

Abstract
A number of pro-fibrogenic stimuli, such as growth factors, cytokines and extracellular matrix (ECM) proteins, involve Akt and its downstream substrates in mediating their effects. We previously reported that absence of Akt1, which is the predominant isoform of the three gene Akt family in vascular cells, resulted in impaired ECM remodeling in skin and vasculature. In the current study, we investigated the importance of Akt1 in TGFβ-and bleomycin-induced synthesis and secretion of ECM proteins by fibroblasts. We observed that both TGFβ and bleomycin stimulated the synthesis of ECM proteins in a dose- and time-dependent manner. Treatment with TGFβ and bleomycin also resulted in increased phosphorylation of Akt, mammalian target of rapamycin (mTOR) and their downstream signaling partners, p70S6 Kinase, Ribosomal S6 protein and 4E-BP1, resulting in the activation of this pathway. The effects of TGFβ and bleomycin on ECM synthesis were blunted by pre-treatment with an mTOR inhibitor rapamycin. Whereas mTOR is responsible for the transcriptional regulation of a number of ECM proteins, adhesion molecules and matrix metalloproteases (MMPs), synthesis of major ECM proteins such as fibronectin and collagens (types I, II and V) by fibroblasts in response to TGFβ and bleomycin is regulated by mTOR at the translational level. These findings indicate the importance of the Akt-mTOR signaling pathway in TGF-mediated fibrogenic events in vivo.
Keywords: TGFβ, bleomycin, Akt, mTOR, extracellular matrix, fibronectin, collagen
Introduction
The process of fibrogenesis involves a series of events coordinated in a highly sequential manner (Kadler et al., 2008). This involves increased production of extracellular matrix (ECM) components (Makinde et al., 2007), proliferation of mesenchymal cells and fibroblasts (Goodwin and Jenkins, 2009; Guarino et al., 2009), migration (Somanath et al., 2007), assembly (Somanath and Byzova, 2009; Somanath et al., 2007) and formation of a fibrotic scar (Eckes et al., 2010). Among these, synthesis and secretion of ECM proteins in response to physiological or pathological stimuli is the first step in any fibrogenic cascade (Hynes, 2009). Fibroblasts are the major secretors of ECM proteins such as collagens and fibronectin (Eyden, 2005) and a number of growth factors and cytokines are known to stimulate this process (LeRoy et al., 1990). Among these, transforming growth factor-β (TGFβ) is the major stimulus, regulating both the physiological fibrogenesis and pathological fibrosis (Pohlers et al., 2009; Rosenbloom et al., 2010). Studies also indicate that bleomycin, an antibiotic known to induce systemic sclerosis, also mediates its effects through enhanced synthesis of TGFβ (Oi et al., 2004). Although the molecular mechanism underlying TGFβ-and bleomycin-induced ECM synthesis are not well understood, it is likely that these stimuli might use common pathways to mediate their effects.
The serine/threonine kinase Akt (Protein kinase B) is known to phosphorylate a wide spectrum of substrates in the regulation of various cellular and tissue specific processes. Versatility of its role in vivo is as complex as the cellular network that Akt bridges in multiple cell types (Somanath et al., 2007; Somanath et al., 2006). Akt1 is the major isoform of the three gene Akt family in vascular cells (Chen et al., 2005). Many of our previous studies using Akt1 knockout mice have revealed the importance of Akt and its substrates in the regulation of integrin activation in endothelial cells and fibroblasts (Somanath et al., 2007), ECM assembly (Somanath and Byzova, 2009; Somanath et al., 2007) and angiogenesis (Chen et al., 2005; Somanath et al., 2008; Somanath et al., 2006). While Akt1−/− mice exhibited impaired expression and organization of collagen in skin and laminin in blood vessels in vivo (Chen et al., 2005; Somanath et al., 2008), Akt1−/− mouse embryonic fibroblasts (MEFs) exhibited impaired assembly of fibronectin in vitro via impaired Rac-p21 activated kinase signaling (Somanath and Byzova, 2009).
Since Akt is well known for the regulation of transcription and protein synthesis (Kandel and Hay, 1999; Somanath et al., 2006), we hypothesized that Akt may also be necessary for the synthesis and secretion of ECM proteins such as fibronectin and collagens. While Akt regulates the activities of a number of transcription factors (Kandel and Hay, 1999; Somanath et al., 2006), it mediates protein synthesis through regulation of mammalian target of rapamycin (mTOR) activity (Hay, 2005), the major protein translational machinery in cells (Hay and Sonenberg, 2004). A recent study also indicates that rapamycin treatment inhibits bleomycin-induced fibrosis in a mouse model (Yoshizaki et al., 2010). In this study, we investigated the role of the Akt1-mTOR pathway in the TGFβ-and bleomycin-induced synthesis and secretion of the ECM proteins, fibronectin and collagens. Our study indicates a significant role of Akt-mTOR signaling in ECM secretion, consequently, initiating the pro-fibrogenic response.
Materials and methods
Animals
Akt1−/− mice were generated as previously described (Chen et al., 2001) and were maintained in the 129 R1/C57BL/6 background. We used sex- and age-matched wild-type and Akt1−/− littermates for the study. The animals were 8–12 weeks of age at the time of cell isolation. All procedures were approved by the Cleveland Clinic and VAMC-Augusta Institutional Animal Care and Use Committees.
Antibodies
Anti-Akt, phosphoS473-Akt, phosphoT389-p70S6K, phosphoT37/46-4E-BP1 and phosphoS2448-mTOR antibodies were purchased from Cell signaling (Boston, MA). Anti-fibronectin and β-actin antibodies were purchased from Sigma (St Louis, MO). Anti-Procollagen type I, type II and type V antibodies were purchased from Rockland (Gilbertsville, PA).
Western blot analysis
Cell lysates were prepared using lysis buffer (20mM Tris-HCl, pH7.4; 1% Triton X-100, 3mM EGTA, 5mM EDTA, phosphatase inhibitors [10mM sodium pyrophosphate, 5mM sodium orthovanadate, 5mM sodium fluoride, and 10μM okadaic acid], protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland) and 1mM PMSF. SDS-PAGE and western blotting were performed as described previously (Somanath et al., 2007).
Cell Lines and Cell Culture
NIH 3T3 fibroblasts were obtained from ATCC (Manassas, VA) and were transfected using FuGENE 6 transfectionreagent as per the manufacturer’s protocol or through retro-viral infections using pBabe vector clones with puromycin resistance. Approximately 70–80% transfection efficiency was obtained in NIH 3T3 fibroblasts through transient transfections as observed by GFP staining.
Isolation of primary mouse embryonic fibroblasts
MEFs were collected from 11–14 day old embryos as standardized in the lab (Somanath and Byzova, 2009; Somanath et al., 2007). Briefly, fetuses were removed from amniotic sacs and cleared of the heads, viscera and limbs. Embryos were transferred to a sterile container containing D-MEM pre-warmed to 37°C. Embryos were then washed and chopped to 3mm3 cubes until the washes were clear of blood cells. Cubes were digested using a 3mg/ml collagenase-dispase mixture for 4 hours at room temperature. After enzymatic digestion, the supernatant was collected through a tissue strainer (Fisher). The filtrate was centrifuged and the pellet was washed in PBS. The cells were then re-suspended in D-MEM culture medium with 10% FBS and plated on uncoated culture dish.
Analysis of ECM secretion
A modification of previously described protocol was used for the analysis of secreted fibronectin (Somanath et al., 2007). Fibroblasts were grown in 6-well plates, and the conditioned serum free media were collected at respective time points and cells were lysed using RIPA buffer after treatment with control vehicle, bFGF (20ng/ml), TGFβ (25, 50, 100 and 250pM) and Bleomycin (1, 2.5 and 5 mU). Protein from the conditioned media was concentrated by TCA precipitation and was re-dissolved in normal saline with traces of NaOH. Once dissolved, pH of the solution was brought back to 7.4 using 1N HCl. Cell lysates and conditioned media were used for the western analyses of secreted ECM proteins using specific antibodies. Protein bands were normalized to the coommassie stained membranes.
ELISA
Fibronectin secretion was analyzed by ELISA as standardized in the lab. Briefly, after culturing the WT and Akt1−/− fibroblasts in a 12 well plate (1×105/well), the medium was aspirated out and the cells were fixed using 1% paraformaldehyde to determine the concentration of fibronectin deposited on the monolayer. Measured volumes of conditioned media were added to different wells in a 96 well plate (performed in quadruplicates) and were allowed to plate to plastic. After washing, each well was then blocked with 2% BSA and incubated overnight with anti-fibronectin antibody (1:10000) (tested for no cross reactivity with other ECM proteins; data not shown) and subsequently washed and incubated with HRP-conjugated secondary antibody (1:25000). Peroxidase activity was quantified using TMB as a substrate.
Real Time-PCR arrays
Cells were treated with TGFβ in the absence (control) or presence (test) of rapamycin (25nM) for 24h and RNA was isolated. Arrays were performed using qRT-PCR array kit (Cat # PAMM-013A; Mouse ECM and Adhesion Molecules; SA Biosciences) and the data was analyzed according to the manufacturer’s instructions.
Statistical analysis
All the data are presented as means ± SD. We performed all the analyses using two-sample t-tests with a two-tailed distribution and the significance was set at 0.05 levels (marked with an asterisk wherever data is statistically significant).
Results
TGFβ and bleomycin stimulated Akt-mTOR signaling and synthesis of ECM proteins in a dose- and time-dependent manner
TGFβ and bleomycin are known mediators of ECM secretion. While TGFβ is a physiological stimulator of early fibrogenic events such as ECM secretion and assembly (Pohlers et al., 2009; Rosenbloom et al., 2010), bleomycin treatment, which was earlier used for breast cancer therapy, is known to induce systemic sclerosis via hyperactivation of fibroblasts and enhanced ECM synthesis (Oi et al., 2004). In this study, we first determined whether TGFβ and bleomycin could activate Akt-mTOR pathway, thus regulating ECM synthesis. To do this, we incubated NIH 3T3 fibroblasts (grown to confluence) with different doses of TGFβ (0, 25, 50, 100 and 250 pM) in serum free medium at 37oC in a CO2 incubator for different time points. After 6, 12 and 24 hours, conditioned media was collected and cells were lysed. Our data indicated that treatment with TGFβ resulted in enhanced fibronectin secretion in a dose- and time-dependent manner (Figure 1A). We noticed a peak in the secretion of fibronectin to the culture medium by the fibroblasts upon stimulation with 250pM TGFβ. Enhanced levels of endogenous fibronectin in cell lysates upon treatment with TGFβ were seen at 24 hours after the treatment. In another set of experiments, we incubated NIH 3T3 fibroblasts with different doses of bleomycin (0, 1, 2.5 and 5mU). After 6, 12 and 24 hours, conditioned media was collected and cells were lysed. Both conditioned media and cell lysates were subjected for western analysis for fibronectin. Cell lysates were further analyzed for changes in phosphorylation of signaling molecules in the Akt-mTOR pathway such as Akt, p70 S6 Kinase, 4E-BP1 and S6 ribosomal protein using specific antibodies (Figure 1B). We noticed a peak in the secretion of fibronectin to the culture medium by fibroblasts upon stimulation with 1mU of Bleomycin. Western analyses indicated that both TGFβ and Bleomycin induced activation of Akt and mTOR in a time and dose-dependent manner. Together, these results indicate that TGFβ and Bleomycin stimulate Akt-mTOR signaling, consequently resulting in the synthesis and secretion of ECM proteins such as fibronectin and collagens.
Figure 1. TGFβ and bleomycin activates Akt-mTOR pathway and synthesis of ECM proteins in a dose- and time-dependent manner.

(A) NIH3T3 fibroblasts were grown to confluence, treated with TGFβ (0, 25, 50, 100 and 250pM) in serum free medium and further incubated at 37°C in a CO2 incubator. After 6, 12 and 24 hours, conditioned media was collected and cells were lysed using RIPA buffer with protease and phosphatase inhibitors. (B) NIH3T3 fibroblasts were grown to confluence, treated with Bleomycin (0, 1, 2.5 and 5mU) in serum free medium and further incubated at 37°C in a CO2 incubator. After 6, 12 and 24 hours, conditioned media was collected and cells were lysed using RIPA buffer. Both the conditioned media and cell lysates were subjected for western analysis for fibronectin and the cell lysates were further analyzed for changes in phosphorylation of signaling molecules in the Akt-mTOR pathway.
Deficiency of Akt1 in fibroblasts results in decreased secretion of ECM proteins
Our previous studies indicated that ECM remodeling in skin and deposition of laminin in blood vessels is impaired in Akt1−/− mice (Chen et al., 2005; Somanath et al., 2008; Somanath et al., 2006). Further studies indicated that Akt, involving a Rac-p21 activated signaling, is necessary for the fibroblast-mediated ECM assembly (Somanath and Byzova, 2009; Somanath et al., 2007). Moreover, the amount of collagen in the skin, as measured using a hydroxy-proline assay, is also reduced in Akt1−/− mice (Chen et al., 2005). This suggested that Akt may also be necessary for the synthesis and/or secretion of ECM proteins by fibroblasts. In order to validate this, we performed western analysis of the conditioned media collected from the cultured plates with WT and Akt1−/− fibroblasts, and probed with antibodies specific for fibronectin and collagen type I (Figure 2A). These westerns further subjected for densitometry (using Image J Software) and statistical analyses. Densitometry analyses indicated that Akt1−/− fibroblasts secreted a significantly less amount of fibronectin (p<0.001) and collagen type I (p<0.0003) compared to WT fibroblasts (Figure 2B). These results were corroborated with an ELISA for fibronectin in the conditioned medium secreted by the WT and Akt1−/− fibroblasts. This experiment showed significantly lesser amount of fibronectin in the conditioned media collected from Akt1−/− fibroblasts, compared to WT (p<0.008) (Figure 2C). In order to further confirm our findings, we transfected NIH 3T3 fibroblasts with WT, constitutively active (myr-Akt) and dominant negative (DN-Akt, K179M mutation in the kinase domain) variants of Akt1, and the conditioned media were analyzed for fibronectin content. Our data indicated that while expression with WT Akt and myr-Akt resulted in enhanced secretion of fibronectin by NIH 3T3 cells, expression with DN-Akt resulted in significant reduction in the secretion of fibronectin (Figure 2D). Collectively our data indicates that inhibition of Akt activity results in decreased synthesis and secretion of ECM proteins such as collagens and fibronectins.
Figure 2. Akt1 deficiency in fibroblasts results in decreased secretion of ECM proteins.

(A) Western analysis of conditioned media secreted by WT and Akt1−/− MEFs for fibronectin and collagen type I. Also shown are the expression levels of fibroectin and β-actin in the corresponding cell lysate. (B) Bar graph showing densitometry analysis of 4 different samples of conditioned media for the secreted fibronectin and collagen type I by WT and Akt1−/− MEFs. (C) Bar graph showing ELISA for secreted fibronectin in the conditioned media secreted by WT and Akt1−/− MEFs. (D) Western analysis of conditioned media secreted by NIH 3T3 fibroblasts transfected with WT-, DN- and myr-Akt1 probed for fibronectin. Also shown are the expression levels of β-actin in the corresponding cell lysates.
Akt-mTOR signaling regulates secretion of ECM proteins by fibroblasts
mTOR is an in vivo target for the complex of rapamycin with its intracellular receptor FKBP12 (Hay, 2005) and its activation is highly under the control of Akt (Hay and Sonenberg, 2004). mTOR regulates a number of functions triggered by Akt activation, such as cell cycle, proliferation, cytoskeletal organization and cell migration (Laplante and Sabatini, 2009; Skeen et al., 2006). Our data showed a decrease in mTOR activity in Akt1−/− fibroblasts (Figures 3A and 3B) and suggests that impaired mTOR signaling could be the reason for the observed 2-fold decrease in fibronectin and collagen type I secretion, compared to WT (Figure 2B). Western analysis of mTOR/p70S6Kinase/4E-BP-1/eIF4E pathways revealed that absence of Akt1 resulted in a decrease in pS2448 mTOR as well a pT37/46 4EBP-1, mTOR substrate (Figure 3A). In order to further confirm this observation, we treated NIH 3T3 fibroblasts with 25nM rapamycin, a highly specific inhibitor of mTOR-Raptor complex, in the absence of FBS; and the conditioned media and the lysates were collected at various time intervals (between 0 and 48 hours). Our data showed that the addition of rapamycin resulted in decreased secretion of fibronectin into the medium starting from 6h after treatment, with a 70–80% decrease in fibronectin secretion at 12h and 24h after rapamycin treatment (Figures 3B and 3D). Additionally, the secreted amounts of collagens type I, type II and type V were also reduced in the conditioned media collected from rapamycin treated NIH 3T3 cells, compared to untreated controls (Figures 3C and 3D). Cells did not survive in serum free medium for more than 24h. Coommassie staining showed equal amounts of proteins in both the samples indicating that these results were not due to a general defect in secretion as a result of rapamycin treatment. Overall these results demonstrate that Akt1-mTOR signaling is necessary for the synthesis and secretion of ECM proteins by fibroblasts.
Figure 3. Akt1-mTOR signaling regulates secretion of ECM proteins by fibroblasts.

(A) Western analysis of cell lysates from NIH 3T3 fibroblasts transfected with WT-, DN- and myr-Akt1 as well as WT and Akt1−/− MEFs in the presence and absence of bFGF. (B) Western analysis of conditioned media collected from NIH 3T3 fibroblasts at various time points (6, 12 and 24h) after treatment with rapamycin for fibronectin. Also shown are the phosphorylation levels of pS2448 mTOR, pT389-p70S6K and pT37/46 4EBP-1 normalized to total mTOR and β-actin levels. (C) Western analysis of conditioned media collected from NIH 3T3 fibroblasts at various time points (12 and 24h) after treatment with rapamycin for collegens type I, type II and type V, normalized to coommassie blue stained membrane. (D) Bar graph showing densitometry analysis of 4 different samples of conditioned media for the secreted fibronectin as well as collagen types I, II and V by NIH 3T3 cells collected after 24h treatment with DMSO (control) and rapamycin.
mTOR inhibition has modest effects on ECM synthesis at the transcriptional level
Akt-mTOR signaling is known to regulate the synthesis of proteins both at the transcriptional (Basu et al., 2010; Sun et al., 2010) and translational levels (Hay, 2005; Hay and Sonenberg, 2004). Hence, we investigated whether any of the mTOR-mediated effects on ECM synthesis are regulated at the transcriptional level. In order to do this we treated NIH 3T3 cells with TGFβ in the presence and absence of mTOR inhibitor, rapamycin for 24h; RNA was isolated and subjected for qRT-PCR arrays. Our data indicated that a number of ECM proteins and adhesion molecules were up- (Table 1) or down-regulated (Table 2) with rapamycin treatment. However, there were no significant changes in the mRNA levels of major ECM proteins such as collagens and fibronectins with rapamycin treatment. Interestingly, as we had shown before (Chen et al., 2005), expression of mRNA for thrombospondins (TSP-1, TSP-2 and TSP-3) were reduced with mTOR inhibition. Expression levels of ADAMTS2, ADAMTS1, Tumstatin, matrix metalloproteinases (MMP-3 and MMP-7) and laminins (alpha-1 and gamma-1) mRNA were also reduced in NIH 3T3 cells treated with rapamycin (Table 2). In contrast mRNA levels of several genes that include adhesion molecules such as integrins (alpha-4 and beta-3), laminins (alpha-3 and beta-3), MMPs (MMP-8, MMP-10, MMP-15), and Tenascin C were reduced in NIH 3T3 cells with mTOR inhibition (Table 1). However, no changes in the mRNA levels of major vascular integrins, except beta-3 and alpha-4 integrins, and ECM proteins (e.g. fibronectins, collagens and SPARC) were observed with rapamycin treatment (Supplemental Table). Thus, our studies demonstrated that at transcriptional level, although mTOR regulates expression of many ECM proteins, integrins and MMPs, synthesis of major ECM proteins such as fibronectins and collagens were not regulated at the transcriptional level. This suggests that Akt-mTOR regulates synthesis and secretion of major ECM proteins via modulation of transcriptional activity involving p70S6 kinase-S6 ribosomal protein and/or 4E-BP1-eIF4E pathways.
Table 1.
ECM proteins and adhesion molecules up-regulated by rapamycin in fibroblasts at the transcriptional level
| Gene Bank | Gene Name | Description | X fold (Increase) |
|---|---|---|---|
| NM_010576 | CD49D | Integrin alpha 4 | 3.4 ↑ |
| NM_016780 | CD61, GP3A, INGRB3, MGC159221, MGC159223 | Integrin beta 3 | 1.8 ↑ |
| XM_140451 | Lama3B, [a]3B | Laminin, alpha 3 | 3.2 ↑ |
| NM_008484 | - | Laminin, beta 3 | 1.4 ↑ |
| NM_019471 | AV377895 | Matrix metallopeptidase 10 | 1.6 ↑ |
| NM_008609 | AI503551, MT2-MMP | Matrix metallopeptidase 15 | 1.5 ↑ |
| NM_008611 | BB138268 | Matrix metallopeptidase 8 | 2.2 ↑ |
| NM_008816 | C85791, Cd31, MGC102160, PECAM-1, Pecam | Platelet/endothelial cell adhesion molecule 1 | 2.1 ↑ |
| NM_011346 | AI528707, CD62L, L-selectin, LECAM-1, Lnhr, Ly-22, Ly-m22, Lyam-1, Lyam1 | Selectin, lymphocyte | 2.2 ↑ |
| NM_011347 | CD62P, Grmp, MGC129336, MGC129337, P-selectin, PADGEM | Selectin, platelet | 3.6 ↑ |
| NM_009306 | AW124717, G630098F17Rik | Synaptotagmin I | 5.2 ↑ |
| NM_011595 | Timp-3 | Tissue inhibitor of metalloproteinase 3 | 1.8 ↑ |
| NM_011607 | AI528729, C130033P17Rik, Hxb, MGC144208, MGC144209, TN, TN-C, Ten, tenascin-C | Tenascin C | 1.5 ↑ |
Table 2.
ECM proteins and adhesion molecules down-regulated by rapamycin in fibroblasts at the transcriptional level
| Gene Bank | Gene Name | Description | X fold (decrease) |
|---|---|---|---|
| NM_175643 | 4732482M10, A430089F14, ADAM-TS2, ADAMTS-3, KIAA4060, PCINP, hPCPNI, mKIAA4060 | A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 2 | 3.9 ↓ |
| NM_011782 | 9530092O11Rik, ADAM-TS5, ADAMTS1, ADAMTS11, AI481094, ASMP-2 | A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 5 (aggrecanase-2) | 1.5 ↓ |
| NM_007727 | AW495098, CNTN, F3cam | Contactin 1 | 2.2 ↓ |
| NM_007734 | [a]3(IV), alpha3(IV), tumstatin | Collagen, type IV, alpha 3 | 2.1 ↓ |
| NM_133918 | 5830419M17Rik, AW229038, BB105748, EMILIN-1, gp115 | Elastin microfibril interfacer 1 | 2.0 ↓ |
| NM_010180 | - | Fibulin 1 | 2.1 ↓ |
| NM_010406 | C5, C5a, He | Hemolytic complement | 1.6 ↓ |
| NM_008404 | 2E6, AI528527, Cd18, LAD, LCAMB, Lfa1, MF17 | Integrin beta 2 | 1.5 ↓ |
| NM_008481 | 5830440B04, KIAA4087, dy, mKIAA4087, mer, merosin | Laminin, alpha 2 | 1.9 ↓ |
| NM_010683 | Lamb2 | Laminin, gamma 1 | 1.9 ↓ |
| NM_010809 | SLN-1, SLN1, STR-1, Stmy1, Str1 | Matrix metallopeptidase 3 | 1.5 ↓ |
| NM_010810 | MAT | Matrix metallopeptidase 7 | 1.5 ↓ |
| NM_011580 | TSP-1, TSP1, Thbs-1, tbsp1 | Thrombospondin 1 | 2.7 ↓ |
| NM_011581 | TSP2, Thbs-2 | Thrombospondin 2 | 1.5 ↓ |
| NM_013691 | TSP3, Thbs-3 | Thrombospondin 3 | 2.4 ↓ |
| NM_011594 | D11Bwg1104e, Timp-2 | Tissue inhibitor of metalloproteinase 2 | 4.3 ↓ |
TGFβ-and bleomycin-stimulated ECM syntheses was inhibited by treatment with rapamycin
Thus far, our results indicated that both TGFβ and bleomycin activate Akt and mTOR signaling pathways and stimulate synthesis and secretion of ECM proteins fibronection and collagens. We next sought to determine if a causal relationship exists between activation of Akt-mTOR signaling and ECM synthesis by fibroblasts. In order to test this, we independently stimulated NIH 3T3 cells with TGFβ and Bleomycin in the presence and absence of inhibitors of PI3 kinase (LY294002), Akt (SH-5; Akt inhibitor II) or mTOR (rapamycin). After 12h incubation, conditioned media was analyzed for fibronectin and cell lysates were subjected for western analysis to detect the phosphorylation changes in Akt, and mTOR substrates such as p70 S6 kinase, ribosomal S6 protein and eIF4E. Our data indicated that inhibition of PI3 kinase, Akt and mTOR resulted in significant reduction in TGF –induced fibronectin synthesis and secretion into the conditioned medium (Figure 4A). In addition, treatment with MEK inhibitor (U0126) also inhibited TGFβ-mediated secretion of fibronectin and collagen type I by NIH 3T3 cells (data not shown). However, it is not clear whether inhibition of TGFβ-stimulated ECM synthesis by the MEK-ERK pathway inhibitor is regulated at the transcriptional or post-transcriptional level. Only a modest decrease was seen in the secretion of fibronectin with PI3 kinase, Akt and mTOR inhibition in the bleomycin treated fibroblasts.
Figure 4. Specific inhibition of mTOR with rapamycin treatment inhibits TGFβ and bleomycin stimulated ECM secretion by the fibroblasts.

(A) NIH3T3 fibroblasts were grown to confluence, treated with 100pM TGFβ in serum free medium in the presence and absence of inhibitors of PI3 Kinase (LY294002), Akt (SH-5) and mTOR (Rapamycin), and further incubated at 37°C in a CO2 incubator. After 12 hours, conditioned media was collected and cells were lysed using RIPA buffer with protease and phosphatase inhibitors. (B) NIH3T3 fibroblasts were grown to confluence, treated with 1 mU Bleomycin in serum free medium in the presence and absence of inhibitors of PI3 Kinase (LY294002), Akt (SH-5) and mTOR (Rapamycin), and further incubated at 37°C in a CO2 incubator. After 12 hours, conditioned media was collected and cells were lysed using RIPA buffer. Both the conditioned media and cell lysates were subjected for western analysis for fibronectin and the cell lysates were further analyzed for changes in phosphorylation of signaling molecules in the Akt-mTOR pathway.
In a second approach, we tested if (1) expression with myr-Akt or DN-Akt in NIH 3T3 cells can modulate TGFβ and bleomycin stimulated ECM synthesis and (2) whether rapamycin treatment can inhibit enhanced ECM synthesis in fibroblasts due to the expression of constitutively active Akt (myr-Akt). Our data showed that while expression with myr-Akt further enhanced TGFβ and bleomycin mediated effects on collagen type I synthesis, expression with DN-Akt resulted in reduced synthesis of collagen type I (Figure 4B). Furthermore, treatment with rapamycin blunted the effects of myr-Akt expression in NIH 3T3 fibroblasts on collagen expression in control, TGFβ and bleomycin treated samples (Figure 4B). Therefore, our data indicate a causal relationship between Akt-mTOR signaling on TGFβ-and bleomcyin-induced ECM synthesis by fibroblasts.
Bleomycin-stimulated ECM synthesis was inhibited by treatment with TGFβ inhibitor, SB431542
Since we observed a delay in the activation of Akt and secretion of ECM proteins by fibroblasts treated with bleomycin, we hypothesized that bleomycin-induced effects on fibroblasts may be secondary to the activation of TGFβ pathway. In order to confirm our hypothesis, fibroblasts were co-treated with bleomycin (1mU) and TGFβ inhibitor SB431542 (10μM) and cell lysates were prepared at 12 hours after treatment. Our results indicated that effects of bleomycin on Akt activation and ECM secretion was blunted by co-treatment with SB431542 (Figure 5). An increase in Akt phosphorylation and secretion of fibronectin and collagen type I into the conditioned medium was observed with bleomycin treatment. Treatment with SB431542 was observed to decrease secretion of ECM proteins and Akt phosphorylation even in control cells not treated with bleomycin (Figure 5). Thus, the effects of bleomycin on ECM secretion were inhibited by co-treatment with SB431542.
Figure 5. Bleomycin-induced Akt activation and secretion of ECM proteins is inhibited by TGF inhibitor SB431542.
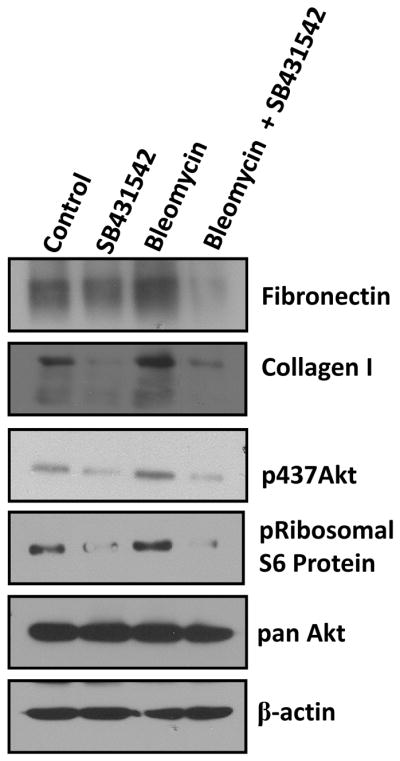
NIH3T3 fibroblasts were grown to confluence and were treated with 1mU bleomycin and/or TGFβ inhibitor SB431542), and further incubated at 37°C in a CO2 incubator. After 12 hours, conditioned media was collected and cells were lysed using RIPA buffer with protease and phosphatase inhibitors. Figure shows phosphorylation status of Akt and S6 ribosomal protein as well as secreted fibronectin and collagen type I.
Discussion
We previously reported that vasculature in Akt1−/− mice exhibited reduced deposition of ECM proteins such as laminin and collagen in the basement membrane (Chen et al., 2005; Somanath et al., 2008). Due to this, vascular maturation and the process of wound healing was impaired in Akt1−/− mice. Akt utilizes multiple mechanisms to activate mTOR. Akt is known to directly phosphorylate Ser2448 for activation of mTOR (Hay and Sonenberg, 2004) and inhibit tuberous sclerosis complex 2 (TSC2) and AMP Kinase, which otherwise inhibits mTOR activity (Laplante and Sabatini, 2009; Skeen et al., 2006). mTOR exists in two different complexes with ‘Rictor’ and ‘Raptor’, each with specific substrate specificities and functions (Laplante and Sabatini, 2009; Skeen et al., 2006). The mTOR-Raptor complex is a known protein translation unit involving eIF4E and P70 S6 Kinase (P70S6K) (Hay and Sonenberg, 2004). Our data show that mTOR activation is impaired in Akt1−/− mice. Since deposition of laminin, fibronectin and collagen is reduced in Akt1−/− mice (Chen et al., 2005; Somanath et al., 2008), we hypothesized that Akt-mTOR pathway may be necessary for the secretion of ECM proteins, vascular stabilization and tissue remodeling by fibroblasts in vivo.
In the current study, we demonstrate that physiological stimuli such as TGFβ and pathological conditions such as systemic sclerosis (as mimicked by treatment of fibroblasts with bleomycin), utilizes the PI3 kinase-Akt-mTOR signaling pathway for the enhanced synthesis of extracellular matrix (ECM) proteins such as fibronectin and collagens. Both TGFβ and bleomycin exhibited a time- and dose-dependent effect on ECM protein synthesis by NIH 3T3 fibroblasts. Deficiency of Akt1 (Somanath and Byzova, 2009; Somanath et al., 2007) as well as expression of NIH 3T3 cells with DN-Akt resulted in significant reduction in the synthesis and secretion of fibronectin, compared to WT fibroblasts or vector control NIH 3T3 cells. In contrast, expression of NIH 3T3 cells with myr-Akt resulted in enhanced secretion of fibronectin and collagen type I in control cells and further enhanced the ECM secretion into the conditioned media when treated with TGFβ or bleomycin. Moreover, inhibition of mTOR resulted in the up-regulation of mRNA for many adhesion molecules such as integrins alpha-4 and beta-3, tenascin as well as matrix metalloproteinases such as MMP-8, MMP-10 and MMP-15. Expression of a few ECM proteins such as laminin alpha-3 and beta-3 were also found to be up-regulated during mTOR inhibition. In contrast, mRNA expression of various ADAMTS proteins, MMPs such as MMP-3 and MMP-7 as well as thrombospondins (TSP-1, TSP-2 and TSP-3) were down-regulated by rapamycin treatment. Interestingly, expression levels of major ECM proteins such as fibronectins and collagens were not regulated at the transcriptional level. Treatment of NIH 3T3 fibroblasts with an mTOR-specific inhibitor blunted the growth factor-mediated synthesis of fibronectin as well as collagen types I, II and V in a time-dependent manner. Treatment with specific inhibitors of PI3 kinase, Akt or mTOR also blunted the effects of TGFβ and bleomycin on ECM synthesis by the fibroblasts. Overall, we show that mTOR is necessary for the Akt-mediated synthesis and secretion of ECM proteins by fibroblasts, in turn, is necessary for the vascular maturation and tissue re-modeling in vivo.
Akt is known to regulate synthesis of an array of proteins both at the transcriptional and translational levels (Kandel and Hay, 1999) by controlling various processes (Somanath et al., 2006). At the transcriptional level, Akt is known to utilize transcription factors such as FoxO, β-catenin, ETS factors, AP-1 and HIF1α etc. (Kandel and Hay, 1999). Transcriptional activity of FoxOs have been shown to be inhibited in Akt1−/− mice (Peng et al., 2003). At the same time, mTOR can also regulate activity of transcription factors such as HIF1 (Basu et al., 2010; Sun et al., 2010) and regulate expression of pro-angiogenic growth factors such as VEGF (Kazi et al., 2009). A very recent study showed that Akt-mTOR signaling is necessary for the synthesis of fibronectin via alternative splicing of the extra type III domain A (EDA) (White et al., 2010). Thus, it is important to see if enhanced synthesis of ECM proteins in general by Akt-mTOR signaling is regulated at the transcriptional and/or the translation levels. Our study showed that mTOR is necessary for the synthesis of a number of ECM proteins.
Rapamycin treatment resulted in decreased mRNA for Fibulin-1 and ECM proteins such as laminin alpha-2 and gamma-1. Fibulins are microfibrillar proteins that bind tropoelastin, interact with integrins, and localize to elastin fibers; tropoelastin and microfibrillar proteins constitute functional elastic fibers (de Vega et al., 2009). Fibulins also interact with ECM proteins. ECM proteins such as laminin alpha-2 and gamma-1, along with other laminins and collagen type IV, constitute more than 80% of ECM proteins in vascular basement membranes (Rowe and Weiss, 2008). TSPs and SPARC are matricellular proteins with pleiotropic effects on vascular cells (Bornstein, 2000; Chlenski and Cohn, 2010). SPARC, TSP-1 and TSP-2 have been implicated in the regulation of collagen and fibronectin matrix assembly in cutaneous wounds and myocardial tissue (Bornstein et al., 2004; Frangogiannis et al., 2005; Seet et al.). In our previous study, we had shown that protein expression levels and promoter activity of TSP-1 and TSP-2 were significantly reduced in Akt1−/− mouse endothelial cells and tissues (Chen et al., 2005). In the current study using fibroblasts, our data showed that mTOR is necessary for the transcriptional regulation of genes encoding ECM proteins such as TSP-1, TSP-2 and TSP-3. ADAMTS proteins are novel extracellular proteases that are structurally related to the ADAM protein family (ADAMTS proteins lack EGF-like domain found normally in ADAM family of proteins) (Apte, 2009). They contain a disintegrin, a metalloproteinase domain and a thrombospondin type 1 motif (Paulissen et al., 2009). In our study, mTOR is shown to regulate mRNA expression of ADAMTS-1 and ADAMTS-2. Furthermore, inhibition of mTOR resulted in reduced mRNA levels of MMPs such as MMP-3 and MMP-7 as well tissue MMP inhibitor, TIMP-2, all of which have been implicated in the ECM remodeling in tissues (Vanhoutte and Heymans, 2010). Our studies indicate that mTOR is necessary for the positive regulation of mRNA expression of several ECM proteins, MMPs and adhesion molecules.
Akt and associated transcription factors such as FoxO and β-catenin, are also known for the transcriptional repression of several genes (Taddei et al., 2008). However, to date, it is not known whether mTOR is involved in the transcriptional repression of any proteins. Our gene array analyses indicated that mTOR inhibition results in up-regulation of certain genes that code for ECM proteins (laminin alpha-3 and beta-3), adhesion molecules (integrin alpha-4, integrin beta-3 and Tenascin C), matrix metallo proteases (MMP-8, MMP-10 and MMP-11) and tissue inhibitor of MMPs-3 (TIMP-3). While these results show that mTOR is also involved in the transcriptional repression of several genes in fibroblasts, how mTOR mediates these effects requires further investigation.
Interestingly, in our gene array analyses, we observed that the mRNA levels of major ECM proteins that are involved in TGFβ-mediated tissue remodeling, such as collagens and fibronectin were not modulated by mTOR inhibition. This indicated that mRNA expression of these ECM proteins is not under transcriptional regulation by mTOR. However, inhibition of mTOR does significantly inhibit synthesis and secretion of collagens and fibronectin by fibroblasts, thus indicating that synthesis of collagens and fibronectin is regulated by mTOR at the translational levels. mTOR exists in two different conformations in mammalian cells; mTOR complex-1 (mTORC1; mTOR-Raptor complex) and mTOR complex-2 (mTORC2; mTOR-Rictor complex) (Laplante and Sabatini, 2009). Regulation of protein synthesis is one of the major functions of mTORC1, which is sensitive to rapamycin (Hay, 2005). mTORC2 is known to function like a so called PDK-2 in the feed-back activation of Akt via phosphorylation of the latter at Ser473 (Hay and Sonenberg, 2004; Sarbassov et al., 2005). Our data demonstrated that Akt-mTOR signaling is necessary for the synthesis and secretion of major ECM proteins such as fibronectin and collagen via regulation of protein synthesis as well as in the transcriptional regulation of various ECM proteins, MMPs, ADAMTS proteases, adhesion molecules, integrins and TIMPs.
In conclusion, our study suggests that while activation of Akt-mTOR activity may play a role in enhncing tissue repair following injury via modulation of expression of many genes involved in the process (Figure 6), this pathway can be a suitable target for therapeutic interventions for many ECM-related clinical conditions such as tissue fibrosis and systemic sclerosis.
Figure 6. Schematic representation of the working hypothesis.

Both TGFβ and bleomycin activates PI3 Kinase-Akt-mTOR signaling pathway. While mTOR modulate the expression of various adhesion molecules, integrins, MMPs and laminins via transcriptional regulation, expression of major ECM proteins such as fibronectin and collagens were regulated by mTOR at the stage of protein synthesis involving P70 S6 Kinase and 4E-BP1 pathway.
Supplementary Material
Acknowledgments
Funding: We acknowledge support from the American Heart Association via a Scientist Development Grant (0830326N), University of Georgia Research Foundation and Wilson Pharmacy Foundation to Somanath P.R., and National Institutes of Health (HL071625Z) to Byzova T.V.
We acknowledge support from the American Heart Association via a Scientist Development Grant (0830326N) to Somanath P.R. and US National Institutes of Health (HL071625Z) to Byzova T.V. Authors thank Dr. Nissim Hay, University of Illinois at Chicago, IL for Akt1−/− mice. We also that the technical assistance provided by Miroslava Tischenko and Shuaib Mohammed.
Abbrevations
- ECM
Extracellular Matrix
- TGFβ
Transforming Growth Factor-β
- bFGF
basic Fibroblast Growth Factor
- mTOR
Mammalian Target of Rapamycin
- DMSO
Dimethyl Sulphoxide
- MMPs
Matrix Metalloproteases
- TIMPs
Tissue Inhibitor of Metalloproteases
- MEFs
Mouse Embryonic Fibroblasts
- RT-PCR
Real-Time Polymerase Chain Reaction
- eIF4E
Eukaryotic Initiation Factor 4E
- TSPs
Thrombospondins
- SPARC
Secreted Protein Acidic and Rich in Cysteine
- ADAMTS
A Disintegrin-like And Metallopeptidase (reprolysin type) with Thrombospondin type 1 motif
References
- Apte SS. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. The Journal of biological chemistry. 2009;284(46):31493–31497. doi: 10.1074/jbc.R109.052340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Datta D, Zurakowski D, Pal S. Altered vascular endothelial growth factor mRNA stability following treatments with immunosuppressive agents: Implications for cancer development. The Journal of biological chemistry. doi: 10.1074/jbc.M110.119446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein P. Matricellular proteins: an overview. Matrix Biol. 2000;19(7):555–556. doi: 10.1016/s0945-053x(00)00103-7. [DOI] [PubMed] [Google Scholar]
- Bornstein P, Agah A, Kyriakides TR. The role of thrombospondins 1 and 2 in the regulation of cell-matrix interactions, collagen fibril formation, and the response to injury. The international journal of biochemistry & cell biology. 2004;36(6):1115–1125. doi: 10.1016/j.biocel.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P, Byzova TV. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nature medicine. 2005;11(11):1188–1196. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, Kadowaki T, Hay N. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes & development. 2001;15(17):2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlenski A, Cohn SL. Modulation of matrix remodeling by SPARC in neoplastic progression. Seminars in cell & developmental biology. 21(1):55–65. doi: 10.1016/j.semcdb.2009.11.018. [DOI] [PubMed] [Google Scholar]
- de Vega S, Iwamoto T, Yamada Y. Fibulins: multiple roles in matrix structures and tissue functions. Cell Mol Life Sci. 2009;66(11–12):1890–1902. doi: 10.1007/s00018-009-8632-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckes B, Nischt R, Krieg T. Fibrogenesis & tissue repair. Vol. 3. Cell-matrix interactions in dermal repair and scarring; p. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyden B. The myofibroblast: a study of normal, reactive and neoplastic tissues, with an emphasis on ultrastructure. Part 1--normal and reactive cells. Journal of submicroscopic cytology and pathology. 2005;37(2):109–204. [PubMed] [Google Scholar]
- Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, Winkelmann K, Michael LH, Lawler J, Entman ML. Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005;111(22):2935–2942. doi: 10.1161/CIRCULATIONAHA.104.510354. [DOI] [PubMed] [Google Scholar]
- Goodwin A, Jenkins G. Role of integrin-mediated TGFbeta activation in the pathogenesis of pulmonary fibrosis. Biochemical Society transactions. 2009;37(Pt 4):849–854. doi: 10.1042/BST0370849. [DOI] [PubMed] [Google Scholar]
- Guarino M, Tosoni A, Nebuloni M. Direct contribution of epithelium to organ fibrosis: epithelial-mesenchymal transition. Human pathology. 2009;40(10):1365–1376. doi: 10.1016/j.humpath.2009.02.020. [DOI] [PubMed] [Google Scholar]
- Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer cell. 2005;8(3):179–183. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes & development. 2004;18(16):1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Hynes RO. The extracellular matrix: not just pretty fibrils. Science (New York, NY) 2009;326(5957):1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Current opinion in cell biology. 2008;20(5):495–501. doi: 10.1016/j.ceb.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ES, Hay N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Experimental cell research. 1999;253(1):210–229. doi: 10.1006/excr.1999.4690. [DOI] [PubMed] [Google Scholar]
- Kazi AA, Molitoris KH, Koos RD. Estrogen rapidly activates the PI3K/AKT pathway and hypoxia-inducible factor 1 and induces vascular endothelial growth factor A expression in luminal epithelial cells of the rat uterus. Biology of reproduction. 2009;81(2):378–387. doi: 10.1095/biolreprod.109.076117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling at a glance. Journal of cell science. 2009;122(Pt 20):3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoy EC, Trojanowska MI, Smith EA. Cytokines and human fibrosis. European cytokine network. 1990;1(4):215–219. [PubMed] [Google Scholar]
- Makinde T, Murphy RF, Agrawal DK. The regulatory role of TGF-beta in airway remodeling in asthma. Immunology and cell biology. 2007;85(5):348–356. doi: 10.1038/sj.icb.7100044. [DOI] [PubMed] [Google Scholar]
- Oi M, Yamamoto T, Nishioka K. Increased expression of TGF-beta1 in the sclerotic skin in bleomycin-‘susceptible’ mouse strains. Journal of medical and dental sciences. 2004;51(1):7–17. [PubMed] [Google Scholar]
- Paulissen G, Rocks N, Gueders MM, Crahay C, Quesada-Calvo F, Bekaert S, Hacha J, El Hour M, Foidart JM, Noel A, Cataldo DD. Role of ADAM and ADAMTS metalloproteinases in airway diseases. Respiratory research. 2009;10:127. doi: 10.1186/1465-9921-10-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng XD, Xu PZ, Chen ML, Hahn-Windgassen A, Skeen J, Jacobs J, Sundararajan D, Chen WS, Crawford SE, Coleman KG, Hay N. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes & development. 2003;17(11):1352–1365. doi: 10.1101/gad.1089403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlers D, Brenmoehl J, Loffler I, Muller CK, Leipner C, Schultze-Mosgau S, Stallmach A, Kinne RW, Wolf G. TGF-beta and fibrosis in different organs - molecular pathway imprints. Biochimica et biophysica acta. 2009;1792(8):746–756. doi: 10.1016/j.bbadis.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Rosenbloom J, Castro SV, Jimenez SA. Narrative review: fibrotic diseases: cellular and molecular mechanisms and novel therapies. Annals of internal medicine. 152(3):159–166. doi: 10.7326/0003-4819-152-3-201002020-00007. [DOI] [PubMed] [Google Scholar]
- Rowe RG, Weiss SJ. Breaching the basement membrane: who, when and how? Trends in cell biology. 2008;18(11):560–574. doi: 10.1016/j.tcb.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (New York, NY) 2005;307(5712):1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Seet LF, Su R, Barathi VA, Lee WS, Poh R, Heng YM, Manser E, Vithana EN, Aung T, Weaver M, Sage EH, Wong TT. SPARC deficiency results in improved surgical survival in a novel mouse model of glaucoma filtration surgery. PloS one. 5(2):e9415. doi: 10.1371/journal.pone.0009415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeen JE, Bhaskar PT, Chen CC, Chen WS, Peng XD, Nogueira V, Hahn-Windgassen A, Kiyokawa H, Hay N. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer cell. 2006;10(4):269–280. doi: 10.1016/j.ccr.2006.08.022. [DOI] [PubMed] [Google Scholar]
- Somanath PR, Byzova TV. 14-3-3beta-Rac1-p21 activated kinase signaling regulates Akt1-mediated cytoskeletal organization, lamellipodia formation and fibronectin matrix assembly. Journal of cellular physiology. 2009;218(2):394–404. doi: 10.1002/jcp.21612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somanath PR, Chen J, Byzova TV. Akt1 is necessary for the vascular maturation and angiogenesis during cutaneous wound healing. Angiogenesis. 2008;11(3):277–288. doi: 10.1007/s10456-008-9111-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somanath PR, Kandel ES, Hay N, Byzova TV. Akt1 signaling regulates integrin activation, matrix recognition, and fibronectin assembly. The Journal of biological chemistry. 2007;282(31):22964–22976. doi: 10.1074/jbc.M700241200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somanath PR, Razorenova OV, Chen J, Byzova TV. Akt1 in endothelial cell and angiogenesis. Cell cycle (Georgetown, Tex) 2006;5(5):512–518. doi: 10.4161/cc.5.5.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Ge Y, Drnevich J, Zhao Y, Band M, Chen J. Mammalian target of rapamycin regulates miRNA-1 and follistatin in skeletal myogenesis. The Journal of cell biology. 189(7):1157–1169. doi: 10.1083/jcb.200912093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S, Dejana E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nature cell biology. 2008;10(8):923–934. doi: 10.1038/ncb1752. [DOI] [PubMed] [Google Scholar]
- Vanhoutte D, Heymans S. TIMPs and cardiac remodeling: ‘Embracing the MMP-independent-side of the family’. Journal of molecular and cellular cardiology. 48(3):445–453. doi: 10.1016/j.yjmcc.2009.09.013. [DOI] [PubMed] [Google Scholar]
- White ES, Sagana RL, Booth AJ, Yan M, Cornett AM, Bloomheart CA, Tsui JL, Wilke CA, Moore BB, Ritzenthaler JD, Roman J, Muro AF. Control of fibroblast fibronectin expression and alternative splicing via the PI3K/Akt/mTOR pathway. Experimental cell research. doi: 10.1016/j.yexcr.2010.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizaki A, Yanaba K, Yoshizaki A, Iwata Y, Komura K, Ogawa F, Takenaka M, Shimizu K, Asano Y, Hasegawa M, Fujimoto M, Sato S. Treatment with rapamycin prevents fibrosis in tight-skin and bleomycin-induced mouse models of systemic sclerosis. Arthritis and rheumatism. doi: 10.1002/art.27498. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.