

Abstract
The advancement of non-viral gene therapy hinges on the ability to exert highly specific spatial and temporal control of gene delivery systems to enable localized release of DNA. In this work, we have developed a system capable of promoting localized delivery of a plasmid by utilizing peptide nucleic acid (PNA) technology to bind DNA to a substrate via an enzymatically-labile peptide sequence. The successful immobilization of the DNA to the model substrate as well as the specificity of the binding was confirmed with atomic force microscopy (AFM) and AFM-confocal overlay imaging. Fluorescence-based quantification revealed that surfaces treated with the conjugates had 49±22 ng of DNA/cm2, while there were 4.2±2.1 ng of DNA/cm2 on surfaces treated with unfunctionalized DNA. When NIH/3T3 cells were grown on the modified substrates, a significantly higher percentage of cells were transfected when the peptide tether was protease-sensitive as compared with when it was not labile. These results indicated that the peptide must be cleaved to release the DNA. In addition to providing cell-triggered release, this system decouples the properties of the complexation agent and the substrate from the method of immobilization/release to provide a model system that can be tailored to specific applications.
Keywords: Substrate-Mediated Gene Delivery, Peptide Nucleic Acid (PNA), Matrix Metalloproteinase, Self-Assembled Monolayer (SAM), Poly(ethylenimine) (PEI)
Introduction
Gene delivery enables mammalian cell manipulation necessary for disease treatment, tissue development, and comprehension of biochemical functions and cellular response.1-3 It has been shown that delivery of biochemical cues such as growth factors into a wound can enhance regeneration and repair.4,5 Given the inherent instability of many recombinant proteins,6 protein-encoding pDNA has been packaged into viral and non-viral vehicles to promote more efficient therapeutic delivery. The process of evolutionary selection has made viral vehicles more efficient at gene delivery, yet safety concerns have limited their use.1 Thus, non-viral delivery has been increasingly explored due to its versatility and the ability to control the chemical and physical properties of the vehicle. However, currently most non-viral gene delivery is conducted as a bolus delivery leading to inefficient transfection due to mass transport issues in addition to degradation, aggregation, and clearance of the pDNA-containing particles from the system.7
For many applications, these delivery inefficiencies may be overcome by surface immobilization of the vehicles.7 Substrate-mediated gene delivery of non-viral vehicles mimics the natural delivery mechanism of viruses.8 Surface delivery enables localized cellular internalization and avoids undesired gene expression in neighboring tissues.1 Furthermore, aggregation of pDNA-containing polyplexes has been shown to be inhibited by polyplex immobilization, demonstrating that immobilization maintains vehicle activity9 and reduces systemic removal.10 The preservation of vector activity and increased pDNA concentration in the cellular microenvironment elevates the efficiency of gene transfer, facilitating transgene expression levels comparable to or better than those achieved with bolus delivery, while delivering lower quantities of surface-immobilized pDNA.7,11 In addition, substrate-mediated systems have shown the ability to spatially target cells7,12,13 and may enable sustained gene delivery.14 As such, gene delivery from a biomaterial has been shown to augment tissue repair and regeneration by enabling localized production of therapeutic proteins and allowing biochemical manipulation of cells in a three-dimensional scaffold that supports cell adhesion and tissue growth.8,15
pDNA immobilization to a biomaterial has been accomplished via non-specific and specific interactions. Non-specific surface adsorption occurs by hydrophobic interactions, van der Waals forces, and electrostatics, depending on the molecular composition of the vehicle and surface.7 Non-specific immobilization approaches typically utilize the interaction of pDNA with cationic agents, including poly-L-lysine, polyethylenimine (PEI),7 chitosan,16 N,N-(dimethylamino)ethylmethacrylate (DMAEMA),17 or dendrimers such as polyamidoamine.18 For instance, multi-layered thin films of pDNA with poly(2-aminoethyl propylene phosphate)19 and pDNA with side-chain functionalized poly(β-amino ester)s have shown the ability to promote localized gene delivery and sustained release. Moreover, the pDNA/side-chain functionalized poly(β-amino ester) films have enabled sequential delivery of multiple different plasmids from a surface.20 Strategies for specific immobilization include the use of complementary functional groups on the pDNA and surface, antigen-antibody interactions, and biotin-avidin interactions.1 For example, viral vectors have been designed that specifically interact with natural and synthetic biomaterials through the use of antibodies or covalent coupling to allow for site-specific gene delivery.21 Histidine-tagged PEI polyplexes have been immobilized to a nickel nitrilotriacetic acid self-assembled monolayer (SAM) on gold.22 Additionally, biotinylated-PEI has been used to complex pDNA and sequester the complexes on avidin-functionalized surfaces.23 While these systems provide many delivery advantages, efficient transfection using these immobilized vehicles requires careful control to balance the binding of the vehicle to the substrate with the ability to release the vehicle for cellular uptake.7 Manipulation of the rate of vehicle release thus inherently requires alteration of the properties of the vehicle and/or substrate. Furthermore, release is not cell-specific, and will occur for any cell that comes into contact with the vehicle.
Due to these limitations, we have designed a surface-mediated gene delivery system that decouples the physical and chemical properties of the vehicle and substrate from the release mechanism and also enables on-demand, cell-triggered release. In our system, pDNA is bound to a substrate via an enzymatically-labile peptide sequence, as shown in Figure 1. This system allows for surface immobilization, control over payload, and overall greater control over cellular transfection. Our design focuses on chemical functionalization of pDNA using a peptide nucleic acid (PNA) clamp and a peptide-based surface tethering system. The PNA clamp, a nucleic acid analogue with a neutral polyamide backbone, sequence-specifically binds to a complementary DNA sequence to form a nuclease- and protease-resistant triplex that does not interfere with the transcriptional activity of the plasmid. Furthermore, by the use of a maleimide-functionalized PNA clamp, pDNA is functionalized to allow direct covalent coupling of peptides and oligonucleotides.24
Figure 1.

DNA Immobilization and cell-responsive release from a model substrate. (A) A SAM-modified (orange lines) gold-coated surface is covalently attached to pDNA-PNA-peptide (DPP) conjugates. (B) The SAM-bound conjugates are complexed with a polycation (+). (C) Upon cellular migration along the substrate and the release of matrix metalloproteinase (MMP) (lightning bolts), the surface is capable of cell-responsively releasing pDNA and transfecting cells.
In our system, we couple a peptide that includes cell adhesive and matrix metalloproteinase (MMP) degradable sequences to promote the release and uptake of the plasmid in a cell-responsive manner. It has been shown that the incorporation of biologically-functional peptide sequences in artificial materials will enable the materials to respond to cellular stimuli. For example, upon exposure to the MMP enzyme, bulk gel degradation of PEG-based hydrogels crosslinked with MMP-cleavable peptides occurred and promoted release of proteins or pDNA sequestered within the gels.15,25-27 For our work, MMP was chosen as a model enzymatic system due to the importance of the MMP family of enzymes in tissue growth, development, and remodeling. MMPs are upregulated in wound sites as well as in cells involved in illnesses such as arthritis and cancer.28 After pDNA-PNA-peptide (DPP) conjugate formation, the DPP conjugates may be directly linked to a variety of biomaterial surfaces. We chose to establish the system by attaching the conjugates to a gold surface modified with a self-assembled monolayer (SAM). The ability to form well-defined surfaces that are easily manipulated motivated the use of SAM technology in our study.29 In this work, we show evidence of surface immobilization of the conjugates and the ability to transfect cells in a cell-responsive manner. This represents the first example of the application of PNA technology to surface-mediated gene delivery, and importantly, provides a new means for substrate-independent, cell-triggered DNA delivery for a variety of therapeutic applications.
Materials and Methods
Materials
25 kDa PEI, N-(3-dimethylpropyl)-N-ethylcarbodiimide hydrochloride (EDC), N-hydroxysulfo-succinimide sodium salt (sulfo-NHS), and 11-mercaptoundecanoic acid (MUA) were purchased from Sigma-Aldrich (St. Louis, MO). 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and ethylenediaminetetraacetic acid (EDTA) were obtained from Acros Organics (Morris Plains, NJ). The gWiz plasmid, containing 10 sequential copies of the PNA-binding site 5′-AGAGAGAG-3′ and encoding for green fluorescent protein (GFP), was purchased from Genlantis (San Diego, CA). The plasmid was amplified in DH5α competent E. coli, purified from bacterial culture using Qiagen reagents (Santa Clara, CA), and stored in Tris-EDTA buffer solution (pH=8.0). The bis-PNA (maleimide-OOTCTCTCTC-OOO-JTJTJTJT-CONH2, where O = 8-amino-3,6-dioxaoctanoic acid and J = pseudoisocytosine) was synthesized by solid-phase synthesis and HPLC-purified to >95% purity by Panagene (Daejeon, Korea). The MMP-labile peptide sequence (NH2-GKGGPQG↓IWGQGRGDSPGDRCK-COOH, where ↓ denotes the MMP cleavage site) and the non-MMP-labile peptide sequence (NH2-KGDQGIAGFKGRGDSPGDRCG-CONH2)26 were synthesized by solid-phase synthesis and HPLC-purified to >95% by Pi Proteomics (Huntsville, AL). The non-MMP-labile peptide sequence of GDQGIAGF was chosen due to its inability to be cleaved by collagenase and the precedence for its use as a negative control for MMP protease degradation.26,27 Bacterial collagenase, type 1 was obtained from Worthington (Lakewood, NJ). YOYO®-1 Iodide and Quant-iT™ PicoGreen® dyes were purchased from Molecular Probes (Eugene, OR). All other reagents were obtained from Fisher Scientific (Fairlawn, NJ) unless otherwise noted.
Formation of pDNA-PNA-Peptide (DPP) Conjugates
As detailed in Figure 2A, the formation of the DPP conjugates occurred in two steps, and was conducted according to a slight modification of established protocols.30 First, a 0.05 μM solution of recombinant gWiz was mixed with PNA in a 32.5:1 molar ratio of PNA to pDNA in 20 mM HEPES buffer (pH=6.0) and incubated overnight at 37°C. The pDNA-PNA solution was diluted with an equivalent volume of 20 mM HEPES buffer containing 150 mM NaCl (pH=7.4). MMP-labile or non-MMP-labile peptides were added in a 20:1 molar ratio of peptide to PNA, and the pDNA-PNA-peptide solution was incubated overnight at 37°C. The DPP conjugates were purified and concentrated with YM-30 centrifugal filtration devices (Millipore, Billerica, MA). The pDNA concentration of the final product was determined using a Thermo Scientific NanoDrop Spectrophotometer (Wilmington, DE). As previously established, the stoichiometry of the DPP product was 1:8:7 pDNA:PNA:peptide.30,31
Figure 2.

Scheme for DPP immobilization to a model substrate. (A) The DPP conjugates are formed in two steps including: (i) the addition of maleimide-PNA (blue) to pDNA (black circles) to form a stable triplex followed by (ii) the binding of thiol-terminated peptide (green) to the pDNA-PNA by thiol-maleimide reaction. The gold-coated surface is activated by (B) MUA (orange) treatment of the gold-coated surface to form a SAM with terminal carboxylic acid functionalities. The DPP conjugates are attached to the surface by the (C) reaction of the amine functionality of the N-terminus of the peptide with the surface carboxylic acid in the presence of NHS and EDC to form stable amide bonds between the conjugates and the surface.
SAM Formation
For atomic force microscopy (AFM) and plate reader assays, 500 Å gold-coated mica slides were obtained from Asylum Research (Santa Barbara, CA) and punched into 12.7 mm discs. For AFM-confocal overlay analyses and transfection studies, 100 Å gold-coated glass discs (15 mm diameter) were purchased from Platypus Technologies (Madison, WI). SAM formation was conducted by established protocols.32 Briefly, the gold-coated substrates were washed with acidic ethanol (pH=2). The surfaces were then incubated in 1 mM MUA in acidic ethanol under nitrogen gas for 24 h at 4°C (Figure 2B). The substrates were then washed with acidic ethanol and ethanol. A FTÅ125 system (First Ten Ångstroms, Portsmouth, VA) was used to confirm SAM formation via contact angle measurements (data not shown).
DPP immobilization
The DPP conjugates were immobilized onto the SAM-modified gold-coated surfaces by incubation of the conjugates with the surfaces in the presence of EDC and sulfo-NHS (Figure 2C). A solution of DPP conjugate in 2 mM EDC and 5 mM sulfo-NHS was placed onto each modified surface. As the C-terminal lysine will be proximal to the DNA, thus limiting its ability to react, amide bond formation should take place between the N-terminus of the peptide or the ε-amine of the lysine at position 2 and the terminal carboxylic acid functionality of the SAM on the surface. The surfaces were washed extensively with 20 mM HEPES buffer (pH=6.0) to remove unbound conjugates, and were subsequently treated with PEI at an N/P ratio of 10 to complex the immobilized DNA. Unbound PEI was removed by further washing with 20 mM HEPES buffer (pH=6.0).
DPP Surface Characterization
AFM images were taken by tapping in air on a NanoScope IIIa multimode scanning probe microscope (SPM) (Digital Instruments, Santa Barbara, CA). Etched silicon RTESP tips (Veeco, Santa Barbara, CA) with a nominal spring constant of 40 N/m were used. The surfaces for this analysis were prepared as described on the 500 Å gold-coated mica surfaces. Image processing was performed using the Nanoscope software v6.13 (Digital Instruments/Veeco) and ImageJ (NIH, Bethesda, MD).
AFM-confocal overlay imaging was performed by tapping mode in air on a Bioscope II SPM (Veeco) and by imaging of the same areas on an LSM 510 NLO multiphoton microscope (Carl Zeiss, Inc., Thornwood, NY) with a 100x objective. To facilitate dual imaging, the multiphoton stage was removed and replaced with the SPM stage. Upon the direction of the SPM software, confocal images of the surface were taken and input into the SPM software to allow for sample alignment between the two systems. Following alignment, 40 μm × 40 μm AFM images and 92.1 μm × 92.1 μm confocal images of the surfaces were taken. For AFM imaging, etched silicon RTESP tips (Veeco) with a nominal spring constant of 40 N/m were used at a scan rate of 1 Hz. The surfaces for this analysis were prepared on 100 Å gold-coated glass discs as described, except that prior to PEI complexation, the surfaces were stained with YOYO®-1 iodide in 1X Dulbecco’s phosphate buffered saline (DPBS) by incubation at room temperature for 1 h. Image processing was performed using Zeiss LSM Image Examiner (Carl Zeiss, Inc.), Nanoscope software v7.3 (Veeco), and Adobe® Photoshop® (Adobe Systems, Inc., San Jose, CA). The AFM image was rotated 90° and vertically inverted to show the same perspective of the sample as imaged by the confocal. The confocal image size was increased so the scale bars in both images indicated equal distances and then the confocal image was cropped to 40 μm × 40 μm (the same size as the AFM scan).
Fluorescent plate reader assays were performed with the area scan function on a BioTek Synergy 2 Multi-Mode Microplate Reader (Winooski, VT). The 500 Å gold-coated mica surfaces were prepared as above but excluding the PEI-complexation step. The surfaces were immobilized in an opaque 6-well dish and treated with Quant-iT™ PicoGreen®. The measured fluorescent surface intensities were correlated to pDNA solution concentrations using a standard curve. The solution concentrations of pDNA were then used to calculate the density of pDNA on the modified surfaces.
Cell culture and transfection
NIH/3T3 murine fibroblasts (CRL-1658, American Type Culture Collection, Manassas, VA) were cultured in a humidified 5% CO2 atmosphere at 37°C in Dulbecco’s Modified Eagle Medium (DMEM) (Mediatech, Manassas, VA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Mediatech) and 1% penicillin-streptomycin solution (Mediatech). When confluent, the cells were passaged by washing with 1X DPBS (Mediatech) and treatment with a solution containing 0.25% trypsin and 2.21 mM EDTA (Mediatech).
For the transfection and cytotoxicity studies, surfaces were created as described on 100 Å gold-coated glass discs. Cells were seeded at a density of 10,000 cells per surface. In the absence of other stimuli, the media was supplemented with 0.05 U/μL collagenase, which is approximately 10 to 300-fold more concentrated than the bulk MMP concentration in wound healing scenarios in vivo.33 For the cytotoxicity assays, cell culture media was removed and the cells were washed with 1X DPBS (Mediatech) and 4 μM ethidium homodimer-1 (AnaSpec, Fremont, CA) was added. Cytotoxicity and cell transfection were investigated by fluorescent imaging of the cells with a Leica AF6000 microscope (Bannockburn, IL). To determine the cell transfection efficiency, the cells were collected via trypsinization, washed, and resuspended in 1X DPBS (Mediatech) buffer prior to quantification of GFP expression with a Becton Dickinson FACSCalibur flow cytometer (Franklin Lakes, NJ). Between 800 and 1000 cell events were counted per sample. Processing of the data was performed using the WinMdi software package (The Scripps Research Institute, La Jolla, CA).
Results and Discussion
DPP-Modified Surface Topography
AFM imaging (Figure 3) indicated the successful immobilization of the conjugates onto the model substrates. Samples created by treatment with 0.02 ng/μL (Figure 3B) or 2 ng/μL (Figure 3C) of the DPP conjugates showed discrete and significant features on their surfaces, whereas the SAM-modified surfaces (Figure 3A) and the surfaces treated with 0.2 μg/μL of unconjugated pDNA (Figure 3D) did not. The SAM surface had a maximum feature height of 4.8±1.3 nm, and the unconjugated pDNA-treated surface also had a maximum feature height of 4.8±0.8 nm. In comparison, treatment with 0.02 ng/μL or 2 ng/μL DPP produced surface protrusions with maximum heights of 17.1±6.8 nm and 15.0±2.7 nm, respectively. These height values were statistically different from those of the SAM-modified surface (p≤0.03), suggesting that surface binding of the DPP conjugates was occurring. Although the feature height on the DPP surfaces was smaller than the diameter of a typical pDNA polyplex or pDNA-PEI complex (ca. 100 – 150 nm),23,34 the differences are most likely caused by the method of pDNA immobilization that was used in this work. As the pDNA was tethered to the substrate and then treated with PEI, the complex formation process was constrained by the surface, potentially causing the pDNA to be uniformly spread out on the surface.
Figure 3.

Topography of the modified surfaces. All samples were created by SAM modification followed by additional treatments. Negative controls include (A) an unmodified SAM surface and (D) a SAM surface treated with 0.2 μg/μL pDNA. Samples include (B) a SAM surface treated with 0.02 ng/μL DPP conjugates and (C) a SAM surface treated with 2 ng/μL DPP conjugates. All surfaces were treated with PEI. Each sample was prepared and analyzed 10 times with representative images shown.
The height of the features on the surfaces treated with 0.02 ng/μL of the DPP conjugate was not found to be statistically different from the feature height on the surfaces treated with 2 ng/μL of the DPP conjugate (p≥0.03). This result suggested that multi-layers of pDNA were not forming on the substrates, consistent with the covalent mode of DPP-surface attachment. Further evidence of the formation of covalent bonds between the DPP conjugates and the surface was provided by experiments with unmodified pDNA. The feature height observed for the surfaces prepared in the presence of unmodified pDNA was not statistically different from that found for the SAM-treated surface (p≥0.03), signifying that there was little to no adsorption of unconjugated pDNA to the surfaces. This suggested that amide bond formation was necessary for immobilization. The larger DPP conjugate concentration did produce an 8-fold increase in the fractional surface coverage attained by treatment. Although one might expect a larger increase in surface coverage, given the 100-fold increase in the treatment concentration, the surfaces appeared to be saturated by treatment with 2 ng/μL of the DPP conjugate, which was confirmed by plate reader assays (Figure 5 and Figure 6). Thus, it is likely that the conjugation of additional DPP conjugates to the surface was impossible due to steric limitations.
Figure 5.

Density of surface-bound DPP as a function of the DPP treatment concentration. Samples include SAM-modified gold-coated mica surfaces treated with 1 ng/μL, 2.5 ng/μL, 5 ng/μL, or 10 ng/μL DPP. The samples were stained with PicoGreen®, excited at 485 nm, and analyzed by measuring the fluorescence emission intensity at 528 nm. The error bars represent one standard deviation from the mean of ≥7 separate samples. The values are not statistically different.
Figure 6.
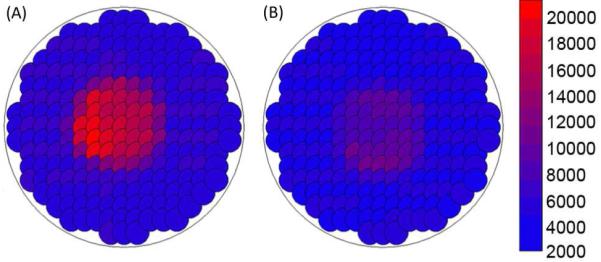
Quantification of DPP binding by plate reader area scanning. Samples include (A) a SAM-modified gold-coated mica surface treated with 0.1 μg/μL DPP, and (B) a SAM-modified gold-coated mica surface treated with 0.1 μg/μL unconjugated pDNA. The samples were stained with PicoGreen®, excited at 485 nm, and analyzed by measuring the fluorescence emission intensity at 528 nm.
Although the AFM images indicated modification of the surfaces with the DPP conjugates, confirmation of the presence of pDNA in the observed features was necessary. To determine if the protrusions observed in the AFM images of the DPP surfaces corresponded to surface-bound pDNA, AFM-confocal overlay analyses were completed on modified surfaces treated with YOYO®-1 Iodide to specifically stain the pDNA on the samples. Scratches in the images (Figure 4) are due to the removal of gold by handling of the surfaces. Surfaces that were treated with unconjugated pDNA instead of with the DPP conjugates did not display significant fluorescent features (Figure 4D). This observation confirmed that minimal non-specific DNA binding was occurring and demonstrated that the fluorescent staining that was observed corresponded to DNA. The images obtained for the DPP conjugate-treated surfaces showed that the significant surface features did contain pDNA, as the fluorescent features observed in the confocal images (Figure 4B) and the topographical features observed in the AFM images (Figure 4A) were colocalized. These experiments also showed that the DPP conjugates were stable under the mild shearing conditions used for surface preparation.
Figure 4.
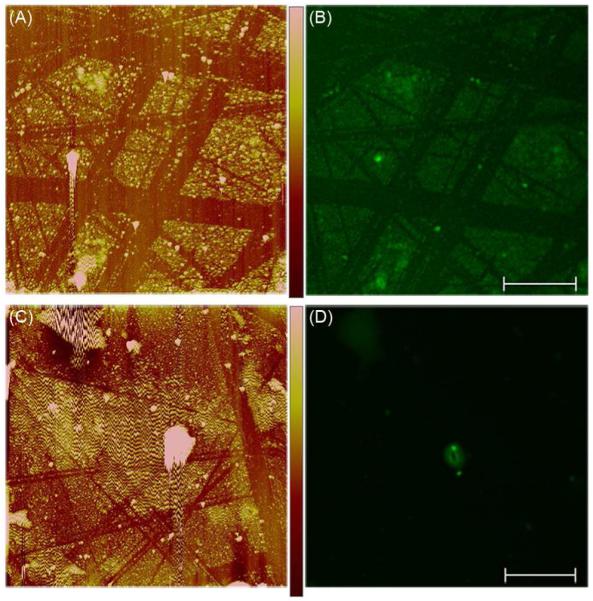
Localization of pDNA on modified surfaces. The samples were prepared by treating SAM-modified gold-coated glass surfaces with 0.1 μg/μL DPP [(A) and (B)] or 0.1 μg/μL pDNA [(C) and (D)] followed by staining with YOYO®-1 Iodide. (A) and (C) The surface modifications observed by AFM imaging. (B) and (D) The fluorescent features observed by confocal microscopy. Color-coded height scale [shown in (A)] = 100 nm and scale bar [shown in (B)] = 10 μm. Each sample was prepared and analyzed 10 times with representative images shown.
Quantification of Surface-Immobilized DPP
Following verification of the surface immobilization of pDNA via the PNA- and peptide-based tethering mechanism, the amount of pDNA present on DPP-modified surfaces and surfaces treated with non-conjugated pDNA was quantified with a fluorescent assay. Prior to completion of the experiment, theoretical calculations were completed to predict the quantity of pDNA necessary to saturate the surface. The radius of gyration of a 5.4 kb pDNA was determined to be ~100 nm by the use of a worm-like polymer model35 with a persistence length of 50 nm.36 Ignoring electrostatic repulsion of the DPP, it was determined that a ~127 mm2 surface would hold a maximum of ~20 ng of pDNA. Experimentally, it was determined that the DPP surface at saturation had 62±27 ng of pDNA and that saturation occurred between incubation concentrations of 1 ng/μL and 2.5 ng/μL of DPP (Figure 5). As the theoretical and experimental values for surface saturation were of the same order of magnitude, the experimental results appeared to be accurate. In addition, area scans of saturated DPP-treated surfaces (Figure 6A) and the corresponding pDNA-treated surfaces (Figure 6B) were also performed. By comparison with plate reader scans of known quantities of pDNA in solution, it was determined that the saturated DPP-treated surface had 49±22 ng of pDNA/cm2, while there were 4.2±2.1 ng of pDNA/cm2 on the pDNA-treated surface. The results of this study (Figure 6) gave further evidence of the bond specificity, as the DPP surface had 12-fold more bound pDNA as compared to the pDNA-treated surface.
Cellular Transfection & Cytotoxicity
Subsequent to surface characterization and validation of the DPP-tethering mechanism, the biological properties of the system were explored by the completion of transfection and cytotoxicity studies. To demonstrate the cell-responsive nature of the system, cellular transfection studies were completed in NIH/3T3 cells growing on surfaces that had been saturated with DPP conjugates containing either MMP-labile peptides or non-MMP-labile peptides (DPP or sDPP conjugates, respectively). Again, scratches in the images (Figure 7) are due to handling of the surfaces causing removal of some of the gold coating on the substrate. As assessed by fluorescence microscopy, neither the SAM surface (Figure 7A) nor the sDPP surface (Figure 7C) were able to produce GFP-expressing cells. In comparison, the DPP surface (Figure 7B) induced GFP expression within a fraction of the cells. Quantification of gene expression was performed via flow cytometry (Figure 7D), and revealed transfection efficiencies of 23.5±7.5% for the DPP surface and 7.4±3.4% for the sDPP surface. In addition, cytotoxicity studies (Figure 8) revealed 94±2%, 88±3%, and 86±4% of cells remained viable when plated on the SAM, DPP, and sDPP surfaces, respectively, indicating that the modified surfaces did not show significant toxicity to cells.
Figure 7.

Transfection with the modified surfaces. The transfection efficiencies of (A) a SAM-modified gold-coated glass surface, (B) a 0.1 μg/μL DPP-treated SAM-modified surface, and (C) a 0.1 μg/μL sDPP-treated SAM-modified surface were assessed in NIH/3T3 cells 24 h post-transfection by fluorescence microscopy (scale bar [shown in (A)] = 100 μm). GFP expression was further quantified by flow cytometry (D). “Auto” indicates untransfected NIH/3T3 cells. The error bars represent one standard deviation from the mean of 3 separate samples. Statistically significant differences were observed between the samples indicated with an asterisk (p≤0.04).
Figure 8.

Cytotoxicity of the modified surfaces. The viability of NIH/3T3 cells cultured on a SAM-modified gold-coated glass surface, a 0.1 μg/μL DPP-treated SAM-modified surface, and a 0.1 μg/μL sDPP-treated SAM-modified surface were assessed 24 h post-transfection by fluorescence microscopy of ethidium homodimer-1 stained samples. The error bars represent one standard deviation from the mean of 4 separate samples. Statistically significant differences were observed between the samples indicated with an asterisk or a double asterisk (p≤0.025).
The transfection studies gave evidence that the DPP surfaces were able to transfect cells with a very small quantity of pDNA. By comparison, typical in vitro transfection experiments with PEI-DNA complexes in solution usually use ~2 μg of DNA for ~50,000 cells.37 In contrast, in this work ~60 ng of DNA is used for ~10,000 cells, or an order of magnitude less pDNA than is typically used to accomplish bolus delivery. Presumably, these enhancements in the pDNA utilization efficiency resulted from the immobilization of the pDNA, and the resulting high local pDNA concentration at the surface. The DPP-modified surfaces demonstrated comparable efficiency in comparison with other substrate-mediated delivery systems. For example, lipoplexes immobilized at surface concentrations ranging from 0.1 μg of pDNA/cm2 to 0.25 μg of pDNA/cm2 on FBS-treated polystyrene accomplish up to 75% transfection efficiency.23 Lipoplexes immobilized on MUA SAMs at 63 ng/cm2 have achieved a 40% transfection efficiency.13 By comparison, the DPP surfaces created in this work contained 49±22 ng of pDNA/cm2 and transfected 23.5±7.5% of the cells. This slight decrease in transfection efficiency observed in the DPP system as compared to the lipoplex systems discussed above may be ascribed to the use of PEI, which has been reported to be less efficient at nuclear delivery than Lipofectamine.38 Furthermore, during DPP-surface preparation, unbound PEI is washed away after the complexation step, which may account for the low cytotoxicity of the surfaces (Figure 8) as well as a somewhat reduced transfection efficiency, since free PEI in solution has been found to significantly enhance the transfection efficiency of pDNA-PEI complexes.39,40 Overall, the efficiency of transfection with the DPP modified surfaces could easily be increased, either via alteration in the number of surface tethers per pDNA (in the current study, 7–8 tethers existed per conjugate), or via alteration in the transfection agent. Future work will explore the effects of reductions in the tether number and different polycations on the efficiency of pDNA release and cell transfection.
The sDPP surface had a statistically significant decrease (p≤0.04) in the amount of transfection as compared to the DPP surface (Figure 7D). The low level of transfection observed with the sDPP surface indicates that transfection is minimal when cleavage of the peptide does not occur. The specificity (Figure 6) and stability of the tethering mechanism used for pDNA immobilization make it improbable that spontaneous detachment of the formulation is the cause for this difference. The gold-thiol bond used to form the SAM13 as well as the covalent (thioether and amide) and hydrogen bonds (PNA-DNA triplex24) used to create the DPP conjugate are stable under the physiological conditions used in the completion of this work. In addition, the physicochemical properties of the DPP and sDPP peptides are essentially identical (net charge of +2, an isoelectric point of 9.9), making it unlikely that they will react differently in any matter excluding their enzymatic lability. Thus, the difference between the cell transfection efficiencies observed between the labile (DPP) and non-labile (sDPP) conjugate samples are most logically ascribed to cleavage of the peptide tether by MMP enzyme. Interestingly, in cell-free analyses, no detectable release of pDNA was observed via agarose gel electrophoretic analysis when a 0.1 U/μL collagenase solution was incubated with PEI-complexed DPP surfaces (data not shown). In addition, preliminary results using fluorescence-based assays showed that only 12 ± 2% of the surface-bound DNA was released over a period of 72 hours when DPP surfaces were incubated with 0.05 U/μL collagenase in the absence of both PEI and cells (data not shown). Given that pDNA release from PEI-complexed surfaces was precipitated by enzymatic treatment only in the presence of cells, the data may further suggest that cellular traction forces in combination with MMP activity are required to initiate pDNA release. Collectively, these data indicate both the role of the cells in the pDNA release mechanism and the fact that complexation of pDNA with PEI limits the accessibility of the peptide tether for enzymatic cleavage. Furthermore, the results suggest that this system has the potential to provide temporal control of gene delivery as well as to target pDNA delivery to specific cell lines.
Conclusions
Current methods for substrate-mediated gene delivery are effective at localized delivery, but would be improved by enhanced spatial and temporal control over the transfection process. This work details the formation of plasmid conjugates that are tethered to a model substrate followed by treatment with a polycation in such a way that the pDNA is released in a cell-responsive manner. The post-complexation of the pDNA lends uniformity to the immobilized DNA-containing structures on the surface, which is not currently attainable with polyplex immobilization due to the inherent particle size distribution.39 As polyplex particles tend to contain multiple pDNA molecules, the method of pDNA immobilization prior to complexation may also introduce more control over the delivery dosage. Additionally, the pDNA-PNA-peptide delivery system represents a significant advance in surface-mediated delivery in that it decouples the mode of pDNA-surface attachment from the properties of the delivery material and the substrate. Cell-triggered gene delivery in response to the secretion of biochemical cues that are characteristic of remodeling tissue has been accomplished using this system. Significant delivery has been demonstrated from surfaces containing an order of magnitude less pDNA than is typical for non-viral gene delivery systems. Although the transfection efficiencies observed were somewhat low, the ability to tailor the design will enable improvement in the release kinetics. This system may be modified by the variation of the enzymatically-labile peptide sequence as well as the number of surface-tether bonds. Exploration of how variations in the number of DPP tethers as well as the valency of the transfection agent impact the DNA release profiles of the system will be necessary. In the future, the tunability of this design will provide localized gene delivery with highly specific spatial and temporal control of the plasmid release.
ACKNOWLEDGMENT
The authors thank Elizabeth Adams, Kirk Czymmek, and Peter Millili for technical assistance with AFM, Confocal microscopy, and Flow cytometry. The AFM and Confocal Microscopy were performed at the Core Microscopy facility of the Delaware Biotechnology Institute (University of Delaware). In addition, the authors would like to acknowledge Julie Albert for completing the contact angle measurements. The project described was partially supported by NSF IGERT and Grant Number 2 P20 RR016472-07 under the INBRE Program of the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH).
REFERENCES
- (1).Pannier AK, Shea LD. Molecular Therapy. 2004;10:19–26. doi: 10.1016/j.ymthe.2004.03.020. [DOI] [PubMed] [Google Scholar]
- (2).Gersbach CA, Phillips JE, Garcia AJ. Annual Review of Biomedical Engineering. 2007;9:87–119. doi: 10.1146/annurev.bioeng.9.060906.151949. [DOI] [PubMed] [Google Scholar]
- (3).Han S, Mahato RI, Sung YK, Kim SW. Molecular Therapy. 2000;2:302–317. doi: 10.1006/mthe.2000.0142. [DOI] [PubMed] [Google Scholar]
- (4).Bunge MB. Neuroscientist. 2001;7:325–339. doi: 10.1177/107385840100700409. [DOI] [PubMed] [Google Scholar]
- (5).Bregman BS, Coumans JV, Dai HN, Kuhn PL, Lynskey J, McAtee M, Sandhu F. Prog Brain Res. 2002;137:257–73. doi: 10.1016/s0079-6123(02)37020-1. [DOI] [PubMed] [Google Scholar]
- (6).Bonadio J. Journal of Molecular Medicine-Jmm. 2000;78:303–311. doi: 10.1007/s001090000118. [DOI] [PubMed] [Google Scholar]
- (7).Bengali Z, Shea LD. Mrs Bulletin. 2005;30:659–662. doi: 10.1557/mrs2005.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).De Laporte L, Shea LD. Advanced Drug Delivery Reviews. 2007;59:292–307. doi: 10.1016/j.addr.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Segura T, Volk MJ, Shea LD. Journal of Controlled Release. 2003;93:69–84. doi: 10.1016/j.jconrel.2003.08.003. [DOI] [PubMed] [Google Scholar]
- (10).Gersbach CA, Coyer SR, Le Doux JM, Garcia AJ. Biomaterials. 2007;28:5121–5127. doi: 10.1016/j.biomaterials.2007.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Luo D, Saltzman WM. Nature Biotechnology. 2000;18:893–895. doi: 10.1038/78523. [DOI] [PubMed] [Google Scholar]
- (12).Ziauddin J, Sabatini DM. Nature. 2001;411:107–110. doi: 10.1038/35075114. [DOI] [PubMed] [Google Scholar]
- (13).Pannier AK, Anderson BC, Shea LD. Acta Biomaterialia. 2005;1:511–522. doi: 10.1016/j.actbio.2005.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jang JH, Rives CB, Shea LD. Molecular Therapy. 2005;12:475–483. doi: 10.1016/j.ymthe.2005.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Trentin D, Hall H, Wechsler S, Hubbell JA. Proc Natl Acad Sci U S A. 2006;103:2506–11. doi: 10.1073/pnas.0505964102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).MacLaughlin FC, Mumper RJ, Wang JJ, Tagliaferri JM, Gill I, Hinchcliffe M, Rolland AP. Journal of Controlled Release. 1998;56:259–272. doi: 10.1016/s0168-3659(98)00097-2. [DOI] [PubMed] [Google Scholar]
- (17).Jiang TT, Chang JB, Wang CM, Ding Z, Chen JN, Zhang JF, Kang ET. Biomacromolecules. 2007;8:1951–1957. doi: 10.1021/bm0700486. [DOI] [PubMed] [Google Scholar]
- (18).KukowskaLatallo JF, Bielinska AU, Johnson J, Spindler R, Tomalia DA, Baker JR. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:4897–4902. doi: 10.1073/pnas.93.10.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lu ZZ, Wu J, Sun TM, Ji J, Yan LF, Wang J. Biomaterials. 2008;29:733–741. doi: 10.1016/j.biomaterials.2007.10.033. [DOI] [PubMed] [Google Scholar]
- (20).Jewell CM, Lynn DM. Adv Drug Deliv Rev. 2008;60:979–99. doi: 10.1016/j.addr.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Fishbein I, Stachelek SJ, Connolly JM, Wilensky RL, Alferiev I, Levy RJ. Journal of Controlled Release. 2005;109:37–48. doi: 10.1016/j.jconrel.2005.09.031. [DOI] [PubMed] [Google Scholar]
- (22).Wang C-HK, Jiang S, Pun SH. Langmuir. 26:15445–15452. doi: 10.1021/la1025203. [DOI] [PubMed] [Google Scholar]
- (23).Bengali Z, Pannier AK, Segura T, Anderson BC, Jang JH, Mustoe TA, Shea LD. Biotechnology and Bioengineering. 2005;90:290–302. doi: 10.1002/bit.20393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zelphati O, Liang X, Nguyen C, Barlow S, Sheng S, Shao Z, Felgner PL. Biotechniques. 2000;28:304. doi: 10.2144/00282rr01. + [DOI] [PubMed] [Google Scholar]
- (25).Anderson SM, Chen TT, Iruela-Arispe ML, Segura T. Biomaterials. 2009;30:4618–4628. doi: 10.1016/j.biomaterials.2009.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5413–5418. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lutolf MP, Hubbell JA. Nature Biotechnology. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- (28).Sternlicht MD, Werb Z. Annual Review of Cell and Developmental Biology. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Whitesides GM, Kriebel JK, Love JC. Sci Prog. 2005;88:17–48. doi: 10.3184/003685005783238462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Millili PG, Yin D, Fan H, Naik UP, Sullivan MO. Bioconjugate Chemistry. doi: 10.1021/bc900328j. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zelphati O, Liang XW, Hobart P, Felgner PL. Human Gene Therapy. 1999;10:15–24. doi: 10.1089/10430349950019156. [DOI] [PubMed] [Google Scholar]
- (32).Graham D, Dingman S. Material Matters. 2006;1:18–19. [Google Scholar]
- (33).Trengove NJ, Stacey MC, Macauley S, Bennett N, Gibson J, Burslem F, Murphy G, Schultz G. Wound Repair and Regeneration. 1999;7:442–452. doi: 10.1046/j.1524-475x.1999.00442.x. [DOI] [PubMed] [Google Scholar]
- (34).Tseng SJ, Chuang CJ, Tang SC. Acta Biomaterialia. 2008;4:799–807. doi: 10.1016/j.actbio.2008.01.018. [DOI] [PubMed] [Google Scholar]
- (35).Latulippe DR, Ager K, Zydney AL. Journal of Membrane Science. 2007;294:169–177. [Google Scholar]
- (36).Manning GS. Biophysical Journal. 2006;91:3607–3616. doi: 10.1529/biophysj.106.089029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Boussif O, Lezoualch F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Varga CM, Tedford NC, Thomas M, Klibanov AM, Griffith LG, Lauffenburger DA. Gene Ther. 2005;12:1023–1032. doi: 10.1038/sj.gt.3302495. [DOI] [PubMed] [Google Scholar]
- (39).Boeckle S, von Gersdorff K, van der Piepen S, Culmsee C, Wagner E, Ogris M. Journal of Gene Medicine. 2004;6:1102–1111. doi: 10.1002/jgm.598. [DOI] [PubMed] [Google Scholar]
- (40).Millili PG, Selekman JA, Blocker KM, Johnson DA, Naik UP, Sullivan MO. Microscopy Research and Technique. 2010;73:866–877. doi: 10.1002/jemt.20839. [DOI] [PMC free article] [PubMed] [Google Scholar]