, Iohom G, Kiely EA, Shorten G. Effects of transurethral resection of prostate on the quality of life of patients with benign prostatic hyperplasia. J Am Coll Surg. 2004;198:394–403. doi: 10.1016/j.jamcollsurg.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 12.Bjorling DE, Wang ZY, Boldon K, Bushman W. Bacterial cystitis is accompanied by increased peripheral thermal sensitivity in mice. J Urol. 2008;179:759–763. doi: 10.1016/j.juro.2007.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hopkins WJ, Gendron-Fitzpatrick A, Balish E, Uehling DT. Time course and host responses to Escherichia coli urinary tract infection in genetically distinct mouse strains. Infect Immun. 1998;66:2798–2802. doi: 10.1128/iai.66.6.2798-2802.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birder LA. TRPs in bladder diseases. Biochim Biophys Acta. 2007;1772:879–884. doi: 10.1016/j.bbadis.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cruz F, Dinis P. Resiniferatoxin and botulinum toxin type A for treatment of lower urinary tract symptoms. Neurourol Urodyn. 2007;26:920–927. doi: 10.1002/nau.20479. [DOI] [PubMed] [Google Scholar]
- 16.Dinis P, Charrua A, Avelino A, Cruz F. Intravesical resiniferatoxin decreases spinal c-fos expression and increases bladder volume to reflex micturition in rats with chronic inflamed urinary bladders. BJU Int. 2004;94:153–157. doi: 10.1111/j.1464-4096.2004.04855.x. [DOI] [PubMed] [Google Scholar]
- 17.Cayan S, Coşkun B, Bozlu M, Acar D, Akbay E, Ulusoy E. Botulinum toxin type A may improve bladder function in a rat chemical cystitis model. Urol Res. 2003;30:399–404. doi: 10.1007/s00240-002-0291-0. [DOI] [PubMed] [Google Scholar]
- 18.Krhut J, Zvara P. Intravesical instillation of botulinum toxin A: an in vivo murine study and pilot clinical trial. Int Urol Nephrol. 2010 Jun 20; doi: 10.1007/s11255-010-9790-z. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 19.Osbaldiston GW, Taussig Clinical report on 46 cases of feline urological syndrome. Vet Med Small Anim Clinician. 1970;65:461–468. [PubMed] [Google Scholar]
- 20.Fabricant CG. Urolithiasis: A review with recent viral studies. Feline Pract. 1973;3:22–30. [Google Scholar]
- 21.Fabricant CG. Herpesvirus induced feline urolithiasis. A review. Comp Immunol Microbiol Infect Dis. 1979;9:460–466. doi: 10.1016/0147-9571(79)90038-9. [DOI] [PubMed] [Google Scholar]
- 22.Fabricant CG, Gillespie JH. Identification and characterization of a second feline herpesvirus. Infect Immun. 1974;9:460–466. doi: 10.1128/iai.9.2.460-466.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martens JG, McConnell S, Swanson CL. The role of infectious agents in naturally occurring feline urologic syndrome. Vet Clin North Am Small Anim Pract. 1984;14:503–511. doi: 10.1016/s0195-5616(84)50057-6. [DOI] [PubMed] [Google Scholar]
- 24.Barsanti JA, Finco DR, Shotts EB, et al. Feline urologic syndrome: further investigation into etiology. J Am Anim Hosp Assoc. 1982;18:391–395. [Google Scholar]
- 25.Kruger JM, Osborne CA, Goyal SM, et al. Clinical evaluation of cats with lower urinary tract disease. J Am Vet Med Assoc. 1991;199:211–216. [PubMed] [Google Scholar]
- 26.Lees GE. Epidemiology of naturally occurring bacterial urinary tract infection. Vet Clin North Am Small Anim Pract. 1984;14:471–479. doi: 10.1016/s0195-5616(84)50054-0. [DOI] [PubMed] [Google Scholar]
- 27.Buffington CAT, Blaisdell JL, Binnns SP, Woodworth BE. Decreased urine glycosaminoglycan (GAG) excretion in cats with inflammatory bladder syndrome. J Urol. 1993;149:509A. [PubMed] [Google Scholar]
- 28.Gao X, Buffington CA, Au JL. Effect of interstitial cystitis on drug absorption from urinary bladder. J Pharmacol Exp Ther. 1994;271:818–823. [PubMed] [Google Scholar]
- 29.Birder LA. Urothelial signaling. Auton Neurosci. 2010;153:33–40. doi: 10.1016/j.autneu.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sahbaie P, Shi X, Guo TZ, Qiao Y, Yeomans DC, Kingery WS, Clark JD. Role of substance P signaling in enhanced nociceptive sensitization and local cytokine production after incision. Pain. 2009;145:341–349. doi: 10.1016/j.pain.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–38. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- 32.Birder LA, Wolf-Johnston AS, Chib MK, Buffington CA, Roppolo JR, Hanna-Mitchell AT. Beyond neurons: Involvement of urothelial and glial cells in bladder function. Neurourol Urodyn. 2010;29:88–96. doi: 10.1002/nau.20747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marchand JE, Sant GR, Kream RM. Increased expression of substance P receptor-encoding mRNA in bladder biopsies from patients with interstitial cystitis. Br J Urol. 1998;81:224–228. doi: 10.1046/j.1464-410x.1998.00507.x. [DOI] [PubMed] [Google Scholar]
- 34.Lowe EM, Anand P, Terenghi G, Williams-Chestnut RE, Sinicropi DV, Osborne JL. Increased nerve growth factor levels in the urinary bladder of women with idiopathic sensory urgency and interstitial cystitis. Br J Urol. 1997;79:572–577. doi: 10.1046/j.1464-410x.1997.00097.x. [DOI] [PubMed] [Google Scholar]
- 35.Birder LA, Ruan HZ, Chopra B, Xiang Z, Barrick S, Buffington CA, Roppolo JR, Ford AP, de Groat WC, Burnstock G. Alterations in P2X and P2Y purinergic receptor expression in urinary bladder from normal cats and cats with interstitial cystitis. Am J Physiol Renal Physiol. 2004;287:F1084–1091. doi: 10.1152/ajprenal.00118.2004. [DOI] [PubMed] [Google Scholar]
- 36.Birder LA, Barrick SR, Roppolo JR, Kanai AJ, de Groat WC, Kiss S, Buffington CA. Feline interstitial cystitis results in mechanical hypersensitivity and altered ATP release from bladder urothelium. Am J Physiol Renal Physiol. 2003;285:F423–429. doi: 10.1152/ajprenal.00056.2003. [DOI] [PubMed] [Google Scholar]
- 37.Sun Y, Keay S, Lehrfeld TJ, Chai TC. Changes in adenosine triphosphate-stimulated ATP release suggest association between cytokine and purinergic signaling in bladder urothelial cells. Urology. 2009;74:1163–1168. doi: 10.1016/j.urology.2009.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Y, Chai TC. Augmented extracellular ATP signaling in bladder urothelial cells from patients with interstitial cystitis. Am J Physiol Cell Physiol. 2006;290:C27–34. doi: 10.1152/ajpcell.00552.2004. [DOI] [PubMed] [Google Scholar]
- 39.Westropp JL, Buffington CA. In vivo models of interstitial cystitis. J Urol. 2002;167:694–702. doi: 10.1016/S0022-5347(01)69129-8. [DOI] [PubMed] [Google Scholar]
- 40.Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–210. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- 41.Lundberg JM, Brodin E, Hua X, Saria A. Vascular permeability changes and smooth muscle contraction in relation to capsaicin-sensitive substance P afferents in the guinea-pig. Acta Physiol Scand. 1984;120:217–227. doi: 10.1111/j.1748-1716.1984.tb00127.x. [DOI] [PubMed] [Google Scholar]
- 42.Abelli L, Nappi F, Perretti F, Maggi CA, Manzini S, Giachetti A. Microvascular leakage induced by substance P in rat urinary bladder: involvement of cyclo-oxygenase metabolites of arachidonic acid. J Auton Pharmacol. 1992;12:269–276. doi: 10.1111/j.1474-8673.1992.tb00341.x. [DOI] [PubMed] [Google Scholar]
- 43.Kowalski ML, Sliwinska-Kowalska M, Kaliner MA. Neurogenic inflammation, vascular permeability, and mast cells. II. Additional evidence indicating that mast cells are not involved in neurogenic inflammation. J Immunol. 1990;145:1214–1221. [PubMed] [Google Scholar]
- 44.Sharkey KA. Substance P and calcitonin gene-related peptide (CGRP) in gastrointestinal inflammation. Ann N Y Acad Sci. 1992;664:425–442. doi: 10.1111/j.1749-6632.1992.tb39781.x. [DOI] [PubMed] [Google Scholar]
- 45.Yonehara N, Imai Y, Shibutani T, Inoki R. Participation of substance P in inflammatory responses. Adv Exp Med Biol. 1989;247B:529–534. doi: 10.1007/978-1-4615-9546-5_87. [DOI] [PubMed] [Google Scholar]
- 46.Pan XQ, Gonzalez JA, Chang S, Chacko S, Wein AJ, Malykhina AP. Experimental colitis triggers the release of substance P and calcitonin gene-related peptide in the urinary bladder via TRPV1 signaling pathways. Exp Neurol. 2010;225:262–273. doi: 10.1016/j.expneurol.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trevisani M, Campi B, Gatti R, André E, Materazzi S, Nicoletti P, Gazzieri D, Geppetti P. The influence of alpha1-adrenoreceptors on neuropeptide release from primary sensory neurons of the lower urinary tract. Eur Urol. 2007;52:901–908. doi: 10.1016/j.eururo.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 48.Holzer P. Local effector functions of capsaicin-sensitive sensory nerve endings: involvement of tachykinins, calcitonin gene-related peptide and other neuropeptides. Neuroscience. 1988;24:739–768. doi: 10.1016/0306-4522(88)90064-4. [DOI] [PubMed] [Google Scholar]
- 49.Donnerer J, Amann R, Schuligoi R, Skofitsch G. Complete recovery by nerve growth factor of neuropeptide content and function in capsaicin-impaired sensory neurons. Brain Res. 1996;741:103–108. doi: 10.1016/s0006-8993(96)00905-5. [DOI] [PubMed] [Google Scholar]
- 50.Geppetti P, Nassini R, Materazzi S, Benemei S. The concept of neurogenic inflammation. BJU Int. 2008;101(Suppl 3):2–6. doi: 10.1111/j.1464-410X.2008.07493.x. [DOI] [PubMed] [Google Scholar]
- 51.Avelino A, Cruz F. TRPV1 (vanilloid receptor) in the urinary tract: expression, function and clinical applications. Naunyn Schmiedebergs Arch Pharmacol. 2006;373:287–299. doi: 10.1007/s00210-006-0073-2. [DOI] [PubMed] [Google Scholar]
- 52.Mourtzoukou EG, Iavazzo C, Falagas ME. Resiniferatoxin in the treatment of interstitial cystitis: a systematic review. Int Urogynecol J Pelvic Floor Dysfunct. 2008;19:1571–1576. doi: 10.1007/s00192-008-0663-2. [DOI] [PubMed] [Google Scholar]
- 53.Dimitrakov J, Kroenke K, Steers WD, Berde C, Zurakowski D, Freeman MR, Jackson JL. Pharmacologic management of painful bladder syndrome/interstitial cystitis: a systematic review. Arch Intern Med. 2007;167:1922–1929. doi: 10.1001/archinte.167.18.1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lecci A, Giuliani S, Patacchini R, Maggi CA. Evidence against a peripheral role of tachykinins in the initiation of micturition reflex in rats. J Pharmacol Exp Ther. 1993;264:1327–1332. [PubMed] [Google Scholar]
- 55.Maggi CA, Patacchini R, Santicioli P, Giuliani S. Tachykinin antagonists and capsaicin-induced contraction of the rat isolated urinary bladder: evidence for tachykinin-mediated cotransmission. Br J Pharmacol. 1991;103:1535–1541. doi: 10.1111/j.1476-5381.1991.tb09823.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bjorling DE, Saban MR, Saban R. Neurogenic inflammation of Guinea-pig bladder. Mediators Inflamm. 1994;3:189–197. doi: 10.1155/S0962935194000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saban MR, Saban R, Bjorling D, Haak-Frendscho M. Involvement of leukotrienes, TNF-alpha, and the LFA-1/ICAM-1 interaction in substance P-induced granulocyte infiltration. J Leukoc Biol. 1997;61:445–451. doi: 10.1002/jlb.61.4.445. [DOI] [PubMed] [Google Scholar]
- 58.Saban R, Franz J, Bjorling DE. Spontaneously released substance P and bradykinin from isolated guinea-pig bladder. Br J Urol. 1997;79:516–524. doi: 10.1046/j.1464-410x.1997.00092.x. [DOI] [PubMed] [Google Scholar]
- 59.Saban MR, Saban R, Bjorling DE. Kinetics of peptide-induced release of inflammatory mediators by the urinary bladder. Br J Urol. 1997;80:742–747. doi: 10.1046/j.1464-410x.1997.00415.x. [DOI] [PubMed] [Google Scholar]
- 60.Dundore PA, Schwartz AM, Semerjian H. Mast cell counts are not useful in the diagnosis of nonulcerative interstitial cystitis. J Urol. 1996;155:885–887. [PubMed] [Google Scholar]
- 61.Sant GR, Kempuraj D, Marchand JE, Theoharides TC. The mast cell in interstitial cystitis: role in pathophysiology and pathogenesis. Urology. 2007;69(Suppl 4):34–40. doi: 10.1016/j.urology.2006.08.1109. [DOI] [PubMed] [Google Scholar]
- 62.Larsen MS, Mortensen S, Nordling J, Horn T. Quantifying mast cells in bladder pain syndrome by immunohistochemical analysis. BJU Int. 2008;102:204–207. doi: 10.1111/j.1464-410X.2008.07576.x. [DOI] [PubMed] [Google Scholar]
- 63.Elbadawi AE, Light JK. Distinctive ultrastructural pathology of nonulcerative interstitial cystitis: new observations and their potential significance in pathogenesis. Urol Int. 1996;56:137–162. doi: 10.1159/000282832. [DOI] [PubMed] [Google Scholar]
- 64.Letourneau R, Pang X, Sant GR, Theoharides TC. Intragranular activation of bladder mast cells and their association with nerve processes in interstitial cystitis. Br J Urol. 1996;77:41–54. doi: 10.1046/j.1464-410x.1996.08178.x. [DOI] [PubMed] [Google Scholar]
- 65.Saban R, Keith IM, Bjorling DE. Neuropeptide-mast cell interaction in interstitial cystitis. In: Sant GR, editor. Interstitial Cystitis. Lippincott-Raven; Philadelphia: 1997. pp. 53–66. [Google Scholar]
- 66.Theoharides TC, Sant GR, el-Mansoury M, Letourneau R, Ucci AA, Jr, Meares EM., Jr Activation of bladder mast cells in interstitial cystitis: a light and electron microscopic study. J Urol. 1995;153:629–636. doi: 10.1097/00005392-199503000-00021. [DOI] [PubMed] [Google Scholar]
- 67.Renzetti LM, Paciorek PM, Tannu SA, Rinaldi NC, Tocker JE, Wasserman MA, Gater PR. Pharmacological evidence for tumor necrosis factor as a mediator of allergic inflammation in the airways. J Pharmacol Exp Ther. 1996;278:847–853. [PubMed] [Google Scholar]
- 68.Theoharides TC, Sant GR. The mast cell as a neuroimmunoendocrine effector in interstitial cystitis. In: Sant GR, editor. Interstitial Cystitis. Lippincott-Raven; Philadelphia: 1997. pp. 101–108. [Google Scholar]
- 69.Welker P, Grabbe J, Grützkau A, Henz BM. Effects of nerve growth factor (NGF) and other fibroblast-derived growth factors on immature human mast cells (HMC-1) Immunology. 1998;94:310–317. [PMC free article] [PubMed] [Google Scholar]
- 70.Ishizaka T, Ishizaka K. Activation of mast cells for mediator release through IgE receptors. Prog Allergy. 1984;34:188–235. [PubMed] [Google Scholar]
- 71.Bjorling DE, Jerde TJ, Zine MJ, Busser BW, Saban MR, Saban R. Mast cells mediate the severity of experimental cystitis in mice. J Urol. 1999;162:231–236. doi: 10.1097/00005392-199907000-00073. [DOI] [PubMed] [Google Scholar]
- 72.Feinberg SM, Malkiel S, McIntire FC. The effect of stress factors of asthma induced in guinea pigs by aerosolized antigens. J Allergy. 1953;24:302–308. doi: 10.1016/0021-8707(53)90173-7. [DOI] [PubMed] [Google Scholar]
- 73.Saban R, Christensen M, Keith I, Graziano FM, Undem BJ, Aagaard J, Bjorling DE, Bruskewitz RC. Experimental model for the study of bladder mast cell degranulation and smooth muscle contraction. Semin Urol. 1991;9:88–101. [PubMed] [Google Scholar]
- 74.Saban R, Saban MR, Nguyen NB, Hammond TG, Wershil BK. Mast cell regulation of inflammation and gene expression during antigen-induced bladder inflammation in mice. Physiol Genomics. 2001;7:35–43. doi: 10.1152/physiolgenomics.00044.2001. [DOI] [PubMed] [Google Scholar]
- 75.McDonald DM, Bowden JJ, Baluk P, Bunnett NW. Neurogenic inflammation. A model for studying efferent actions of sensory nerves. Adv Exp Med Biol. 1996;410:453–462. [PubMed] [Google Scholar]
- 76.Baluk P, Thurston G, Murphy TJ, Bunnett NW, McDonald DM. Neurogenic plasma leakage in mouse airways. Br J Pharmacol. 1999;126:522–528. doi: 10.1038/sj.bjp.0702323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McDonald DM, Thurston G, Baluk P. Endothelial gaps as sites for plasma leakage in inflammation. Microcirculation. 1999;6:7–22. [PubMed] [Google Scholar]
- 78.Saban R, Saban MR, Nguyen NB, Lu B, Gerard C, Gerard NP, Hammond TG. Neurokinin-1 (NK-1) receptor is required in antigen-induced cystitis. Am J Pathol. 2000;156:775–780. doi: 10.1016/S0002-9440(10)64944-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bjorling DE, Saban MR, Bruskewitz RC, Saban R. Response of the isolated guinea pig bladder to exogenous and endogenous leukotrienes. J Urol. 1994;152:1281–1286. doi: 10.1016/s0022-5347(17)32568-5. [DOI] [PubMed] [Google Scholar]
- 80.Saban R, Undem BJ, Keith IM, Saban MR, Tengowski MW, Graziano FM, Bjorling DE. Differential release of prostaglandins and leukotrienes by sensitized guinea pig urinary bladder layers upon antigen challenge. J Urol. 1994;152:544–549. doi: 10.1016/s0022-5347(17)32790-8. [DOI] [PubMed] [Google Scholar]
- 81.Koziol JA. Epidemiology of interstitial cystitis. Urol Clin North Am. 1994;21:7–20. [PubMed] [Google Scholar]
- 82.Jasmin L, Carstens E, Basbaum AI. Interneurons presynaptic to rat tail-flick motoneurons as mapped by transneuronal transport of pseudorabiesvirus: few have long ascending collaterals. Neuroscience. 1997;76:859–876. doi: 10.1016/s0306-4522(96)00384-3. [DOI] [PubMed] [Google Scholar]
- 83.Jasmin L, Janni G, Manz HJ, Rabkin SD. Activation of CNS circuits producing a neurogenic cystitis: evidence for centrally induced peripheral inflammation. J Neurosci. 1998;18:10016–10029. doi: 10.1523/JNEUROSCI.18-23-10016.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rudick CN, Schaeffer AJ, Klumpp DJ. Pharmacologic attenuation of pelvic pain in a murine model of interstitial cystitis. BMC Urol. 2009;9:16. doi: 10.1186/1471-2490-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rudick CN, Bryce PJ, Guichelaar LA, Berry RE, Klumpp DJ. Mast cell-derived histamine mediates cystitis pain. PLoS One. 2008;3(5):e2096. doi: 10.1371/journal.pone.0002096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rudick CN, Chen MC, Mongiu AK, Klumpp DJ. Organ cross talk modulates pelvic pain. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1191–1198. doi: 10.1152/ajpregu.00411.2007. [DOI] [PubMed] [Google Scholar]
- 87.Chen MC, Keshavan P, Gregory GD, Klumpp DJ. RANTES mediates TNF-dependent lamina propria mast cell accumulation and barrier dysfunction in neurogenic cystitis. Am J Physiol Renal Physiol. 2007;292:F1372–1379. doi: 10.1152/ajprenal.00472.2006. [DOI] [PubMed] [Google Scholar]
- 88.Doggweiler R, Jasmin L, Schmidt RA. Neurogenically mediated cystitis in rats: an animal model. J Urol. 1998;160:1551–1556. [PubMed] [Google Scholar]
- 89.Silk MR. Bladder antibodies in interstitial cystitis. J Urol. 1970;103:307–309. doi: 10.1016/s0022-5347(17)61948-7. [DOI] [PubMed] [Google Scholar]
- 90.Anderson JB, Parivar F, Lee G, Wallington TB, MacIver AG, Bradbrook RA, Gingell JC. The enigma of interstitial cystitis--an autoimmune disease? Br J Urol. 1989;63:58–63. doi: 10.1111/j.1464-410x.1989.tb05124.x. [DOI] [PubMed] [Google Scholar]
- 91.Jokinen EJ, Alfthan OS, Oravisto KJ. Antitissue antibodies in interstitial cystitis. Clin Exp Immunol. 1972;11:333–339. [PMC free article] [PubMed] [Google Scholar]
- 92.Mattila J, Harmoinen A, Hällström O. Serum immunoglobulin and complement alterations in interstitial cystitis. Eur Urol. 1983;9:350–352. doi: 10.1159/000474122. [DOI] [PubMed] [Google Scholar]
- 93.Ochs RL, Stein TW, Jr, Peebles CL, Gittes RF, Tan EM. Autoantibodies in interstitial cystitis. J Urol. 1994;151:587–592. doi: 10.1016/s0022-5347(17)35023-1. [DOI] [PubMed] [Google Scholar]
- 94.Lee KL, Chen MY, Yeh JH, Huang SW, Tai HC, Yu HJ. Lower urinary tract symptoms in female patients with rheumatoid arthritis. Scand J Rheumatol. 2006;35:96–101. doi: 10.1080/03009740500395278. [DOI] [PubMed] [Google Scholar]
- 95.Leppilahti M, Tammela TL, Huhtala H, Kiilholma P, Leppilahti K, Auvinen A. Interstitial cystitis-like urinary symptoms among patients with Sjogren’s syndrome: a population-based study in Finland. Am J Med. 2003;115:62–65. doi: 10.1016/s0002-9343(03)00257-2. [DOI] [PubMed] [Google Scholar]
- 96.van de Merwe JP. Interstitial cystitis and systemic autoimmune diseases. Nat Clin Pract Urol. 2007;4:484–491. doi: 10.1038/ncpuro0874. [DOI] [PubMed] [Google Scholar]
- 97.Peeker R, Atanasiu L, Logadottir Y. Intercurrent autoimmune conditions in classic and non-ulcer interstitial cystitis. Scand J Urol Nephrol. 2003;37:60–63. doi: 10.1080/00365590310008721. [DOI] [PubMed] [Google Scholar]
- 98.Bullock AD, Becich MJ, Klutke CG, Ratliff TL. Experimental autoimmune cystitis: a potential murine model for ulcerative interstitial cystitis. J Urol. 1992;148:1951–1956. doi: 10.1016/s0022-5347(17)37091-x. [DOI] [PubMed] [Google Scholar]
- 99.Phull H, Salkini M, Purves T, Funk J, Copeland D, Comiter CV. Angiotensin II plays a role in acute murine experimental autoimmune cystitis. BJU Int. 2007;100:664–667. doi: 10.1111/j.1464-410X.2007.07035.x. [DOI] [PubMed] [Google Scholar]
- 100.Lin YH, Liu G, Kavran M, Altuntas CZ, Gasbarro G, Tuohy VK, Daneshgari F. Lower urinary tract phenotype of experimental autoimmune cystitis in mouse: a potential animal model for interstitial cystitis. BJU Int. 2008;102:1724–1730. doi: 10.1111/j.1464-410X.2008.07891.x. [DOI] [PubMed] [Google Scholar]
- 101.Luber-Narod J, Austin-Ritchie T, Banner B, Hollins C, 3rd, Maramag C, Price H, Menon M. Experimental autoimmune cystitis in the Lewis rat: a potential animal model for interstitial cystitis. Urol Res. 1996;24:367–373. doi: 10.1007/BF00389795. [DOI] [PubMed] [Google Scholar]
- 102.Liu W, Deyoung BR, Chen X, Evanoff DP, Luo Y. RDP58 inhibits T cell-mediated bladder inflammation in an autoimmune cystitis model. J Autoimmun. 2008;30:257–265. doi: 10.1016/j.jaut.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu W, Evanoff DP, Chen X, Luo Y. Urinary bladder epithelium antigen induces CD8+ T cell tolerance, activation, and autoimmune response. J Immunol. 2007;178:539–546. doi: 10.4049/jimmunol.178.1.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cox PJ. Cyclophosphamide cystitis – identification of acrolein as the causative agent. Biochem Pharmacol. 1979;28:2045–2049. doi: 10.1016/0006-2952(79)90222-3. [DOI] [PubMed] [Google Scholar]
- 105.Lanteri-Minet M, Bon K, de Pommery J, Michiels JF, Menetrey D. Cyclophosphamide cystitis as a model of visceral pain in rats: model elaboration and spinal structures involved as revealed by the expression of c-Fos and Krox-24 proteins. Exp Brain Res. 1995;105:220–232. doi: 10.1007/BF00240958. [DOI] [PubMed] [Google Scholar]
- 106.Jerde TJ, Bjorling DE, Steinberg H, Warner T, Saban R. Determination of mouse bladder inflammatory response to E. coli lipopolysaccharide. Urol Res. 2000;28:269–273. doi: 10.1007/s002400000114. [DOI] [PubMed] [Google Scholar]
- 107.Saban MR, Saban R, Hammond TG, Haak-Frendscho M, Steinberg H, Tengowski MW, Bjorling DE. LPS-sensory peptide communication in experimental cystitis. Am J Physiol Renal Physiol. 2002;282:F202–210. doi: 10.1152/ajprenal.0163.2001. [DOI] [PubMed] [Google Scholar]
- 108.Olsson LE, Wheeler MA, Sessa WC, Weiss RM. Bladder instillation and intraperitoneal injection of Escherichia coli lipopolysaccharide up-regulate cytokines and iNOS in rat urinary bladder. J Pharmacol Exp Ther. 1998;284:1203–1208. [PubMed] [Google Scholar]
- 109.Stein PC, Pham H, Ito T, Parsons CL. Bladder injury model induced in rats by exposure to protamine sulfate followed by bacterial endotoxin. J Urol. 1996;155:1133–1138. [PubMed] [Google Scholar]
- 110.Meyer-Siegler KL, Ordorica RC, Vera PL. Macrophage migration inhibitory factor is upregulated in an endotoxin-induced model of bladder inflammation in rats. J Interferon Cytokine Res. 2004;24:55–63. doi: 10.1089/107999004772719918. [DOI] [PubMed] [Google Scholar]
- 111.Weng TI, Chen WJ, Liu SH. Bladder instillation of Escherichia coli lipopolysaccharide alters the muscle contractions in rat urinary bladder via a protein kinase C-related pathway. Toxicol Appl Pharmacol. 2005;208:163–169. doi: 10.1016/j.taap.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 112.Wille PR, Vitor R, Gabilan NH, Nicolau M. Plasma extravasation mediated by lipopolysaccharide-induction of kinin B1 receptors in rat tissues. Mediators Inflamm. 2001;10:163–167. doi: 10.1080/09629350124164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tapping RI, Akashi S, Miyake K, Godowski PJ, Tobias PS. Toll-like receptor 4, but not toll-like receptor 2, is a signaling receptor for Escherichia and Salmonella lipopolysaccharides. J Immunol. 2000;165:5780–5787. doi: 10.4049/jimmunol.165.10.5780. [DOI] [PubMed] [Google Scholar]
- 114.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 115.Schilling JD, Mulvey MA, Vincent CD, Lorenz RG, Hultgren SJ. Bacterial invasion augments epithelial cytokine responses to Escherichia coli through a lipopolysaccharide-dependent mechanism. J Immunol. 2001;166:1148–1155. doi: 10.4049/jimmunol.166.2.1148. [DOI] [PubMed] [Google Scholar]
- 116.Rouanet C, Locht C. Boosting BCG to protect against TB. Expert Rev Respir Med. 2010;4:339–348. doi: 10.1586/ers.10.25. [DOI] [PubMed] [Google Scholar]
- 117.Sexton WJ, Wiegand LR, Correa JJ, Politis C, Dickinson SI, Kang LC. Bladder cancer: a review of non-muscle invasive disease. Cancer Control. 2010;17:256–268. doi: 10.1177/107327481001700406. [DOI] [PubMed] [Google Scholar]
- 118.Saban MR, Simpson C, Davis C, Wallis G, Knowlton N, Frank MB, Centola M, Gallucci RM, Saban R. Discriminators of mouse bladder response to intravesical Bacillus Calmette-Guerin (BCG) BMC Immunol. 2007 May 16;8:6. doi: 10.1186/1471-2172-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.McMahon SB, Abel C. A model for the study of visceral pain states: chronic inflammation of the chronic decerebrate rat urinary bladder by irritant chemicals. Pain. 1987;28:109–127. doi: 10.1016/0304-3959(87)91065-7. [DOI] [PubMed] [Google Scholar]
- 120.Rivas DA, Chancellor MB, Shupp-Byrne S, Shenot PJ, McHugh K, McCue P. Molecular marker for development of interstitial cystitis in rat model: isoactin gene expression. J Urol. 1997;157:1937–1940. doi: 10.1016/s0022-5347(01)64905-x. [DOI] [PubMed] [Google Scholar]
- 121.Hauser PJ, Buethe DA, Califano J, Sofinowski TM, Culkin DJ, Hurst RE. Restoring barrier function to acid damaged bladder by intravesical chondroitin sulfate. J Urol. 2009;182:2477–2482. doi: 10.1016/j.juro.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fraser MO, Chuang YC, Tyagi P, Yokoyama T, Yoshimura N, Huang L, De Groat WC, Chancellor MB. Intravesical liposome administration--a novel treatment for hyperactive bladder in the rat. Urology. 2003;61:656–663. doi: 10.1016/s0090-4295(02)02281-1. [DOI] [PubMed] [Google Scholar]
- 123.Birder LA, Nakamura Y, Kiss S, Nealen ML, Barrick S, Kanai AJ, Wang E, Ruiz G, de Groat WC, Apodaca G, Watkins S, Caterina MJ. Altered urinary bladder function in mice lacking the vanilloid receptor TRPV1. Nat Neurosci. 2002;5:856–860. doi: 10.1038/nn902. [DOI] [PubMed] [Google Scholar]
- 124.Arms L, Girard BM, Vizzard MA. Expression and function of CXCL12/CXCR4 in rat urinary bladder with cyclophosphamide-induced cystitis. Am J Physiol Renal Physiol. 2010;298:F589–600. doi: 10.1152/ajprenal.00628.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cheppudira BP, Girard BM, Malley SE, Schutz KC, May V, Vizzard MA. Upregulation of vascular endothelial growth factor isoform VEGF-164 and receptors (VEGFR-2, Npn-1, and Npn-2) in rats with cyclophosphamide-induced cystitis. Am J Physiol Renal Physiol. 2008;295:F826–836. doi: 10.1152/ajprenal.90305.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cheppudira BP, Girard BM, Malley SE, Dattilio A, Schutz KC, May V, Vizzard MA. Involvement of JAK-STAT signaling/function after cyclophosphamide-induced bladder inflammation in female rats. Am J Physiol Renal Physiol. 2009;297:F1038–1044. doi: 10.1152/ajprenal.00110.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Corrow KA, Vizzard MA. Phosphorylation of extracellular signal-regulated kinases in bladder afferent pathways with cyclophosphamide-induced cystitis. Neuroscience. 2009;163:1353–1362. doi: 10.1016/j.neuroscience.2009.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Girard BM, Malley SE, Braas KM, Waschek JA, May V, Vizzard MA. Exaggerated expression of inflammatory mediators in vasoactive intestinal polypeptide knockout (VIP-/-) mice with cyclophosphamide (CYP)-induced cystitis. J Mol Neurosci. 2008;36:188–199. doi: 10.1007/s12031-008-9084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Girard BM, Wolf-Johnston A, Braas KM, Birder LA, May V, Vizzard MA. PACAP-mediated ATP release from rat urothelium and regulation of PACAP/VIP and receptor mRNA in micturition pathways after cyclophosphamide (CYP)-induced cystitis. J Mol Neurosci. 2008;36:310–320. doi: 10.1007/s12031-008-9104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rubin JS, Rubin RT. Cyclophosphamide hemorrhagic cystitis. J Urol. 1966;96:313–316. doi: 10.1016/S0022-5347(17)63260-9. [DOI] [PubMed] [Google Scholar]
- 131.Locher GW, Cooper EH. Repair of rat urinary bladder epithelium following injury by cyclophosphamide. Invest Urol. 1970;8:116–123. [PubMed] [Google Scholar]
- 132.Okamura T, Garland EM, Taylor RJ, Johansson SL, Cohen SM. The effect of cyclophosphamide administration on the kidney of the rat. Toxicol Lett. 1992;63:261–276. doi: 10.1016/0378-4274(92)90089-3. [DOI] [PubMed] [Google Scholar]
- 133.Kalota SJ, Stein PC, Parsons CL. Prevention of acrolein-induced bladder injury by pentosanpolysulfate. J Urol. 1992;148:163–166. doi: 10.1016/s0022-5347(17)36545-x. [DOI] [PubMed] [Google Scholar]
- 134.Sakata T, Smith RA, Garland EM, Cohen SM. Rat urinary bladder epithelial lesions induced by acrolein. J Environ Pathol Toxicol Oncol. 1989;9:159–169. [PubMed] [Google Scholar]
- 135.Fraiser L, Kehrer JP. Murine strain differences in metabolism and bladder toxicity of cyclophosphamide. Toxicology. 1992;75:257–272. doi: 10.1016/0300-483x(92)90007-2. [DOI] [PubMed] [Google Scholar]
- 136.Bjorling DE, Elkahwaji JE, Bushman W, Janda LM, Boldon K, Hopkins WJ, Wang ZY. Acute acrolein-induced cystitis in mice. BJU Int. 2007;99:1523–1529. doi: 10.1111/j.1464-410X.2007.06773.x. [DOI] [PubMed] [Google Scholar]
- 137. NIH website: http://www.niddk.nih.gov/patient/mtops/mtops-q&a.htm.
- 138.Abrams P, Cardozo L, Fall M, Griffiths D, Rosier P, Ulmsten U, van Kerrebroeck P, Victor A, Wein A, Standardisation Sub-committee of the International Continence Society The standardisation of terminology of lower urinary tract function: report from the Standardisation Sub-committee of the International Continence Society. Neurourol Urodyn. 2002;21:167–178. doi: 10.1002/nau.10052. [DOI] [PubMed] [Google Scholar]
- 139.Chapple CR, Roehrborn CG. A shifted paradigm for the further understanding, evaluation, and treatment of lower urinary tract symptoms in men: focus on the bladder. Eur Urol. 2006;49:651–658. doi: 10.1016/j.eururo.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 140.Dmochowski RR. Bladder outlet obstruction: etiology and evaluation. Rev Urol. 2005;7(Suppl 6):S3–S13. [PMC free article] [PubMed] [Google Scholar]
- 141.Knutson T, Edlund C, Fall M, Dahlstrand C. BPH with coexisting overactive bladder dysfunction--an everyday urological dilemma. Neurourol Urodyn. 2001;20:237–247. doi: 10.1002/nau.1001. [DOI] [PubMed] [Google Scholar]
- 142.Mirone V, Imbimbo C, Longo N, Fusco F. The detrusor muscle: an innocent victim of bladder outlet obstruction. Eur Urol. 2007;51:57–66. doi: 10.1016/j.eururo.2006.07.050. [DOI] [PubMed] [Google Scholar]
- 143.Gomes CM, Nunes RV, Araújo RM, Sacomani CR, Trigo-Rocha FE, Bruschini H, Srougi M. Urodynamic evaluation of patients with lower urinary tract symptoms and small prostate volume. Urol Int. 2008;81(2):129–34. doi: 10.1159/000144049. [DOI] [PubMed] [Google Scholar]
- 144.Nickel JC. Prostatic inflammation in benign prostatic hyperplasia - the third component? Can J Urol. 1994;1:1–4. [PubMed] [Google Scholar]
- 145.Krieger JN, Riley DE. Prostatitis: what is the role of infection. Int J Antimicrob Agents. 2002;19:475–479. doi: 10.1016/s0924-8579(02)00086-9. [DOI] [PubMed] [Google Scholar]
- 146.Hochreiter WW, Duncan JL, Schaeffer AJ. Evaluation of the bacterial flora of the prostate using a 16S rRNA gene based polymerase chain reaction. J Urol. 2000;163:127–130. [PubMed] [Google Scholar]
- 147.Weiss J, Wein A, Jacobs J, Hanno P. Use of nitrofurantoin macrocrystals after transurethral prostatectomy. J Urol. 1983;130:479–480. doi: 10.1016/s0022-5347(17)51258-6. [DOI] [PubMed] [Google Scholar]
- 148.Kirby RS, Lowe D, Bultitude MI, Shuttleworth KE. Intra-prostatic urinary reflux: an aetiological factor in abacterial prostatitis. Br J Urol. 1982;54:729–731. doi: 10.1111/j.1464-410x.1982.tb13635.x. [DOI] [PubMed] [Google Scholar]
- 149.Pontari MA, Ruggieri MR. Mechanisms in prostatitis/chronic pelvic pain syndrome. J Urol. 2004;172:839–845. doi: 10.1097/01.ju.0000136002.76898.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Elkahwaji JE, Zhong W, Hopkins WJ, Bushman W. Chronic bacterial infection and inflammation incite reactive hyperplasia in a mouse model of chronic prostatitis. Prostate. 2007;67:14–21. doi: 10.1002/pros.20445. [DOI] [PubMed] [Google Scholar]
- 151.Uehling DT, Hopkins WJ, Jensen J, Balish E. Vaginal immunization against induced cystitis in monkeys. J Urol. 1987;137:327–329. doi: 10.1016/s0022-5347(17)44015-8. [DOI] [PubMed] [Google Scholar]