

Abstract
Increasing evidence indicates that endoplasmic reticulum (ER) stress activates the adaptive unfolded protein response (UPR), but that beyond a certain degree of ER damage, this response triggers apoptotic pathways. The general mechanisms of the UPR and its apoptotic pathways are well characterized. However, the metabolic events that occur during the adaptive phase of ER stress, before the cell death response, remain unknown. Here, we show that, during the onset of ER stress, the reticular and mitochondrial networks are redistributed towards the perinuclear area and their points of connection are increased in a microtubule-dependent fashion. A localized increase in mitochondrial transmembrane potential is observed only in redistributed mitochondria, whereas mitochondria that remain in other subcellular zones display no significant changes. Spatial re-organization of these organelles correlates with an increase in ATP levels, oxygen consumption, reductive power and increased mitochondrial Ca2+ uptake. Accordingly, uncoupling of the organelles or blocking Ca2+ transfer impaired the metabolic response, rendering cells more vulnerable to ER stress. Overall, these data indicate that ER stress induces an early increase in mitochondrial metabolism that depends crucially upon organelle coupling and Ca2+ transfer, which, by enhancing cellular bioenergetics, establishes the metabolic basis for the adaptation to this response.
Key words: Ca2+, Metabolism, Mitochondria, Mitofusin 2 (Mfn2), Unfolded protein response (UPR), Endoplasmic reticulum stress
Introduction
Endoplasmic reticulum (ER) stress is a cellular state in which the folding capacity of the ER is overwhelmed owing to an increase in protein load or disruption in the folding environment (Berridge, 2002). The accumulation of unfolded proteins is detected by transmembrane sensors at the ER surface, which initiate a transduction cascade known as the unfolded protein response (UPR). During this response, the induction of a specific set of nuclear genes is observed, as well as a general arrest of translation, in order to restore the folding capacity of the ER (Ron and Walter, 2007). If protein homeostasis is not re-established, the initially adaptive UPR becomes an inducer of cell death, leading to apoptosis (Nakagawa et al., 2000). The chaperones, foldases and folding quality-control proteins that are induced during ER stress are well characterized (Hetz and Glimcher, 2009), as are the general mechanisms that transduce this response into apoptosis (Rasheva and Domingos, 2009). However, the metabolic adjustments necessary for cell survival during ER stress are poorly understood. From an energetic point of view, the requirements of the cell for metabolic substrates become enhanced in order to adapt to different stress conditions (Liu et al., 2005; Ikesugi et al., 2006; Haga et al., 2010). On the basis of these premises, it is likely that mitochondria participate in the cellular adaptive response to ER stress, possibly determining cell fate after activation of the UPR.
Interactions between the ER and mitochondria occur throughout their networks, both physically and functionally (Lebiedzinska et al., 2009). The molecular foundations of this crosstalk are diverse, and Ca2+ is one of the most important signals that these organelles use for communication (Szabadkai and Duchen, 2008). Ca2+ allosterically increases the activity of matrix dehydrogenases required for mitochondrial respiration and promotes ATP production by disinhibition of the ATP synthase (Brown, 1992; Balaban, 2009). By contrast, mitochondrial Ca2+ overload can result in permeability transition and activation of intrinsic apoptosis (Szabadkai and Rizzuto, 2004; Hajnoczky et al., 2006). Because the channels through which Ca2+ enters mitochondria are of low affinity, it has been proposed that regions of close proximity between mitochondria and the ER are necessary for Ca2+ entry into the mitochondrial matrix (Rizzuto et al., 1998). A major determinant of the ER–mitochondria interface is the distance between their surfaces, controlled by the movement of these organelles along the cytoskeleton (Hollenbeck and Saxton, 2005; Boldogh and Pon, 2006). Ca2+ modulates this distance, as its release from ER channels is a signal that locally arrests mitochondrial motility and promotes their docking at the ER surface, enhancing Ca2+ transfer and energy supply (Yi et al., 2004). Similarly, late phases of ER stress promote mitochondrial immobilization and coupling to the ER surface, leading to mitochondria-dependent apoptosis (Chami et al., 2008). Constitutive Ca2+ transfer from the ER to mitochondria is essential for the maintenance of baseline bioenergetics (Green and Wang, 2010), but whether Ca2+ participates during ER stress as a signal to promote an adaptive mitochondrial response remains unknown.
Here, we focused on the metabolic events that occur during the initial stages of ER stress, before lethal events. We provide evidence that mitochondrial and reticular networks approach and interact in a microtubule-dependent manner during the onset of ER stress. This increased coupling is restricted to the perinuclear region and results in augmented Ca2+ transfer, leading to a localized enhancement in mitochondrial respiration that increases the reductive capacity and ATP production. These results suggest that mitochondria mobilized close to the ER confer a stress-activated bioenergetic response that ultimately contributes favorably to the cellular adaptation to ER stress.
Results
Mitochondria approach perinuclear ER during early phases of ER stress
The antibiotic tunicamycin, derived from Streptomyces lysosuperficus, blocks the synthesis of N-linked glycoproteins (N-glycans), therefore it is widely used in cell biology to induce the UPR (Price and Tsvetanova, 2007). We subjected HeLa cells to tunicamycin treatment and checked for ER stress markers by immunoblotting. After 6 hours of treatment with 0.1 and 1 μg/ml tunicamycin, we observed an increase in the expression of the ER chaperone and UPR effector CHOP (also known as DDIT-3), as well as an increase in the phosphorylation of both the translation initiation factor eIF2α and the UPR transducer JNK (Fig. 1A). Although at the early stage (4–6 hours) the ER stress signaling pathways are active, an increase in cell death was only detectable at 24 hours and became significant after 48 hours (P<0.05; Fig. 1B), suggesting that before triggering cell death, ER stress activates different processes. In order to detect changes in the morphology of the mitochondrial network during tunicamycin-induced ER stress, we quantified the area of the Mitotracker-stained mitochondrial network in relation to the whole-cell area on two-dimensional confocal stack images. After 4 hours of treatment with tunicamycin, a reduction in the cellular area occupied by the mitochondrial network was observed, which returned to control levels after 20 hours (Fig. 1C). This suggested either a rapid decrease in whole mitochondrial volume or, more likely, a subcellular redistribution and/or condensation of the organelle network. Thus, we next performed a study of the distribution of the mitochondrial network in cells subjected to 4 hours of treatment with tunicamycin. An algorithm for the ImageJ software was created to scan the radial fluorescence, measured from the center of the nucleus towards the plasma membrane in a full-angle mode (0–360°), as illustrated in Fig. 1D. Using this tool, we compared changes in the subcellular distribution of the mitochondrial network between untreated controls and tunicamycin-treated cells. The 4-hour tunicamycin treatment led to a significant increase in mitochondrial abundance in the perinuclear zone as compared with that in controls, with a parallel decrease detected in the central and peripheral subcellular areas (Fig. 1E). In order to determine whether the ER behaved in a similar manner, we studied the radial subcellular distribution of ER-targeted red fluorescent protein (ER-RFP) after 4 hours of tunicamycin treatment. A redistribution of the ER towards the perinuclear area was also evident (Fig. 1F), suggesting that the interaction between the mitochondrial network and the ER increases during early ER stress conditions.
Fig. 1.

Mitochondria translocate to perinuclear ER during early phases of ER stress. (A) Western blot analysis of HeLa cells treated for 4 hours with tunicamycin (Tun) as indicated. (B) The quantification of dead cells (PI positive) and cells with a low ΔΨm [low DiOC6(3) staining] after treatment with 1 μg/ml tunicamycin was determined by flow cytometry. (C) Confocal images of Mitotracker-Green-stained mitochondria in control cells or cells treated with 0.5 μg/ml tunicamycin for 4 hours. Quantification of the percentage ratio of mitochondrial area:whole cell area is shown for HeLa cells treated with 0.5 μg/ml tunicamycin as indicated. Co, control (untreated). (D) Example of the radial fluorescence analysis of subcellular zones. (E,F) Quantification of the radial fluorescence of control cells or cells treated with 0.5 μg/ml tunicamycin for 4 hours. Data are means ± s.e.m. *P<0.05 compared with controls within the same subcellular zone or as indicated. Scale bars: 10 μm.
To elucidate further whether the approaching of the organelles correlates with increased coupling between them, we performed colocalization studies on confocal planes and monitored the points of interaction between the ER and mitochondria by quantifying the Manders' coefficient, which is a measure of the fraction of one structure in contact with the other (Manders et al., 1993; Costes et al., 2004). HeLa cells were transfected with ER-RFP and mitochondria were stained with Mitotracker Green (Fig. 2A). The Manders' coefficient M1 indicates the fraction of the ER that colocalizes with mitochondria (Fig. 2B), whereas the Manders' coefficient M2 indicates the fraction of mitochondria that colocalizes with the ER (Fig. 2C). In both cases, we observed that tunicamycin treatment led to increased interaction between the organelles, which was more pronounced in the fraction of mitochondria interacting with the ER. In order to define further the subcellular location where these interactions were enhanced, we quantified the Manders' coefficients within the predefined subcellular regions. We observed that the fraction of ER that colocalizes with mitochondria was enriched in the perinuclear zone (Fig. 2D), and notably the fraction of mitochondria that colocalizes with the ER was most enhanced in the same compartment (Fig. 2E). We also used thapsigargin as an alternative ER stress inducer, in order to validate the data obtained with tunicamycin. Thapsigargin-treated HeLa cells displayed a redistribution of the ER and mitochondria towards the nucleus and also increased their reticular and mitochondrial connection points, as quantified by the Manders' coefficients M1 and M2 within the redistributed networks (supplementary material Fig. S1A–D). To verify further our findings with a different technique we performed electron microscopy (EM) experiments, which showed that there was a significant increase in the number of mitochondria displaying close ER contacts after 4 hours of tunicamycin treatment (P<0.001) and a clear redistribution of both organelles towards the perinuclear zone (Fig. 2F,G). Although the whole mitochondrial network was condensed (Fig. 1C), we additionally observed that, in comparison with control cells, tunicamycin-treated cells displayed an increased area per mitochondrion [controls: 0.27±0.09 μm2, n=13; tunicamycin-treated: 0.37±0.10 μm2, n=13 (means±s.e.m)], suggesting an increase in mitochondrial volume during ER stress. Altogether, these data indicate that, during the early stages of ER stress, the mitochondrial network approaches the ER in regions of close proximity to the nucleus, increasing the coupling points between both organelles.
Fig. 2.

ER–mitochondrial coupling observed during early phases of ER stress. (A) Confocal images of HeLa cells transiently expressing ER-targeted RFP (red) and stained with Mitotracker Green (green), either treated with 0.5 μg/ml tunicamycin (Tun) for 4 hours or left untreated (control). Scale bars: 10 μm. (B,C) Quantification of the Manders' coefficient M1 (fraction of ER in colocalization with mitochondria) or M2 (fraction of mitochondria in colocalization with ER). (D,E) Quantification of the M1 and M2 coefficients within the predefined subcellular regions. (F) Representative transmission electron microscopy images from control cells or cells treated with 0.5 μg/ml tunicamycin for 4 hours. Three different magnifications of the same cell are shown. Close ER–mitochondrial contacts are indicated only in the middle panel by white arrows. Scale bars: 0.5, 1 and 2 μm as indicated. N, nucleus. (G) Quantification of the percentage of mitochondria with close ER contacts per field. Co, control (untreated). Data are means ± s.e.m. *P<0.05; **P<0.01; ***P<0.001 compared with respective controls or controls in the same subcellular zone.
Microtubules coordinate ER–mitochondrial interactions
Interactions between mitochondria and the cytoskeleton are necessary for mitochondrial morphology, motility and distribution (Yi et al., 2004; Hollenbeck and Saxton, 2005; Frederick and Shaw, 2007). Indeed, microtubules have been described as the hub for mitochondrial and ER movements (Friedman et al., 2010). On this basis, we evaluated the contribution of the microtubular network to the organelle rearrangement by employing nocodazole, a blocker of microtubule polymerization (Samson et al., 1979; Modriansky and Dvorak, 2005). By monitoring the mitochondrial and ER distribution within the predefined cellular regions, we observed that microtubule disruption significantly altered the organelle rearrangement profile observed with tunicamycin alone, leading to the retention of mitochondria and ER in the central zone (Fig. 3A,B). In addition, the increase in the coupling points between both organelles was observed primarily in the central and radial zones, rather than in perinuclear area (Fig. 3C). In addition to microtubules, the actin cytoskeleton also contributes to mitochondrial movements (Boldogh and Pon, 2006). Therefore, we studied the effects of tunicamycin in combination with cytochalasin D, which is a potent inhibitor of actin polymerization (Brenner and Korn, 1980; Casella et al., 1981). By monitoring the predefined cellular regions (and principally the perinuclear region), we detected no significant differences in the ER and mitochondrial profiles between cells treated with tunicamycin alone and cells treated with tunicamycin and cytochalasin D (supplementary material Fig. S2A,B). To study further the effect of actin microfibers in this organelle-coupling response, we quantified the Manders' coefficient M2, which again indicates that the fraction of mitochondria that couples with the ER is not affected by actin disassembly (supplementary material Fig. S2C). Taken together, these results suggest that the dynamic microtubular network, and not the actin cytoskeleton, is necessary for the mitochondrial–ER coupling observed during early phases of ER stress.
Fig. 3.

Mitochondrial rearrangement requires an intact microtubular network. (A) Analysis of the radial fluorescence of Mitotracker-stained control HeLa cells or cells treated for 4 hours with 0.5 μg/ml tunicamycin (Tun) in combination with 10 μM nocodazole, as indicated by the grayscale blocks. (B) Analysis of the radial fluorescence of transiently expressing ER-targeted RFP HeLa cells that were untreated (control) or treated with tunicamycin in combination with nocodazole as indicated by grayscale blocks as in A. (C) Quantification of the Manders' coefficient M2 (fraction of mitochondria in colocalization with ER) within the predefined subcellular zones for cells expressing ER-targeted RFP and stained with Mitotracker Green. Grayscale indicates the groups as described in A. Data are means±s.e.m. *P<0.05 compared with control cells within the same subcellular zone.
Localized perinuclear enhancement of mitochondrial function during early phases of ER stress
An increase in mitochondrial function is an adaptive response to various types of stress (Duchen, 2004; Gautier et al., 2008; Addabbo et al., 2009). Therefore, we decided to monitor changes in cellular ATP production upon tunicamycin-induced ER stress. The level of total cellular ATP started to increase as early as 1 hour after tunicamycin treatment and was augmented for 4 hours; however, it displayed a substantial decrease with respect to basal levels after prolonged ER stress (20 hours; Fig. 4A). Increases in ATP levels can result from mitochondrial oxidative phosphorylation (OXPHOS) or cytosolic glycolysis coupled to lactate production and release. 2-Deoxyglucose (2-DG) is a cell-permeant glucose analog that lacks a hydroxy group and hence selectively blocks glycolysis, whereas OXPHOS can be maintained by other substrates in the medium, such as glutamine and pyruvate (DeBerardinis et al., 2007). We measured cellular ATP levels after treatment with tunicamycin combined with 2-DG, and observed no differences with respect to tunicamycin alone, indicating that cytosolic glycolysis does not contribute to the increase in ATP synthesis during ER stress (Fig. 4B). By contrast, by combining tunicamycin treatment with carbonyl cyanide m-chlorophenylhydrazone (CCCP; an uncoupler of the mitochondrial respiratory chain), the effects of tunicamycin upon ATP production were inhibited (Fig. 4B), as they were with the positive control oligomycin (an inhibitor of mitochondrial FO ATP synthase). Moreover, tunicamycin treatment also led to a significant increase (P<0.05) in oxygen consumption (Fig. 4C), highlighting the key role of mitochondrial OXPHOS in these events. In order to study further mitochondrial function during ER stress, we monitored changes in the mitochondrial transmembrane potential (ΔΨm) by quantifying steady state tetramethylrhodamine (TMRM) loading of the mitochondrial matrix. We observed that ΔΨm increased early and reached a maximum at 4 hours of treatment, returning to basal levels after 20 hours (Fig. 4D). On the basis of the observation that the mitochondrial network is mobilized towards the perinuclear zone during ER stress, we next studied changes in ΔΨm within the redistributed network. We observed that the mitochondrial network localized to the perinuclear area displayed an increased ΔΨm, whereas in other subcellular areas ΔΨm did not change significantly (Fig. 4E). By performing an MTT assay, we monitored the amount of reductive power that results from the mitochondrial tricarboxylic acid cycle. Tunicamycin treatment led to a significant increase (P<0.05) in reductive power at 4 hours, falling below baseline after 20 hours (Fig. 4F). These results also indicate that the metabolic responses occur before the decrease in cell viability observed at later times.
Fig. 4.

Increase in cellular bioenergetics during early phases of ER stress. (A) Quantification of intracellular ATP levels in HeLa cells treated with 0.5 μg/ml tunicamycin (Tun) for the times indicated. (B) Quantification of intracellular ATP levels in cells treated for 4 hours with 0.5 μg/ml tunicamycin, in combination with 20 mM 2-DG, 20 μM CCCP or 1 μM oligomycin. (C) Determination of oxygen consumption in control cells, or cells treated with 0.5 μg/ml tunicamycin for 4 hours. Co, control (untreated). (D) Determination of ΔΨm in TMRM-stained HeLa cells treated with 0.5 μg/ml tunicamycin as indicated. (E) Analysis of the radial fluorescence of TMRM-stained HeLa cells treated with 0.5 μg/ml tunicamycin for 4 hours. (F) Determination of reductive power and cell viability, through an MTT reductase activity assay, in HeLa cells treated with 0.5 μg/ml tunicamycin for the indicated times. Data are means±s.e.m. *P<0.05 compared with respective controls or as indicated.
Given the necessary role of microtubules for the movement and coupling between reticular and mitochondrial networks, we wondered whether ER-induced metabolic enhancement depended upon the physical association of mitochondria with the ER. To test this, we subjected cells to tunicamycin treatment in combination with nocodazole, in order to impair the contact between both organelles. Under these conditions, the increases in ATP production, as well as in oxygen consumption, were abrogated (Fig. 5A,B), further highlighting microtubule-dependent organelle rearrangement as a necessary step in the functional coupling between the ER and mitochondria. Accordingly, knockdown of mitofusin 2 (Mfn2), which is an essential component of the mitochondrial–ER coupling molecular complex (de Brito and Scorrano, 2008), impaired the increase in oxygen consumption observed after 4 hours of tunicamycin treatment (Fig. 5C). Taken together, these results indicate that early after the onset of ER stress, a localized enhancement in mitochondrial function provides an increased amount of ATP and reductive power at the perinuclear ER–mitochondrial interface in a microtubule-dependent fashion.
Fig. 5.

Metabolic enhancement requires an intact microtubular network. (A) Intracellular ATP levels were measured in control (untreated) HeLa cells, or cells treated with 0.5 μg/ml tunicamycin (Tun) in combination with 10 μM nocodazole as indicated. (B) Oxygen consumption was determined in control HeLa cells or cells treated with 0.5 μg/ml tunicamycin in combination with 10 μM nocodazole as indicated. (C) Oxygen consumption was determined in HeLa cells transduced with adenovirus encoding antisense mitofusin 2 (AsMfn2) or transduced with the empty vector (EV), and treated with 0.5 μg/ml tunicamycin in combination with 10 μM nocodazole as indicated. Data are means±s.e.m. *P<0.05 compared with untreated controls; #P<0.05 compared with tunicamycin alone (B) or tunicamycin plus EV (C).
Increased mitochondrial Ca2+ drives the adaptive metabolic boost observed during early phases of ER stress
Increases in mitochondrial respiration and ATP production are often consequences of increases in mitochondrial Ca2+ (Green and Wang, 2010). In order to determine whether early phases of ER stress induced by tunicamycin increased mitochondrial Ca2+, we treated cells expressing cytosolic or mitochondrial aequorins with histamine [which evokes PtdIns(3,4,5)P3-dependent Ca2+ release] and compared their mitochondrial Ca2+ uptake. We observed that histamine led to a mitochondrial Ca2+ uptake that was significantly higher in tunicamycin-pretreated cells (P<0.05; 4 hours) than in untreated cells (Fig. 6A). Cytosolic Ca2+ increased similarly in tunicamycin-treated and untreated cells (Fig. 6B). These results indicate that the differences in mitochondrial Ca2+ levels are not due to altered Ca2+ release mediated by the PtdIns(3,4,5)P3 receptor but to an enhanced mitochondrial Ca2+ uptake, presumably due to the increased apposition of ER and mitochondrial Ca2+ channels. By using a different dye, Fura-2, we monitored the peak cytosolic Ca2+ levels after thapsigargin addition, reflecting the kinetics of Ca2+ release after sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) inhibition. After 4 hours of tunicamycin treatment, the thapsigargin-induced Ca2+ peak was increased, and it was further elevated by inhibition of mitochondrial Ca2+ uptake using Ru360 (Fig. 6C). These results suggest that, besides the PtdIns(3,4,5)P3-receptor-mediated direct Ca2+ transfer from the ER to neighboring mitochondria, an additional phenomenon associated with the early phases of ER stress involves Ca2+ leak from the ER, also resulting in mitochondrial Ca2+ uptake. Indeed, no mitochondrial Ca2+ uptake following the thapsigargin-induced Ca2+ leak was observed in Mfn2-knockout cells (Fig. 6D), which is reflected by the lack of effect of Ru360. This result further indicates that juxtaposition of mitochondria with the ER is necessary for the mitochondrial Ca2+ uptake evoked by Ca2+ leak during early phases of ER stress.
Fig. 6.

Augmented mitochondrial Ca2+ uptake during early phases of ER stress. (A) Representative traces of mitochondrial [Ca2+] ([Ca2+]m) obtained from HeLa cells expressing mitochondrial aequorin, either untreated (Co, control) or treated with 1 μg/ml tunicamycin (Tun) for 4 hours prior to histamine addition. Statistical analysis of the peak [Ca2+]m is presented in the bar graph. (B) Representative traces of cytosolic [Ca2+] ([Ca2+]c) obtained from HeLa cells expressing cytosolic aequorin, either untreated or treated with 1 μg/ml tunicamycin for 4 hours prior to histamine addition. Statistical analysis of the peak [Ca2+]c is presented in the bar graph. (C) HeLa cells were treated for 4 hours with 1 μg/ml tunicamycin and loaded with Fura-2 for cytosolic Ca2+ measurements. ER Ca2+ depletion was induced by addition of 1 μM thapsigargin, in the presence of 10 μM Ru360 as indicated. Peak values reflecting the kinetics after ER Ca2+ depletion are presented, normalized to those in control cells. (D) The same protocol as in C was used in wild-type (wt) MEFs or mitofusin2-knockout (Mfn2 ko) MEFs. Peak values reflecting the kinetics after ER Ca2+ depletion are presented, normalized to those in control wild-type MEFs. Data are means±s.e.m. (A and B) and means±s.d. (C and D). *P<0.05 compared with untreated controls or as indicated. **P<0.01 as indicated.
Finally, to test whether mitochondrial Ca2+ levels control the metabolic mitochondrial boost, we measured oxygen consumption rates resulting from OXPHOS in the presence of the PtdIns(3,4,5)P3 receptor inhibitor xestospongin B or the mitochondrial Ca2+ uptake inhibitor RuRed. We observed that both xestospongin B and RuRed decreased the rate of oxygen consumption after tunicamycin treatment (Fig. 7A,B), which confirms that increased mitochondrial Ca2+ uptake, resulting from ER–mitochondrial coupling, is necessary for the metabolic response observed during early phases of ER stress. Therefore, in order to evaluate whether the early metabolic boost forms part of an adaptive response triggered by ER stress, we inhibited mitochondrial Ca2+ uptake and measured cell viability [through propidium iodide (PI) incorporation] and ΔΨm. We observed that the inhibition of mitochondrial Ca2+ uptake during the early phase of ER stress increased cell death (PI-positive cells) and also decreased ΔΨm at 48 hours (Fig. 7C).
Fig. 7.
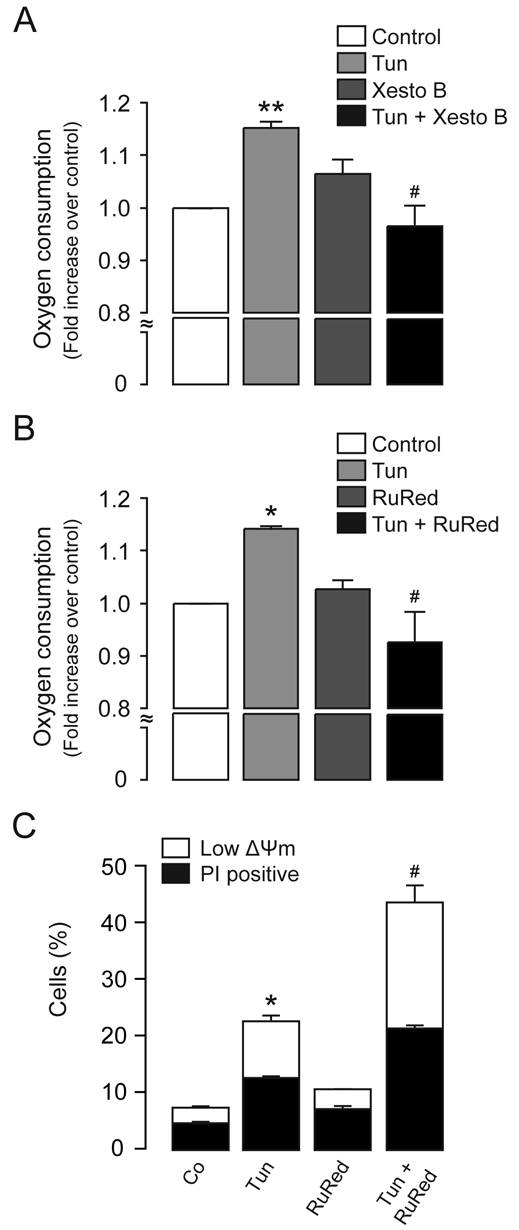
ER–mitochondrial Ca2+ transfer drives the adaptive metabolic response. (A) Oxygen consumption rates were measured in HeLa cells treated for 4 hours with 0.5 μg/ml tunicamycin (Tun) in combination with 2 μM xestospongin B as indicated. (B) Oxygen consumption rates were measured in HeLa cells treated for 4 hours with 0.5 μg/ml tunicamycin in combination with 10 μM RuRed as indicated. (C) The quantification of dead cells (PI positive) and cells with a low ΔΨm [low DiOC6(3) staining] after 4 hours treatment with 1 μg/ml tunicamycin in combination with 10 μM RuRed was determined by flow cytometry. Data are means±s.e.m. *P<0.05; **P<0.01 compared with untreated controls; #P<0.05 compared with tunicamycin alone.
In total, the results presented in this study suggest strongly that Ca2+ transfer resulting from enhanced ER–mitochondrial coupling leads to a localized increase in mitochondrial metabolism, thus providing energetic substrates key for a cellular adaptive response in face of ER stress. Further experiments will determine whether this bioenergetic response is necessary for improving the energetic state of the ER, and therefore its folding capacity, or, as it is restricted to perinuclear zones, for the activation of a specific nuclear transcriptional program that participates in the cellular adaptation to stress.
Discussion
The results from this and previous studies (Rizzuto et al., 1998; Szabadkai et al., 2006) indicate that an intimate molecular interchange exists between the ER and mitochondria, and that these events become increased under stress conditions. Most of the previous studies have concentrated on the role of the ER, and its transfer of Ca2+ to mitochondria, pointing to their implications in mitochondria-dependent cell death (Darios et al., 2005; Csordas et al., 2006; Chami et al., 2008). Here, we focused on mitochondrial events that take place during the adaptive phase of ER stress, before the appearance of cell death. In particular, we have shown that diverse parameters of mitochondrial metabolism are enhanced at the onset of ER stress. These parameters include ATP production, reductive capacity, oxygen consumption, mitochondrial transmembrane potential and, notably, microtubule-dependent movement and coupling of ER and mitochondrial networks. Accordingly, during organelle coupling, increased Ca2+ transfer from the ER to mitochondria drives the metabolic boost, and when the association of these organelles is impaired, all metabolic changes are abrogated, which indicates that physical connection between stressed ER and mitochondria is a necessary first step for the enhancement of mitochondrial metabolism.
Mitochondria are dynamically arranged in a subcellular network and manifest a wide range of microtubule-dependent translocation events (Hajnoczky et al., 2007). It has been proposed that high Ca2+ concentrations influence mitochondrial motility and distribution in the vicinity of signaling ER spots at the surface of the ER, and these microdomains could determine Ca2+-dependent cell survival and cell death mechanisms (Yi et al., 2004; Goetz et al., 2007). As previously reported for later time-points of ER stress (Chami et al., 2008), we observed that, during the early phases of ER stress, Ca2+ leak was provoked from the ER, leading to mitochondrial Ca2+ uptake. Although ER–mitochondrial coupling was essential for Ca2+ transfer, we do not know whether the Ca2+ leak is a signal to drive or target mitochondrial redistribution. However, we observed that the ER–mitochondrial coupling was dependent on an intact microtubular network, which agrees with previous reports in which both Ca2+ and microtubules are essential for reticular and mitochondrial movements (Yi et al., 2004; Friedman et al., 2010). Mitochondria and the ER become more confluent when they are located at the perinuclear region, around the centrosome, whereas in peripheral regions their proximity diminishes (Darios et al., 2005). Similarly, studies on the regulation of the ER–mitochondria interface also show that the death stimulus C2-ceramide leads to organelle clustering at the perinuclear region, concomitant with an increase in the Ca2+ transfer between these organelles (Darios et al., 2005). In the context of ER stress, it has been described that later phases of ER stress lead to tightening of the ER–mitochondria interface, with increased mitochondrial Ca2+ and activation of apoptosis (Csordas et al., 2006; Chami et al., 2008). Here, we observed that ER stress leads to a redistribution of mitochondrial and reticular networks at the perinuclear zone, with increased connection points and Ca2+ transfer; therefore, our results agree with these precedents, but differ in the sense that the observed effects are not associated with cell death mechanisms. As these effects were obtained early during the onset of ER stress, they rather play a role in a metabolic adaptive response. In concordance with this, it has been described that cells with dysfunctional mitochondria display increased toxicity to glucose deprivation and that this sensitivity is associated with an impaired UPR (Haga et al., 2010).
In terms of bioenergetics, ER homeostasis depends upon a permanent supply of ATP and energy substrates, which are necessary for optimal protein folding (Berridge, 2002; Gorlach et al., 2006), as well as for the clearance of aggregated proteins (Hoseki et al., 2010). Therefore, an increase in energy substrates seems essential for the adaptation to ER stress; however, this hypothesis has not been extensively studied. Previous work has demonstrated that, in response to diverse stress conditions, an increase in the cellular metabolic demands is observed (Duchen, 2004; Gautier et al., 2008; Addabbo et al., 2009). For instance, nutritional stress, induced by glucose deprivation, promotes an elevation in intracellular ATP levels (Liu et al., 2005). Glucose deprivation has been described as a condition causing ER stress and apoptosis (Ikesugi et al., 2006), and can be prevented by overexpression of the chaperone Grp75 (Heal and McGivan, 1997; Yang et al., 2008). Interestingly, this chaperone has been described as mediating the functional association of reticular and mitochondrial Ca2+ channels (Szabadkai et al., 2006). Therefore, it is likely that chaperone complexes containing Grp75, and perhaps other proteins such as Rab32 (Bui et al., 2010) or PACS-2 (Simmen et al., 2005), are involved in the adaptive response to ER stress. Similarly, cells lacking Mfn2 display impaired ER–mitochondrial Ca2+ transfer and are more prone to apoptosis (Jahani-Asl et al., 2007; Brooks et al., 2010). Accordingly, we observed that in the absence of Mfn2, both mitochondrial Ca2+ uptake and oxygen consumption were impaired, leading to increased cell death.
The present study demonstrates an important link between cellular stress and mitochondrial responses, establishing the metabolic basis for the adaptation to ER stress. Moreover, it defines the temporal window of the ER stress response to a wider range of events. During chronic ER stress, an increased Ca2+ transfer promoted by the truncated SERCA variant S1T leads to mitochondrial Ca2+ overload, with activation of mitochondrial apoptosis (Chami et al., 2008; Hayashi et al., 2009). Here, we focused on acute ER stress and observed that, during this phase, an increase in ER–mitochondrial Ca2+ transfer enhances the metabolic state of mitochondria. Our results suggest a sequential series of events, in which Ca2+ leak influences the microtubule-dependent physical association between mitochondria and the ER, leading to Ins(1,4,5)P3-receptor-mediated Ca2+ transfer to mitochondria. The increase in mitochondrial Ca2+ constitutes the driving force to increase ΔΨm, by stimulating OXPHOS and oxygen consumption, leading to increased production of ATP and increased reductive power. As we observed that the metabolic increase was restricted to the mitochondrial network close to the ER, our results also suggest that the metabolic needs are increased in specific zones of the ER more affected by stress, whereas mitochondrial metabolism remains at baseline conditions in peripheral zones. These responses could be the consequence of a specialized and compartmentalized mechanism to segregate damage to particular zones of the ER or simply a consequence of random damage constituting an initial attempt to generate energy substrates to cope with the localized stress state. An interesting aspect of these findings is that the ER–mitochondrial association was enriched in the perinuclear zone of cells. A possible explanation for this is based on the assumption that the UPR requires the induction of a specific gene expression program and therefore mitochondria close to the nucleus could supply the bioenergetic demands for this condition. Another possibility is based on the mechanism of action of tunicamycin, which leads to an accumulation of unfolded glycans in the ER, and therefore affects those zones more active during protein synthesis. The ER is, although continuous as a membrane structure, segmented functionally and molecularly from the nuclear envelope sheets to the peripheral tubules (Park and Blackstone, 2010). The rough envelope sheets of the ER are perinuclear, much more dense than the smooth peripheral tubules and, in addition, they are the zones receiving recently synthesized mRNA for translation. The ER close to the nucleus is enriched in translocons, which participate in protein import to the ER (Shibata et al., 2006), whereas peripheral zones are specialized in lipid biosynthesis (Park and Blackstone, 2010), and are enriched in different proteins (Shibata et al., 2008). It therefore seems probable that ER stress caused by an accumulation of misfolded proteins should manifest close to the nucleus, at least at earlier phases, which justifies the localized needs of a bioenergetic supply. We hypothesize that, above a certain threshold of ER damage, mitochondrial responses are no longer controlled, and Ca2+ overload results in permeability transition and the activation of mitochondrial apoptosis, as previously reported (Csordas et al., 2006; Chami et al., 2008).
On the basis of the results shown here, and the previous work of other groups, we propose that ER–mitochondrial coupling, and the consequent increase in mitochondrial Ca2+ uptake, belong to a dual response to ER stress conditions: on one hand, promoting an enhancement of mitochondrial metabolism as an adaptive response and, yet, on the other hand, leading to mitochondrial dysfunction as a cell death mechanism when stress is not resolved. Therefore, rather than proposing a point at which mitochondrial Ca2+ stops being pro-survival, and steps into a pro-death molecular pattern, we propose that earlier events occurring in the ER lead to tightening of the ER–mitochondrial interface and are responsible for promoting either adaptive or deleterious mitochondrial responses.
Materials and Methods
ATP measurements
Cells were plated in 96-well plates and ATP content was determined using a luciferin and luciferase assay (Cell-Titer Glo Kit; Promega), as described previously (Villena et al., 2008).
Cell culture
HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum (FBS). Cells were plated at 50–70% confluence on 60-mm diameter, six-, 24- or 96-well plates, according to the experiment. Cells were plated 24 hours prior to the exposition to tunicamycin for the indicated times, in the presence or absence of the different inhibitors. For transfection experiments, cells were treated with Lipofectamine 2000 at 24 hours after plating, according to manufacturer's specifications. The different treatments were performed 24 hours after transfections. Mfn2-knockout and wild-type mouse embryonic fibroblasts (MEFs) were a gift from David Chan (Caltech, Pasadena, CA) and were maintained in DMEM supplemented with 10% FBS, 2 mM L-glutamine and 0.1mM non-essential amino acids.
Dynamic in vivo [Ca2+] measurements
Basal and histamine-induced cytosolic Ca2+ signals were measured using either the cytosolic version of the recombinant Ca2+ sensor aequorin (cytAEQ) or the mutant variant restricted to the mitochondria (mitAEQ). All measurements were carried out in Krebs–Ringer modified buffer (KRB) (135 mM NaCl, 5 mM KCl, 1 mM MgSO4, 0.4 mM K2HPO4, 5.5 mM glucose and 20 mM HEPES pH 7.4), supplemented with 1 mM CaCl2. HeLa cells transiently expressing cytAEQ or mitAEQ were administrated with coelenterazine and transferred into a perfusion chamber. The light signal was collected in a purpose-built luminometer and calibrated into [Ca2+] values. For the analysis of ER Ca2+ depletion kinetics, HeLa cells, and Mfn2-knockout and wild-type MEFs were loaded with 2 μM Fura-2 (Invitrogen). 2×106 cells were trypsinized and resuspended in DMEM with 10% FBS. Cells were pelleted at 100 g and resuspended in 2 ml Ca2+-free buffer (20 mM HEPES pH 7.4, 6 mM KCl, 143 mM NaCl, 0.1 % glucose, 1 mM MgSO4 and 250 μM sulfinpyrazone). Cell suspensions were monitored for light emission at 510 nm after excitation at 340 and 380 nm on an 814 photomultiplier detection system (PTI, Birmingham, NJ). Ca2+ depletion was triggered by the addition of 1 μM thapsigargin in the presence or absence of 10 μM Ru360 (C2H26N8O5Ru2Cl3).
Flow cytometry analysis of ΔΨm and cell viability
The frequency of adherent and nonadherent cells with a low ΔΨm was determined by flow cytometry. Cells on 24-well plates were trypsinized and resuspended in culture medium with 40 nM 3,30-dihexyloxacarbocyanine iodide [DiOC6(3)] for 15 minutes at 37°C. The vital dye propidium iodide (PI, 1 μg/ml) was added 1 minute before experiments. Cell fluorescence was determined using a FACS Scan system (Becton Dickinson, San Jose, CA), as previously reported (Munoz et al., 2009). Living cells (PI negative) with low ΔΨm [low DiOC6(3) staining] were classified as ‘low ΔΨm’, whereas dead cells (PI positive) with low ΔΨm were classified as ‘PI positive’, as previously described (Tajeddine et al., 2008).
Image processing
For mitochondrial analysis using Mitotracker Green or TMRM, whole cells were imaged on the z axis (8 to 12 focal planes). Images were subsequentially deconvolved and ‘y’ summed using the ImageJ software (NIH). Compiled images obtained were used for mitochondrial area, or ΔΨm and radial profile analysis, as illustrated in Fig. 1D. For mitochondrial network and ER colocalization, one focal plane was analyzed. The images obtained were deconvolved and the background was subtracted using the ImageJ software. Colocalization between organelles was quantified using the Manders' algorithm, as previously described (Manders et al., 1993; Costes et al., 2004; Parra et al., 2008).
Microscopic analysis of ΔΨm
For ΔΨm imaging, cells were plated on 25-mm glass coverslips in six-well plates and treated according to the experimental conditions. Briefly, cells were loaded with 200 nM tetramethylrhodamine (TMRM) for 20 minutes at 37°C, and the fluorescence imaging of cells was conducted in a confocal microscope (excitation 543 nm, emission 560 nm), as previously described (Voronina et al., 2004; Parra et al., 2008).
Mitochondrial network and ER imaging
Cells were plated on 25-mm glass coverslips in six-well plates. For ER and mitochondrial imaging, cells were transfected with a plasmid encoding ER-targeted RFP, and/or treated with 200 nM Mitotracker Green, respectively. Confocal image stacks were captured with a Zeiss LSM-5, Pascal 5 Axiovert 200, equipped with a Plan-Apochromat 63×, 1.4 NA oil DIC objective and with the LSM 5 3.2 software for image capture and analysis.
Oxygen consumption determination
Cells were plated on 60-mm-diameter Petri dishes and treated according to the experiment. Cells were then trypsinized, and the suspension (in PBS) was placed in a chamber at 25°C, coupled to a Clark electrode 5331 (Yellow Springs Instruments). Data obtained correspond to the amount of oxygen remaining in the chamber over a period of time. Cells were maintained in the chamber for 20 minutes in order to calculate the rate of oxygen consumption.
Reagents
DMEM, Trypan Clue, nocodazole, CCCP and oligomycin were purchased from Sigma. Cytochalasin D and 2-deoxyglucose (2DG) were purchased from. FBS was from Hyclone (Rockford, IL). The CellTiter Glo kit for ATP measuring was purchased from Promega. Trypsin, MTT, DiOC6(3), Lipofectamine 2000, Mitotracker Green and tetramethylrhodamine methyl ester (TMRM) were from Invitrogen. Tunicamycin and thapsigargin were from Enzo Life Sciences (Plymouth Meeting, PA). Ru360 and Ruthenium Red (RuRed) were from EMD Chemicals (Gibbstown, NJ). The anti-phosphorylated-eIF2α antibody was from Cell Signaling Technology. Anti-CHOP, anti-JNK and anti-phosphorylated-JNK antibodies were from Santa Cruz Biotechnology.
Statistical analysis
Results are means±s.e.m. unless otherwise indicated. At least three independent experiments were performed in triplicate, in order to test the statistical hypothesis. Data were analyzed using one-way or two-way ANOVA, followed by a Bonferroni post t-test. P<0.05 was considered statistically significant.
Transmission electron microscopy
Cells were fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, embedded in 2% agarose, post-fixed in buffered 1% osmium tetroxide, en-bloc stained in 2% uranyl acetate, dehydrated with a graded series of ethanol and embedded in EMbed-812 resin. Thin sections were cut on a Leica Ultracut UCT ultramicrotome and stained with 2% uranyl acetate and lead citrate. Images were acquired on a FEI Tecnai G2 Spirit electron microscope equipped with a LaB6 source and operating at 120 kV.
Treatments
Early ER stress was induced by treatment with 0.1–1 μg/ml tunicamycin (as indicated) or 1 μM thapsigargin for 0–4 hours. For viability experiments, tunicamycin was further present until 48 hours. Cytoskeleton disruption was induced by treatment with 10 μM nocodazole and 10 μM cytochalasin D. Glycolitic metabolism was impaired by 20 mM 2-DG and mitochondrial metabolism was impaired by 20 μM CCCP and 1 μg/ml oligomycin. Mitochondrial Ca2+ uptake was inhibited by 10 μM Ru360 and 10 μM RuRed. The Ins(1,4,5)P3 receptor was inhibited with 2 μM xestospongin B. Inhibitors were preincubated and maintained during treatments. Adenoviral transduction using AsMfn2, an adenovirus expressing Mfn2 antisense mRNA (Bach et al., 2003), was performed in HeLa cells at a multiplicity of infection (MOI) of 1000, 48 hours before the induction of ER stress. An empty adenovirus was used as control (EV).
Western blot analysis
Equal amounts of protein were separated by SDS-PAGE (10% polyacrylamide gels) and electrotransferred onto nitrocellulose. Membranes were blocked with 5% milk in Tris-buffered saline, pH 7.6, containing 0.1% Tween 20 (TBST). Membranes were incubated with primary antibodies at 4°C and re-blotted with horseradish-peroxidase-linked secondary antibody [1:5000 in 1% (w/v) milk in TBST]. The bands were detected using ECL, with exposure to Kodak film, and quantified by scanning densitometry.
Supplementary Material
Acknowledgments
This work was supported by CONICYT, Chile (FONDECYT grant 1080436 and FONDAP grant 15010006 to S.L.), Parkinson's UK (grant G-0905 to G.S.), the National Institutes of Health (to J.A.H. and B.A.R.), the American Heart Association (to M.I.,J.A.H. and B.A.R.), the American Heart Association-Jon Holden DeHaan Foundation (to J.A.H. and B.A.R.), NSERC grant 386757-2010 (to T.S.), CCSRI grant 2010-700306 (to T.S.), AI-HS scholarship 200500396 (to T.S.) and the Ministerio de Educación y Ciencia (MEC, SAF2008-03803 to A.Z.), grant 2009SGR915 from the Generalitat de Catalunya (to A.Z.) and CIBERDEM (Instituto de Salud Carlos III to A.Z.). We thank CONICYT, Chile for the doctoral fellowships to R.B., V.P., C.Q. and A.E.R., for the postdoctoral support (FONDAP) to J.M.V. and the postdoctoral fellowship 3110114 from to R.T.; we thank Becas Chile for the postdoctoral funding to J.M.V. We thank David Chan (Pasadena, CA) for Mfn2 wild-type and knockout cells. We also thank Carla Ortiz, Cristian Ibarra and Aleck W. Jones for helpful discussions regarding this manuscript. We thank Fidel Albornoz and Ruth Marquez for excellent technical assistance. S.L. is on a sabbatical leave at the University of Texas Southwestern Medical Center, Dallas, Texas, USA. The authors declare no conflicts of interest. Deposited in PMC for release after 12 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/13/2143/DC1
References
- Addabbo F., Ratliff B., Park H. C., Kuo M. C., Ungvari Z., Csiszar A., Krasnikov B., Sodhi K., Zhang F., Nasjletti A., et al. (2009). The Krebs cycle and mitochondrial mass are early victims of endothelial dysfunction: proteomic approach. Am. J. Pathol. 174, 34-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach D., Pich S., Soriano F. X., Vega N., Baumgartner B., Oriola J., Daugaard J. R., Lloberas J., Camps M., Zierath J. R., et al. (2003). Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J. Biol. Chem. 278, 17190-17197 [DOI] [PubMed] [Google Scholar]
- Balaban R. S. (2009). The role of Ca2+ signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim. Biophys. Acta 1787, 1334-1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge M. J. (2002). The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32, 235-249 [DOI] [PubMed] [Google Scholar]
- Boldogh I. R., Pon L. A. (2006). Interactions of mitochondria with the actin cytoskeleton. Biochim. Biophys. Acta 1763, 450-462 [DOI] [PubMed] [Google Scholar]
- Brenner S. L., Korn E. D. (1980). The effects of cytochalasins on actin polymerization and actin ATPase provide insights into the mechanism of polymerization. J. Biol. Chem. 255, 841-844 [PubMed] [Google Scholar]
- Brooks C., Cho S. G., Wang C. Y., Yang T., Dong Z. (2010). Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am. J. Physiol. Cell Physiol. 300, C447-C455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G. C. (1992). Control of respiration and ATP synthesis in mammalian mitochondria and cells. Biochem. J. 284, 1-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui M., Gilady S. Y., Fitzsimmons R. E., Benson M. D., Lynes E. M., Gesson K., Alto N. M., Strack S., Scott J. D., Simmen T. (2010). Rab32 modulates apoptosis onset and mitochondria-associated membrane (MAM) properties. J. Biol. Chem. 285, 31590-31602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casella J. F., Flanagan M. D., Lin S. (1981). Cytochalasin D inhibits actin polymerization and induces depolymerization of actin filaments formed during platelet shape change. Nature 293, 302-305 [DOI] [PubMed] [Google Scholar]
- Chami M., Oules B., Szabadkai G., Tacine R., Rizzuto R., Paterlini-Brechot P. (2008). Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol. Cell 32, 641-651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costes S. V., Daelemans D., Cho E. H., Dobbin Z., Pavlakis G., Lockett S. (2004). Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 86, 3993-4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G., Renken C., Varnai P., Walter L., Weaver D., Buttle K. F., Balla T., Mannella C. A., Hajnoczky G. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915-921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darios F., Muriel M. P., Khondiker M. E., Brice A., Ruberg M. (2005). Neurotoxic calcium transfer from endoplasmic reticulum to mitochondria is regulated by cyclin-dependent kinase 5-dependent phosphorylation of tau. J. Neurosci. 25, 4159-4168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito O. M., Scorrano L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605-610 [DOI] [PubMed] [Google Scholar]
- DeBerardinis R. J., Mancuso A., Daikhin E., Nissim I., Yudkoff M., Wehrli S., Thompson C. B. (2007). Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 104, 19345-19350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen M. R. (2004). Mitochondria in health and disease: perspectives on a new mitochondrial biology. Mol. Aspects Med. 25, 365-451 [DOI] [PubMed] [Google Scholar]
- Frederick R. L., Shaw J. M. (2007). Moving mitochondria: establishing distribution of an essential organelle. Traffic 8, 1668-1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J. R., Webster B. M., Mastronarde D. N., Verhey K. J., Voeltz G. K. (2010). ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules. J. Cell Biol. 190, 363-375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier C. A., Kitada T., Shen J. (2008). Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 105, 11364-11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz J. G., Genty H., St-Pierre P., Dang T., Joshi B., Sauve R., Vogl W., Nabi I. R. (2007). Reversible interactions between smooth domains of the endoplasmic reticulum and mitochondria are regulated by physiological cytosolic Ca2+ levels. J. Cell Sci. 120, 3553-3564 [DOI] [PubMed] [Google Scholar]
- Gorlach A., Klappa P., Kietzmann T. (2006). The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 8, 1391-1418 [DOI] [PubMed] [Google Scholar]
- Green D. R., Wang R. (2010). Calcium and energy: making the cake and eating it too? Cell 142, 200-202 [DOI] [PubMed] [Google Scholar]
- Haga N., Saito S., Tsukumo Y., Sakurai J., Furuno A., Tsuruo T., Tomida A. (2010). Mitochondria regulate the unfolded protein response leading to cancer cell survival under glucose deprivation conditions. Cancer Sci. 101, 1125-1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnoczky G., Csordas G., Das S., Garcia-Perez C., Saotome M., Sinha Roy S., Yi M. (2006). Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 40, 553-560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnoczky G., Saotome M., Csordas G., Weaver D., Yi M. (2007). Calcium signalling and mitochondrial motility. Novartis Found. Symp. 287, 105-117; discussion 117-121 [DOI] [PubMed] [Google Scholar]
- Hayashi T., Rizzuto R., Hajnoczky G., Su T. P. (2009). MAM: more than just a housekeeper. Trends Cell Biol. 19, 81-88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heal R. D., McGivan J. D. (1997). Induction of the stress protein Grp75 by amino acid deprivation in CHO cells does not involve an increase in Grp75 mRNA levels. Biochim. Biophys. Acta 1357, 31-40 [DOI] [PubMed] [Google Scholar]
- Hetz C., Glimcher L. H. (2009). Fine-tuning of the unfolded protein response: assembling the IRE1alpha interactome. Mol. Cell 35, 551-561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck P. J., Saxton W. M. (2005). The axonal transport of mitochondria. J. Cell Sci. 118, 5411-5419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoseki J., Ushioda R., Nagata K. (2010). Mechanism and components of endoplasmic reticulum-associated degradation. J. Biochem. 147, 19-25 [DOI] [PubMed] [Google Scholar]
- Ikesugi K., Mulhern M. L., Madson C. J., Hosoya K., Terasaki T., Kador P. F., Shinohara T. (2006). Induction of endoplasmic reticulum stress in retinal pericytes by glucose deprivation. Curr. Eye Res. 31, 947-953 [DOI] [PubMed] [Google Scholar]
- Jahani-Asl A., Cheung E. C., Neuspiel M., MacLaurin J. G., Fortin A., Park D. S., McBride H. M., Slack R. S. (2007). Mitofusin 2 protects cerebellar granule neurons against injury-induced cell death. J. Biol. Chem. 282, 23788-23798 [DOI] [PubMed] [Google Scholar]
- Lebiedzinska M., Szabadkai G., Jones A. W., Duszynski J., Wieckowski M. R. (2009). Interactions between the endoplasmic reticulum, mitochondria, plasma membrane and other subcellular organelles. Int. J. Biochem. Cell Biol. 41, 1805-1816 [DOI] [PubMed] [Google Scholar]
- Liu Y., Liu W., Song X. D., Zuo J. (2005). Effect of GRP75/mthsp70/PBP74/mortalin overexpression on intracellular ATP level, mitochondrial membrane potential and ROS accumulation following glucose deprivation in PC12 cells. Mol. Cell. Biochem. 268, 45-51 [DOI] [PubMed] [Google Scholar]
- Manders E. M. M., Verbeek E. J., Aten J. A. (1993). Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 169, 375-382 [DOI] [PubMed] [Google Scholar]
- Modriansky M., Dvorak Z. (2005). Microtubule disruptors and their interaction with biotransformation enzymes. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 149, 213-215 [PubMed] [Google Scholar]
- Munoz J. P., Chiong M., Garcia L., Troncoso R., Toro B., Pedrozo Z., Diaz-Elizondo J., Salas D., Parra V., Nunez M. T., et al. (2009). Iron induces protection and necrosis in cultured cardiomyocytes: Role of reactive oxygen species and nitric oxide. Free Radic. Biol. Med. 48, 526-534 [DOI] [PubMed] [Google Scholar]
- Nakagawa T., Zhu H., Morishima N., Li E., Xu J., Yankner B. A., Yuan J. (2000). Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403, 98-103 [DOI] [PubMed] [Google Scholar]
- Park S. H., Blackstone C. (2010). Further assembly required: construction and dynamics of the endoplasmic reticulum network. EMBO Rep. 11, 515-521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra V., Eisner V., Chiong M., Criollo A., Moraga F., Garcia A., Hartel S., Jaimovich E., Zorzano A., Hidalgo C., et al. (2008). Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc. Res. 77, 387-397 [DOI] [PubMed] [Google Scholar]
- Price N. P., Tsvetanova B. (2007). Biosynthesis of the tunicamycins: a review. J. Antibiot. (Tokyo) 60, 485-491 [DOI] [PubMed] [Google Scholar]
- Rasheva V. I., Domingos P. M. (2009). Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis 14, 996-1007 [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Pinton P., Carrington W., Fay F. S., Fogarty K. E., Lifshitz L. M., Tuft R. A., Pozzan T. (1998). Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763-1766 [DOI] [PubMed] [Google Scholar]
- Ron D., Walter P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519-529 [DOI] [PubMed] [Google Scholar]
- Samson F., Donoso J. A., Heller-Bettinger I., Watson D., Himes R. H. (1979). Nocodazole action on tubulin assembly, axonal ultrastructure and fast axoplasmic transport. J. Pharmacol. Exp. Ther. 208, 411-417 [PubMed] [Google Scholar]
- Shibata Y., Voeltz G. K., Rapoport T. A. (2006). Rough sheets and smooth tubules. Cell 126, 435-439 [DOI] [PubMed] [Google Scholar]
- Shibata Y., Voss C., Rist J. M., Hu J., Rapoport T. A., Prinz W. A., Voeltz G. K. (2008). The reticulon and DP1/Yop1p proteins form immobile oligomers in the tubular endoplasmic reticulum. J. Biol. Chem. 283, 18892-18904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmen T., Aslan J. E., Blagoveshchenskaya A. D., Thomas L., Wan L., Xiang Y., Feliciangeli S. F., Hung C. H., Crump C. M., Thomas G. (2005). PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 24, 717-729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadkai G., Rizzuto R. (2004). Participation of endoplasmic reticulum and mitochondrial calcium handling in apoptosis: more than just neighborhood? FEBS Lett. 567, 111-115 [DOI] [PubMed] [Google Scholar]
- Szabadkai G., Duchen M. R. (2008). Mitochondria: the hub of cellular Ca2+ signaling. Physiology (Bethesda) 23, 84-94 [DOI] [PubMed] [Google Scholar]
- Szabadkai G., Bianchi K., Varnai P., De Stefani D., Wieckowski M. R., Cavagna D., Nagy A. I., Balla T., Rizzuto R. (2006). Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901-911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajeddine N., Galluzzi L., Kepp O., Hangen E., Morselli E., Senovilla L., Araujo N., Pinna G., Larochette N., Zamzami N., et al. (2008). Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene 27, 4221-4232 [DOI] [PubMed] [Google Scholar]
- Villena J., Henriquez M., Torres V., Moraga F., Diaz-Elizondo J., Arredondo C., Chiong M., Olea-Azar C., Stutzin A., Lavandero S., et al. (2008). Ceramide-induced formation of ROS and ATP depletion trigger necrosis in lymphoid cells. Free Radic. Biol. Med. 44, 1146-1160 [DOI] [PubMed] [Google Scholar]
- Voronina S. G., Barrow S. L., Gerasimenko O. V., Petersen O. H., Tepikin A. V. (2004). Effects of secretagogues and bile acids on mitochondrial membrane potential of pancreatic acinar cells: comparison of different modes of evaluating DeltaPsim. J. Biol. Chem. 279, 27327-27338 [DOI] [PubMed] [Google Scholar]
- Yang L., Liu X., Hao J., Yang Y., Zhao M., Zuo J., Liu W. (2008). Glucose-regulated protein 75 suppresses apoptosis induced by glucose deprivation in PC12 cells through inhibition of Bax conformational change. Acta Biochim. Biophys. Sinica 40, 339-348 [DOI] [PubMed] [Google Scholar]
- Yi M., Weaver D., Hajnoczky G. (2004). Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J. Cell Biol. 167, 661-672 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.