

Abstract
Merosin-deficient congenital muscular dystrophy 1A (MDC1A) is a devastating neuromuscular disease that results in children being confined to a wheelchair, requiring ventilator assistance to breathe and premature death. MDC1A is caused by mutations in the LAMA2 gene, which results in the partial or complete loss of laminin-211 and laminin-221, the major laminin isoforms found in the basal lamina of skeletal muscle. MDC1A patients exhibit reduced α7β1 integrin; however, it is unclear how the secondary loss of α7β1 integrin contributes to MDC1A disease progression. To investigate whether restoring α7 integrin expression can alleviate the myopathic phenotype observed in MDC1A, we produced transgenic mice that overexpressed the α7 integrin in the skeletal muscle of the dyW−/− mouse model of MDC1A. Enhanced expression of the α7 integrin restored sarcolemmal localization of the α7β1 integrin to laminin-α2-deficient myofibers, changed the composition of the muscle extracellular matrix, reduced muscle pathology, maintained muscle strength and function and improved the life expectancy of dyW−/− mice. Taken together, these results indicate that enhanced expression of α7 integrin prevents muscle disease progression through augmentation and/or stabilization of the existing extracellular matrix in laminin-α2-deficient mice, and strategies that increase α7 integrin in muscle might provide an innovative approach for the treatment of MDC1A.
Key words: MDC1A, α7 integrin, Transgenic mice, Muscular dystrophy
Introduction
Merosin-deficient congenital muscular dystrophy 1A (MDC1A) is caused by mutations in the LAMA2 gene resulting in the production of a truncated form or complete loss of laminin-α2 protein (Helbling-Leclerc et al., 1995; Naom et al., 1997). The most severe form of MDC1A is associated with mutations that result in a complete loss of laminin-α2 protein and therefore the absence of laminin-211 and laminin-221 heterotrimers in the basal lamina. MDC1A patients exhibit severe weakness, dysmyelinating neuropathy, hypotonia, muscle atrophy and limited eye movement (Philpot et al., 1999b; Jones et al., 2001; Muntoni and Voit, 2004). Patients also exhibit feeding problems and/or respiratory difficulties and often require feeding tube placement or ventilator assistance (Philpot et al., 1999a). Most MDC1A patients are unable to walk without assistance and are confined to a wheelchair (Muntoni and Voit, 2004). A unique characteristic of MDC1A are changes in brain white-matter that are observed after 6 months of age. These changes might be associated with an increased likelihood of seizure-like activity (Philpot et al., 1999b; Jones et al., 2001; Muntoni and Voit, 2004). There is currently no cure or effective treatment for MDC1A, and patients die as early as the first decade of life (Muntoni and Voit, 2004; Mendell et al., 2006).
Laminin-211 and laminin-221 are the predominant laminin isoforms in the basal lamina of adult skeletal muscle. Laminin-211 is present at the extrajunctional sarcolemma, and laminin-221 is enriched at the myotendinous and the neuromuscular junctions (Martin et al., 1995; Burkin and Kaufman, 1999; Sasaki et al., 2002). Loss of laminin-211, in MDC1A patients and in mouse models of this disease, results in an inability of muscle fibers to adhere to the basement membrane and as a result laminin-α2-null myotubes are unstable and undergo apoptosis (Vachon et al., 1997; Kuang et al., 1999). Failed regeneration is also observed in laminin-α2-deficient myofibers, indicating the importance of the laminin-rich microenvironment for muscle repair (Kuang et al., 1999; Rooney et al., 2009b). In addition, laminin-α2 is expressed by Schwann cells in the peripheral nervous system, and lack of laminin-211 leads to reduced myelination resulting in impaired conduction velocity and peripheral neuropathy (Miyagoe-Suzuki et al., 2000; Occhi et al., 2005).
The α7β1 integrin is a primary laminin receptor expressed in skeletal, cardiac and vascular smooth muscle (Burkin et al., 1998; Burkin and Kaufman, 1999; Burkin et al., 2000). The α7 integrin transcript undergoes developmentally regulated RNA splicing to produce three alternative cytoplasmic domains, designated α7A, α7B and α7C, and two alternative extracellular domains, designated α7X1 and α7X2 (Collo et al., 1993; Song et al., 1993; Ziober et al., 1993; Hodges et al., 1997). The cytoplasmic domains differ in size, sequence and potential for signal transduction, whereas extracellular domains bind with differential affinities to laminin isoforms (Song et al., 1993; von der Mark et al., 2002). The α7B cytoplasmic domain is the largest and has a number of regions that potentially participate in signal transduction (Song et al., 1993).
The α7β1 integrin can bind to multiple laminin isoforms, including laminin-111, laminin-211, laminin-221, laminin-332, laminin-411, laminin-511, galectin-1 and fibronectin (Gu et al., 1994; Burkin and Kaufman, 1999; Patarroyo et al., 2002). Mutations in the α7-integrin-encoding gene (ITGA7) cause α7 integrin congenital myopathy, and patients exhibit delayed developmental milestones and impaired mobility (Hayashi et al., 1998). Mice that lack the α7 integrin develop muscular dystrophy that affects the myotendious junctions (Mayer et al., 1997; Hayashi et al., 1998; Nawrotzki et al., 2003; Flintoff-Dye et al., 2005). In the adult mouse, the α7β1 integrin is localized at the extrajunctional sarcolemma but is also enriched at neuromuscular and myotendinous junctions, where it mediates the adhesion of the muscle fibers to the extracellular matrix (Martin et al., 1996; Burkin et al., 1998; Burkin et al., 2000).
MDC1A patients, dy−/− and dy2J−/− mouse models have decreased levels of α7 integrin protein in their muscles, and this secondary loss of α7 integrin might contribute to disease progression (Hodges et al., 1997; Vachon et al., 1997; Peat et al., 2008). To determine whether restoring α7 integrin expression could serve as a therapeutic strategy in the treatment of MDC1A, we generated transgenic mice that overexpressed the α7 integrin in the skeletal muscle of the dyW−/− mouse model of MDC1A. Transgenic expression of the α7 integrin increased longevity and reduced the myopathic phenotype of dyW−/− mice. We show that these beneficial changes were achieved through augmenting and/or stabilizing the existing extracellular matrix in laminin-α2-deficient muscle. Our data demonstrate, for what we believe to be the first time, that the α7 integrin is a modifier of disease progression in laminin-α2-deficient mice and might be a novel drug target for the treatment of MDC1A.
Results
The α7B integrin is overexpressed in the muscle of dyW−/−;itga7+ transgenic mice
To determine whether overexpression of the α7 integrin in laminin-α2-deficient muscle could alleviate disease progression, transgenic mice were generated in which rat α7BX2 integrin expression was driven by the muscle creatine kinase (Mck, also known as Ckm) promoter, which we designate as itga7+ mice. These mice were bred with dyW+/− animals to generate dyW−/−;itga7+ mice. Pups were genotyped at 10 days of age for the laminin-α2 mutant and wild-type alleles and the Itga7 transgene, as previously described (Kuang et al., 1998; Burkin et al., 2005; Boppart et al., 2008).
To confirm that the presence of the transgene resulted in increased α7 integrin in dyW muscle quantitative real-time PCR (qRT-PCR) and immunoblotting were performed on the mouse tibialis anterior (TA) and gastrocnemius muscle. qRT-PCR experiments showed a 4.1-fold increase in α7 integrin transcript in the dyW−/− muscle compared with that in wild-type muscle, whereas dyW−/−;itga7+ mice exhibited a 17-fold increase in α7 integrin transcript in muscle compared with that in wild-type (Table 1). Immunoblotting confirmed a 2.8-fold increase in α7B integrin in the muscle of dyW−/−;itga7+ mice (Fig. 1A,C). There was a 67.5% and 83.2% reduction in α7A integrin in the muscles of dyW−/− and dyW−/−;itga7+ animals respectively compared with that in wild-type (Fig. 1A,B). Analysis of other laminin-binding integrins in skeletal muscle revealed a 86% and 61.5% reduction in α3 integrin protein in dyW−/− and dyW−/−;itga7+ animals respectively compared with that in wild-type (Fig. 1D). Finally no significant change in β1D integrin protein was observed in any of the genotypes (Fig. 1E). Together these results indicate the MCK-α7BX2 transgene increased both the transcript and protein level expression of α7BX2 integrin in the skeletal muscle of dyW−/− mice without altering the expression of other laminin-binding integrin isoforms and chains.
Table 1.
Changes in gene expression in dyW−/− mice

Fig. 1.

Enhanced α7B integrin in the muscle of dyW−/−;itga7+ mice. (A) Immunoblot showing protein levels of α7A integrin, α7B integrin, α3 integrin and β1D integrin in wild-type, dyW−/−, and dyW−/−;itga7+ mice. α-tubulin was used to normalize protein loading. (B) Densitometry analysis of α7A integrin indicating that both the dyW−/− animals (*P<0.05) and the dyW−/−;itga7+ animals have significantly reduced levels of α7A integrin (**P<0.01). (C) Densitometry showing that α7B integrin is significantly elevated in the dyW−/−;itga7+ animals when compared with the dyW−/− animals (*P<0.05). (D) Protein quantification for α3 integrin showing a significant reduction in protein levels in the dyW−/− mice compared with the wild-type mice (*P<0.05) with no other significant changes. (E) No differences in β1D protein levels were observed among the three genotypes analyzed.
Transgenic expression of the α7 integrin in dyW−/− muscle restores sarcolemmal localization of the α7β1 integrin
Previous studies have shown that loss of laminin-α2 results in reduced sarcolemmal localization of α7 integrin (Hodges et al., 1997; Vachon et al., 1997; Peat et al., 2008). Immunofluorescence was used to determine whether forced expression of α7 integrin restored sacrolemmal localization of the α7β1 integrin in skeletal muscle. Immunofluorescence analysis confirmed that, whereas wild-type mice exhibited substantial laminin-α2 in the basal lamina, dyW−/− and dyW−/−;itga7+ mice were negative for laminin-α2 and produced only negligible amounts of the laminin-α2 globular domain (Fig. 2A), which is consistent with previous studies (Guo et al., 2003). The localization of β1D integrin was examined in wild-type, dyW−/− and dyW−/−;itga7+ TA muscle (Fig. 2A). Compared with that in wild-type mice, dyW−/− muscle exhibited a reduced sarcolemmal localization of β1D integrin. By contrast, transgenic overexpression of the α7 integrin restored sarcolemmal localization of β1D integrin in TA muscle. Spectrin levels were the same in all three genotypes and were used as a positive control for immunofluorescence.
Fig. 2.

Transgenic expression of α7 integrin restores sarcolemmal location in dyW−/− skeletal muscle. (A) Immunofluorescence using TA muscle from 4-week-old male mice was used to confirm the absence of laminin-α2 protein in dyW−/− muscle. Wild-type TA muscle shows sarcolemmal staining for laminin-α2, which is not present in dyW−/− or dyW−/−;itga7+ animals. Sections were also incubated with anti-β1D antibody to visualize β1D integrin. (B) High-resolution confocal microscopy of TA muscle using rhodamine-labeled WGA, to highlight the sarcolemma, and CDB347 antibody, to visualize α 7B integrin, was performed. The increased yellow colocalization signal in the dyW−/−;itga7+ animals indicates restoration of sarcolemmal localization of the α7 integrin. Scale bars: 20 μm (A); 30 μm (B).
To examine whether transgenic overexpression restored the sarcolemmal localization of the α7 integrin in muscle, TA cryosections were stained for α7B integrin (green) and with rhodamine-labeled wheat germ agglutinin (WGA) (red), which binds N-acetylglucosamine-labeled sugar residues in the basal lamina, and high-resolution confocal microscopy was used to assess sarcolemmal localization of α7 integrin (Fig. 2B). Wild-type and dyW−/−;itga7+ muscle exhibited colocalization of the α7 integrin and WGA (indicated by the yellow staining), as well as sharp sarcolemmal staining for α7 integrin (Fig. 2B). By contrast, the dyW−/− animals showed little colocalization between α7 integrin and WGA, and did not show sharp α7 integrin sarcolemmal localization (Fig. 2B). Together these data indicate that transgenic expression of the α7 integrin in dyW−/− muscle restored sarcolemmal localization of the α7β1 integrin in laminin-α2 deficient muscle.
Transgenic overexpression of α7 integrin increases the longevity of dyW−/− mice
MDC1A patients and the dyW−/− mouse model exhibit severe muscle wasting that results in reduced life expectancy (Muntoni and Voit, 2004; Mendell et al., 2006). We next investigated whether transgenic overexpression of α7 integrin improved the longevity of dyW−/− animals. Kaplan–Meier survival analysis revealed that 50% of dyW−/− mice died by 70 days of age (Fig. 3A). By contrast, 50% of dyW−/−;itga7+ mice survived beyond 140 days of age (Fig. 3A). The oldest dyW−/− mouse lived to 132 days, whereas the oldest dyW−/−;itga7+ animal lived to 318 days, which represents a 2.4-fold increase in life expectancy (Fig. 3A). These results indicate that increasing α7 integrin expression in skeletal muscle substantially improves the life expectancy of laminin-α2-deficient mice.
Fig. 3.

Transgenic expression of α7 integrin in the skeletal muscle of dyW−/− mice increases longevity but not weight gain. (A) Kaplan–Meier survival curve shows dyW−/−;itga7+ (n=9) animals have a median life expectancy that is 2.0-fold longer than their dyW−/− (n=15) littermates (P<0.05). (B) Analysis of body weights shows that wild-type mice are heavier than both dyW−/−;itga7+ and dyW−/− animals at all ages analyzed (***P<0.001).
MDC1A patients exhibit reduced body weight and muscle strength (Muntoni and Voit, 2004; Mendell et al., 2006). We next examined whether transgenic expression of the α7 integrin improved the body weight and muscle strength and function of dyW−/− mice. Compared with that of wild-type mice, both dyW−/− and dyW−/−;itga7+ mice had reduced body weight at 3, 8 and 12 weeks of age with no difference between these laminin-α2-deficient mice (Fig. 3B). This indicates that the transgene is not effective at restoring body weight to that of wild-type animals.
We next examined muscle strength. Forelimb grip strength was measured to analyze overall forelimb muscle strength. Compared with 4-week-old wild-type mice, both dyW−/− and dyW−/−;itga7+ mice exhibited a 36.9% and 30.8% reduction respectively in forelimb grip strength compared with that of wild-type mice, with no difference between these laminin-α2-null animals (Fig. 4A). However, when the animals were re-evaluated at 8 weeks of age, dyW−/− animals showed a 55% reduction in grip strength compared with that of wild-type mice, whereas dyW−/−;itga7+ mice showed only a 24.8% reduction in grip strength compared with that in wild-type (Fig. 4A). This indicates that while the dyW−/− animals progressively got weaker, enhanced levels of α7 integrin in the dyW−/−;itga7+ animals preserved muscle strength over time.
Fig. 4.

The dyW−/−;itga7+ transgenic mice maintain muscle strength and exhibit increased motor activity. (A) Box and whiskers plot of forelimb grip strength at four weeks of age showing that dyW−/− and dyW−/−;itga7+ mice are weaker than the wild-type animals (***P<0.001). At 8 weeks of age wild-type and dyW−/−;itga7+ animals (*P<0.05) are significantly stronger than dyW−/− mice. (B) Box and whiskers plot of activity, as measured by the amount of active time over a 5-minute period. At 4 weeks of age there is no significant difference between any of the genotypes analyzed (P>0.05). At 8 weeks of age wild-type and dyW−/−;itga7+ mice were significantly more active than dyW−/− animals. There was no significant difference in the activity levels of 8 week old wild-type and dyW;itga7+ mice. (C) Box and whiskers plot of stand-up activity over a 5-minute period. At 4 weeks of age there were no significant differences between the genotypes. At 8 weeks of age, wild-type and dyW;itga7+ animals exhibited significantly more stand ups compared to dyW−/− animals. There was no significant difference in stand up activity between the dyW;itga7+ and wild-type animals. *P<0.05; **P<0.01; ****P<0.0001. For each plot the box represents 1st quartile, mean and 3rd quartile and the whiskers represent minimum and maximum values.
We next evaluated two different measures of activity in 4- and 8-week-old animals. Measurement of overall activity over a 5-minute period when a mouse is introduced into a new cage revealed that at 4 weeks of age there was no significant difference between wild-type, dyW−/− and dyW−/−;itga7+ mice (Fig. 4B). By contrast, at 8 weeks of age, wild-type and dyW−/−;itga7+ mice animals exhibited a 1.8-fold and 1.5-fold increase in active time respectively compared with that of dyW−/− animals. There was no significant difference between the wild-type and dyW−/−;itga7+ animals (Fig. 4B). Finally, stand-up activity of animals introduced to a new cage over a 5-minute period was assessed on mice capable of standing up. At four weeks of age there was no significant difference between the genotypes (Fig. 4C). At eight weeks of age wild-type and dyW−/−;itga7+ animals exhibited a 5.2-fold and 4.2-fold increase in stand-up activity compared with that of dyW−/− animals (Fig. 4C). Together these results indicate transgenic expression of the α7 integrin maintained muscle strength and activity in the dyW−/− mouse model of MDC1A.
Transgenic overexpression of α7 integrin reduces skeletal muscle pathology in dyW−/− mice
To determine whether enhanced α7 integrin expression prevented skeletal muscle pathology in laminin-α2-deficient mice, TA muscle cryosections from 4-week-old wild-type, dyW−/− and dyW−/−;itga7+ mice were stained with hematoxylin and eosin. Compared with that in wild-type mice, dyW−/− TA muscle showed greater variation in myofiber size, presence of centrally located nuclei (CLN), fibrosis and inflammatory cell infiltrate (Fig. 5A). The dyW−/−;itga7+ muscle exhibited less muscle pathology, including fewer myofibers with CLN, less inflammatory infiltrate and less variation in myofiber size (Fig. 5A).
Fig. 5.

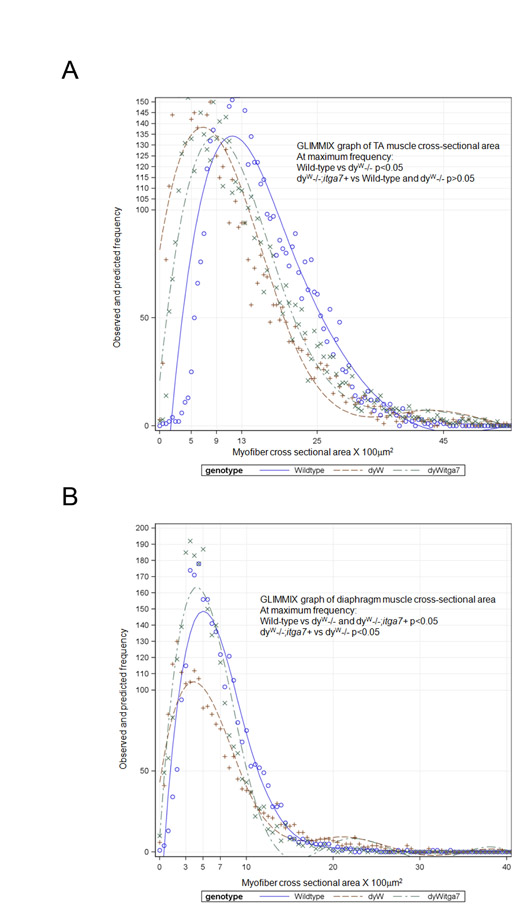
Reduced muscle disease in laminin-α2-deficient mice that transgenically express α7 integrin. (A) Hematoxylin and eosin staining of sections from TA muscles from 4-week-old mice shows more muscle pathology in dyW−/− mice compared with that in wild-type. The TA muscle of dyW−/−;itga7+ mice exhibited reduced muscle pathology. Scale bar: 20 μm. (B) The percentage of myofibers containing CLN were used to evaluate muscle repair in TA muscles from 4-week-old mice. Compared with that in wild-type, dyW−/− showed a 19-fold increase in the percentage of myofibers containing CLN. By contrast, dyW−/−;itga7+ TA muscle showed a 66.5% reduction in the percentage of myofibers with CLN (***P<0.001). (C) Myofiber cross-sectional area revealed dyW−/−;itga7+ animals have a fiber size distribution closer to that of wild-type animals than to dyW−/− mice (supplementary material Fig. S1).
The requirement for muscle repair was determined by evaluating the percentage of myofibers containing CLN. CLN are an indicator of muscle degeneration and regeneration, which requires satellite cell activation to repair damaged myofibers (Xu et al., 2007). Compared with 4-week-old wild-type mice, which showed few myofibers with CLN, 4-week-old dyW−/− TA muscle exhibited a 4.8-fold increase in myofibers with CLN (Fig. 5B). There was a 66.5% reduction in the percentage of CLN in the dyW−/−;itga7+ compared with that in the dyW−/− animals, although the level of CLN in these mice was still elevated compared with that in wild-type mice (Fig. 5B). These results indicate transgenic expression of the α7 integrin in the skeletal muscle results in reduced muscle pathology and fewer myofibers with CLN than in dyW−/− mice.
MDC1A patients and dyW−/− mice exhibit substantial myofiber size variation and the presence of a large number of hypotrophic muscle fibers. Measurements of myofiber cross-sectional area were used to determine whether transgenic expression of the α7 integrin improved this aspect of muscle pathology. Compared with that of wild-type, with a mean myofiber cross-sectional area of 16.1 μm2, dyW−/− TA muscle had a mean myofiber cross-sectional area of 9.5 μm2, with a curve shifted to the left indicating increased numbers of hypotrophic muscle fibers (Fig. 5C). By contrast, TA myofibers in dyW−/−;itga7+ mice had a mean cross-sectional area of 11.5 μm2 (Fig. 5C). These results indicate that transgenic expression of the α7 integrin in skeletal muscle of laminin-α2-null mice reduced the variation in myofiber size and improved the myofiber cross-sectional area. At the maximum frequency of occurrence (i.e. that most commonly observed), the dyW−/− mice had significantly smaller fibers than the wild-type mice. There was no significant difference between the dyW−/− and dyW−/−;itga7+ or between the dyW−/−;itga7+ and wild-type mice (supplementary material Fig. S1). This indicates transgenic mice that overexpress the α7 integrin in muscle have fewer hypotrophic fibers compared with dyW−/− animals and are more similar to wild-type.
Transgenic α7 integrin expression alters the composition of the extracellular matrix in laminin-α2-deficient muscle
The loss of laminin-211 and laminin-221 in the muscle extracellular matrix is the underlying cause of muscle disease in MDC1A. Given that the α7 integrin is the primary laminin receptor in muscle we next determined the mechanism by which increased α7β1 integrin rescued dyW−/− mice in the absence of its laminin-211–laminin-221 ligand. qRT-PCR was used to examine the expression profile of genes encoding an array of extracellular matrix proteins in the gastrocnemius muscle of 4-week-old wild-type, dyW−/− and dyW−/−;itga7+ mice. qRT-PCR revealed that dyW−/− mice exhibited increased levels of several transcripts: disintegrin and metalloproteinase with thrombospondin motifs 5 (Adamts5), agrin (Agrn), collagen 6A1 (Col6a1), galectin-1 (Lgals1), galectin-3 (Lgals3), matrix metalloproteinase 2 (Mmp2), integrin α3 (Itga3), integrin α6 (Itga6), integrin α7 (Itga7), laminin-α4 (Lama4), laminin-α5 (Lama5), nidogen 1 (Nid1), tenascin C (Tnc), tissue inhibitor of metalloproteinase 1 (Timp1) and tissue inhibitor of metalloproteinase 2 (Timp2), compared with that in wild-type (Table 1). Transgenic expression of the α7 integrin in dyW−/−;itga7+ mice resulted in reduced levels of agrin and Mmp2 transcripts compared with dyW−/− mice (Table 1). Transgenic expression of the α7 integrin in dyW−/−;itga7+ mice resulted in increased levels of transcripts for Col6A1, Lgals1, Lgals3, Itga3, Itga6, Itga7, Tnc and Timp1 compared with that in dyW−/− mice (Table 1).
The α7β1 integrin has been shown to interact with galectin-1 in cultured muscle cells (Gu et al., 1994), and we next determined whether transgenic expression of the α7 integrin altered expression of galectin-1 and -3 in the muscle of laminin-α2-null mice. Compared with that in wild-type mice, the level of galectin-1 transcript was increased 9.2-fold in dyW−/− muscle and 12.1-fold in dyW−/−;itga7+ animals (Table 1). This increase in galectin-1 transcript correlated with a 1.8-fold increase in galectin-1 protein in dyW−/−;itga7+ animals compared with that in wild-type (Fig. 6A). These results indicate an increase in galectin-1 protein in the gastrocnemius muscle of dyW−/−;itga7+ animals.
Fig. 6.

Transgenic expression of α7 integrin in the absence of laminin-α2 increased expression of galectin-1 and -3 in the muscle of dyW−/− mice. (A) Immunoblot of galectin-1 protein (50 μg protein lysate was used) showing that 4-week-old dyW−/−;itga7+ animals have a 1.4-fold increase in galectin-1 protein compared with wild-type and dyW−/− mice (*P<0.05). (B) An immunoblot of galectin-3 protein required differential protein loading across the genotypes as follows: wild-type, 100 μg; dyW−/−, 50 μg; and dyW−/−;itga7+, 25 μg. Quantitative analysis revealed that 4-week-old dyW−/− mice have 2-fold more galectin-3 compared with that in wild-type animals (**P<0.01). The dyW−/−;itga7+ animals have 7-fold and 2.9-fold more galectin-3 protein compared with that in both wild-type and dyW−/− animals, respectively (***P<0.001).
Galectin-3 transcript was increased 70-fold and 80-fold in 4-week-old dyW−/− and dyW−/−;itga7+ muscle, respectively, compared with that in wild-type mice (Table 1). This increase in galectin-3 transcript resulted in a 2-fold increase in galectin-3 protein in dyW−/− mice and a 7-fold increase in galectin-3 protein in dyW−/−;itga7+ animals compared with that in wild-type (Fig. 6B). These results indicate that loss of laminin-α2 results in increased galectin-3 in the muscle extracellular matrix of dyW−/− mice and that transgenic expression of α7 integrin further enhanced the levels of galectin-3 in laminin-α2-deficient muscle.
In laminin-α2-deficient muscle, tenascin C is normally localized at the myotendinous junctions and has been shown to be enriched at extrajunctional sites, which correlate with regions of muscle regeneration (Ringelmann et al., 1999). qRT-PCR was used to examine whether transgenic overexpression of the α7 integrin altered the expression of tenascin C in the muscle of dyW−/− mice. qRT-PCR confirmed a 28-fold increase in Tnc transcript in the gastrocnemius muscle of dyW−/− mice and a 49-fold increase in Tnc transcript in dyW−/−;itga7+ gastrocnemius muscle compared with that in wild-type (Table 1). These results indicate transgenic expression of the α7 integrin augmented Tnc transcription in laminin-α2-null muscle.
Immunofluorescence was used to confirm the qRT-PCR and immunoblotting results for several proteins. Immunofluorescence also demonstrated increased extracellular galectin-1, galectin-3 and tenascin C in the extracellular matrix, with galectin-3 and tenascin C being more prevalent in the dyW−/−;itga7+ mice (Fig. 7). Immunostaining demonstrated reduced MMP2 and increased TIMP1 in the extracellular matrix of the dyW−/−;itga7+ mice compared with that in the dyW−/− mice (Fig. 7). These results support the idea that overexpression of the α7 integrin results in both augmentation and stabilization of the existing extracellular matrix in dyW−/−;itga7+ animals.
Fig. 7.

Enhanced expression of α7 integrin augments the extracellular matrix and slows matrix turnover in the dyW−/− mouse. Immunfluorescence was used to evaluate extracellular matrix proteins and associated proteins. Galectin-1 was found to be elevated in both the dyW−/− and dyW−/−;itga7+ animals when compared with that in the wild-type. The dyW−/−;itga7+ animals had the greatest amount of staining for galectin-3 with some staining present in the dyW−/− animals and none in wild-type controls. Tenascin C was found in areas of inflammation in the dyW−/− animals, whereas it was located more dispersedly in the dyW−/−;itga7+ animals (arrows). Matrix metalloproteinase 2 (MMP2) was highest in the dyW−/− animals. Overexpression of α7 integrin led to reduced staining for MMP2 in the dyW−/−;itga7+ animals. Conversely, Tissue inhibitor of metalloproteinase 1 (TIMP1) was found to be elevated by enhanced α7 integrin expression in the dyW−/−; itga7+ mice compared with both dyW−/− and wild-type animals. Scale bar: 20 μm.
Inflammatory infiltrate is reduced in dyW muscle with enhanced α7 integrin
Inflammatory cell infiltration (particularly by the monocyte or macrophage line) is a hallmark of MDC1A (Pegoraro et al., 1996). To determine whether overexpression of the α7 integrin reduced inflammation in dyW−/−;itga7+ muscle, CD11b was used as a marker for the presence of macrophages in skeletal muscle (Fig. 8A). Compared with that in the 4-week-old wild-type TA muscle, dyW−/− TA muscle exhibited an 11.1-fold increase in CD11b-positive cells (Fig. 8B). By contrast, 4-week-old dyW−/−;itga7+ showed a 55.3% decrease in the number of CD11b-positive cells compared with that in dyW−/− TA muscle (Fig. 8B). These results indicate transgenic overexpression of the α7 integrin reduced the level of macrophage-mediated inflammation in dyW−/− muscle.
Fig. 8.

Enhanced expression of α7 integrin reduces inflammation in dyW−/− muscle. (A) Immunofluorescence of TA muscles from 4-week-old mice with anti-CD11b antibody, a marker for macrophages. Compared with wild-type mice, dyW−/− mice have an increased number of CD11b-positive macrophages. The dyW−/−;itga7+ animals have less inflammatory cell infiltration compared with dyW−/− animals. Scale bar: 20 μm. (B) Quantification revealed that dyW−/− mice have a 2.4-fold increase in CD11b-positive macrophages in TA muscle compared with the number in wild-type mice. By contrast, dyW−/−;itga7+ animals exhibited a 55.3% decrease in CD11b positive macrophages in TA muscle compared with the number in dyW−/− mice. ***P<0.001 for all groups.
Transgenic expression of α7 integrin prevents muscle disease progression in the diaphragm of dyW mice
MDC1A patients exhibit severe restrictive respiratory syndrome and require ventilator assistance to breathe as a result of severe diaphragm muscle pathology (Muntoni and Voit, 2004). Histological analysis and measurements of myofiber area were used to examine whether transgenic expression of the α7 integrin prevented the onset of severe diaphragm muscle pathology. Hematoxylin and eosin studies revealed that transgenic expression of the α7 integrin in 4-week-old dyW−/− diaphragm muscle resulted in reduced mononuclear cell infiltrate, hypotrophic muscle fibers, CLN and fibrosis (Fig. 9A).
Fig. 9.

Enhanced α7 integrin expression improves diaphragm pathology in 4-week-old dyW−/− mice. (A) Hematoxylin and eosin staining of diaphragm muscle from 4-week-old dyW−/− mice exhibit variation in myofiber size, presence of CLN and fibrosis. By contrast, the dyW−/−;itga7+ diaphragm shows reduced muscle pathology. Scale bar: 20 μm. (B) Cross-sectional myofiber area of the diaphragm muscle of 4-week-old animals shows that the dyW−/− ;itga7+ mice have a myofiber cross-sectional area more similar to wild-type animals than that observed in dyW−/− mice (see also supplementary material Fig. S1).
Analysis of myofiber cross-sectional areas confirmed the improvement in the muscle pathology observed in the histological studies. Compared with wild-type mice, which have peak myofiber cross-sectional area of between 3.5–4.5 μm2, dyW−/− muscle exhibited a large number of hypotrophic muscle fibers, with a peak myofiber area of only 2 μm2 (Fig. 9B). By contrast, dyW−/−;itga7+ diaphragm myofibers exhibited a peak myofiber area of between 3.5 and 5 μm2 and a curve more similar to that of wild-type (Fig. 9B). At the maximum frequency myofiber area, all three groups were significantly different from one another (supplementary material Fig. S1). These results indicate that transgenic expression of the α7 integrin prevents muscle disease progression in the diaphragm of laminin-α2-null mice.
Discussion
Merosin-deficient congenital muscular dystrophy (MDC1A) is considered one of the most common congenital muscular dystrophies. Classic MDC1A is associated with a complete loss of laminin-α2 and has the most severe clinical signs with severe muscle weakness, hypotonia and muscle atrophy. Feeding and respiratory difficulties are common, and patients often require either feeding tube placement or positive pressure ventilation. Maximal motor activity is often unsupported sitting with few patients achieving independent ambulation (Muntoni and Voit, 2004). There is currently no cure or effective treatment for MDC1A and patients often die at a young age from respiratory insufficiency (Mendell et al., 2006).
The observation of reduced α7 integrin in MDC1A muscle has led to the proposal that the secondary loss of α7 integrin contributes to the myopathic phenotype observed in MDC1A patients and laminin-α2-null mice (Hodges et al., 1997; Vachon et al., 1997; Peat et al., 2008). Because α7 integrin congenital myopathy does not phenocopy MDC1A, disease progression must involve more than loss of the α7 integrin receptor; however, given that the α7β1 integrin binds to multiple ligands, restoring α7β1 integrin receptor in skeletal muscle might improve muscle integrity and reduce the myopathy associated with disease progression.
Previous studies have shown a loss of α7 integrin from the skeletal muscle of dy−/− and dy2J−/− mouse models of MDC1A (Hodges et al., 1997; Vachon et al., 1997; Gawlik et al., 2006). Our results show the dyW−/− muscle exhibits reduced α7 integrin expression and loss of sarcolemmal localization of the α7β1 integrin. Studies have suggested transcriptional regulation between laminin and α7 integrin in muscle (Hodges et al., 1997) and the small amount of truncated laminin-α2 globular domain produced in dyW−/− muscle (Guo et al., 2003), which might be sufficient to promote some α7 integrin expression but insufficient for the correct sarcolemmal localization in muscle. In this study, we show that transgenic overexpression of the α7 integrin in dyW−/− mice resulted in sarcolemmal localization of the α7β1 integrin. Restoring sarcolemmal localization of the α7β1 integrin reduced muscle pathology, maintained muscle strength and increased survival of dyW−/− mice. Taken together, our data support the hypothesis that secondary loss of α7 integrin contributes to muscle disease progression and that restoring sarcolemmal localization might reduce the myopathy in MDC1A.
Studies have indicated that the α7β1 integrin might act to modulate the organization and deposition of the muscle extracellular matrix (Colognato et al., 1999; Rooney et al., 2009a). To investigate the mechanism by which enhanced α7 integrin expression improved the myopathic phenotype in the absence of the laminin-211–laminin-221 ligand, we examined changes in levels of other extracellular matrix proteins and enzymes that regulate the extracellular matrix. MDC1A patients and laminin-α2-deficient mice have increased laminin-α4 and laminin-α5 chains; however, previous studies have indicated that the increase in these laminin isoforms is unlikely to rescue the myopathic phenotype (Patton et al., 1997; Cohn and Campbell, 2000; Gawlik et al., 2004). Transgenic expression of α7 integrin did not further increase α7A integrin isoform or α3 integrin in skeletal muscle, indicating that these laminin and integrin isoforms do not play a role in the rescue of laminin-α2-deficient mice that overexpress α7 integrin in skeletal muscle.
Our analysis of extracellular matrix proteins in the dyW−/− muscle basal lamina revealed that transgenic expression of the α7 integrin resulted in reduced expression of Mmp2 and increased levels of Timp1. Mmp2 is associated with the breakdown of the basal lamina, whereas Timp1 acts to inhibit the activity of matrix metalloproteinases, such as Mmp2 (Chen and Li, 2009). Reduced Mmp2 and increased Timp1 in the muscle of α7 integrin transgenic dyW−/− mice indicate that increased α7β1 integrin acts to stabilize the basal lamina by affecting the expression of enzymes that degrade and remodel the muscle extracellular matrix.
The α7β1 integrin has been shown to directly bind galectin-1 (Gu et al., 1994). Galectin-1 (L-14) is a lactoside-binding protein that has been shown to inhibit myoblast adhesion by blocking the interaction of laminin to integrins. Galectin-3 has also been shown to mediate cell adhesion and regulate the cell signaling pathways involved in apoptosis, cell proliferation, inflammation and gene transcription (Dumic et al., 2006). Galectin-3 binds extracellular matrix proteins, including laminin and tenascin C, and has been shown to bind directly to integrins. In breast carcinoma cells, galectin-3 has been shown to mediate β1 integrin endocytosis (Furtak et al., 2001). In this study, we show that both galectin-1 and -3 are increased in dyW−/− muscle and further increased in dyW−/−;itga7+ skeletal muscle that transgenically overexpress the α7 integrin. These results suggest that, in the absence of laminin-211, expression of galectin-1 and -3 are increased and might serve as biomarkers for disease progression. Because the α7β1 integrin has been previously shown to bind galectin-1, our results suggest that rescue of laminin-α2-deficient muscle by overexpression of the α7 integrin is mediated through interactions of the α7β1 integrin with galectin-1 and/or galectin-3 to stabilize myofibers.
Although tenascin C is normally enriched at myotendinous junctions in wild-type muscle, in dy−/− laminin-α2-deficient mice, tenascin C is found around regenerating myofibers, sites of inflammation and extrajunctional sites (Ringelmann et al., 1999). Our study shows that transgenic expression the α7 integrin resulted in increased tenascin C transcript in dyW−/− muscle. Because transgenic expression of the α7 integrin results in a reduced percentage of myofibers with CLN (and therefore reduced muscle regeneration) and decreased inflammation, these results indicate that increased α7β1 integrin stabilizes the existing extracellular matrix, including tenascin C, in dyW−/− muscle.
Approaches towards the treatment of MDC1A have targeted apoptosis in mouse models, through the inhibition of Bax, transgenic expression of the anti-apoptotic protein Bcl-2 and treatment with the anti-apoptotic drugs doxycycline (Girgenrath et al., 2004; Girgenrath et al., 2009) or omigapil (Erb et al., 2009). Another approach has focused on restoring or improving the muscle extracellular matrix through transgenic expression, in mouse models, of laminin-α1 (Gawlik et al., 2004; Gawlik et al., 2006), laminin-α2 (Kuang et al., 1998), mini-agrin (Bentzinger et al., 2005) and N-acetylgalactosamine transferase (Xu et al., 2007). Our results indicate that increased expression of the α7β1 integrin receptor in muscle improves adhesion of myofibers to the endogenous basal lamina to reduce muscle disease progression and maintain muscle strength in the dyW−/− mouse model of MDC1A. Results from this study indicate that compounds that increase α7 integrin expression in laminin-α2-deficient muscle might provide a novel therapeutic approach for the treatment of MDC1A.
Materials and Methods
Generation of laminin-α2-deficient mice that overexpress α7 integrin in skeletal muscle
All experiments involving mice were approved by the University of Nevada, Reno Institutional Animal Care and Use Committee. Transgenic α7 integrin dyW−/− mice were generated by breeding mice that overexpressed the α7BX2 integrin in skeletal muscle (Burkin et al., 2001) with dyW+/− animals (Kuang et al., 1998). Resultant pups, which were heterozygous for the laminin-α2 mutant allele and positive for the α7BX2 transgene were bred to dyW+/− mice. The male pups from these matings included dyW+/+;itga7–(wild-type), dyW−/−;itga7–(dyW−/−) (laminin-α2-deficient) and dyW−/−;itga7+ (laminin-α2-deficient and overexpressing the α7BX2 integrin) mice. Male littermates were used as controls for all experiments. Genomic DNA was isolated from tail biopsies taken at 10 days of age using the Wizard SV genomic dna purification system (Promega). PCR was used as previously described to detect the laminin-α2 allele and the α7BX2 transgene (Kuang et al., 1998; Burkin et al., 2005; Boppart et al., 2008). The dyW+/− mice were a gift from Eva Engvall via Paul Martin (The Ohio State University, Columbus, OH).
Isolation of skeletal muscle
Four-week-old wild-type, dyW−/− and dyW−/−;itga7+ male mice were killed by CO2 asphyxiation followed by cervical dislocation in accordance with a protocol approved by the University of Nevada Reno Animal Care and Use Committee. Skeletal muscles were dissected and flash frozen in liquid-nitrogen-cooled isopentane. Tissues were stored at −80°C.
Western blot analysis
Gastrocnemius muscles from 4-week-old male mice were pulverized with a mortar and pestle cooled in liquid nitrogen. Protein was extracted in RIPA buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 1 mM Na3VO4, 10 mM NaF, 0.5% Triton X-100, 0.5% NP50, 10% glycerol, 2 mM PMSF and a 1:200 dilution of Protease Inhibitor Cocktail Set III) and quantified using a Bradford assay (Bio-Rad). Proteins were separated by SDS-PAGE. The α7 integrin was detected with a 1:1000 dilution of anti-α7B antibody overnight. Integrin α7A was detected using a 1:1000 dilution of CDB 345 antibody overnight (a gift from Stephen Kauffman, University of Illinois, Urbana-Champaign, IL). Integrin α3 was quantified using the AB1920 antibody (Chemicon). The β1D integrin was visualized using an anti-β1D-antibody (a gift from Woo Keun Song, Gwanju Institute for Science and Technology, South Korea) overnight. All primary antibodies were followed by a goat anti-rabbit-IgG secondary antibody (1:5000, Li-Cor Biosciences) for 1 hour. Galectin-1 was detected with a 1:1000 dilution of H00003956-D01P (Abnova, Walnut, CA). Galectin-3 was detected with a 1:1000 dilution of ab53082 (Abcam, Cambridge, MA). Immunoblots were normalized by using a 1:5000 dilution of an anti-α-tubulin antibody (AbCam) followed by a 1:5000 dilution of goat anti-mouse-IgG secondary antibody. Band intensities were determined with an Odyssey Imaging System.
Immunofluorescence
Cryosections (8 μm) of 4-week-old male tibialis anterior (TA) muscle were cut using a LeicaCM 1850 cryostat and mounted onto pre-cleaned Surgipath slides. Sections were fixed using 4% paraformaldehyde (PFA) for 2 or 5 minutes then rehydrated using PBS. Slides were blocked in 5% BSA in PBS then incubated with antibodies against laminin-α2G or β1D integrin, as previously described (Rooney et al., 2009b). For detection of galectin-1, H00003959-D01P (Abnova) antibody was used. Galectin-3 was visualized using ab53082 (AbCam). Antibody T3413 (Sigma) was used to detect tenascin C. Mmp2 and Timp1 were detected using antibodies ab37150 and ab86482 respectively (AbCam). Slides were then incubated using an appropriate secondary, which was a FITC-conjugated anti-rabbit-IgG antibody in all cases except for tenascin C, where a FITC-conjugated anti-rat-IgG secondary antibody was used. For detection of spectrin, slides were fixed for 1 minute in ice-cold acetone then treated with the M.O.M. kit according to package instructions (FMK-2201, Vector Laboratories). A mouse monocloncal anti-spectrin antibody (Novo Castra NCL-spec2) was then used at 1:100 for 30 minutes followed by a FITC-conjugated anti-mouse-IgG secondary antibody at 1:1000 for 1 hour. Slides were mounted using Vectashield with DAPI and imaged using a Zeiss Axioskop 2 Plus fluorescence microscope. Images were captured using a Zeiss AxioCam HRc digital camera with Axiovision 4.1 software.
Inflammatory cell infiltrate
TA muscle cryosections from 4-week-old mice were fixed in 4% PFA for 5 minutes followed by rehydration with PBS. Slides were incubated with FITC-conjugated rat anti-mouse-CD11b antibody (BD Biosciences) at 1:1000 for 1 hour to detect macrophages in the muscle tissue. Slides were washed with PBS and mounted using Vectashield with DAPI. Muscle sections from five mice of each genotype were analyzed and CD11b-positive cells per 20 fields at 400× magnification were counted. A Zeiss Axioskop 2 Plus fluorescent microscope was used to view the slides and images were captured using a Zeiss AxioCam HRc digital camera with Axiovision 4.1 software.
Confocal microscopy
The TA muscles from 4-week-old male mice from each genotype were sectioned and subjected to immunofluorescence. For detection of α7B integrin, sections were fixed in ice-cold acetone (−20°C) for 1 minute then rehydrated using PBS. Cryosections were blocked in a 5% BSA in PBS solution for 20 minutes followed by incubation with CDB347 (which recognizes the cytoplasmic domain of both mouse and rat α7B integrin) or anti-β1DA2-integrin antibodies for 1 hour. Slides were then washed with 1% BSA in PBS and incubated with FITC-conjugated anti-rabbit-IgG antibody for 1 hour. Slides were again washed with 1% BSA in PBS. To outline the myofibers, sections were incubated with rhodamine-labeled wheat germ agglutinin for 30 minutes. Slides were mounted using Vectashield with DAPI. Images were captured using an Olympus Fluoview confocal scanning system.
Survival and weight gain analysis
Male mice were allowed to age and monitored daily for weight loss and any signs of pain, distress or illness. A weight loss of >10% over a 1-week period was also considered a terminal sign and the animals were humanely euthanized. Weights from animals of each genotype were compared at 3, 8 and 12 weeks of age.
Grip strength and activity assays
The forelimb grip strength of 4- and 8-week-old male wild-type, dyW−/− and dyW−/−;itga7+ mice were measured using a SDI grip strength system and a Chatillon DFE digital force gauge (San Diego Instruments) as per a standard protocol. Five consecutive tests were performed for each mouse and the data averaged for each mouse genotype. In order to assess mobility 4- and 8-week-old male wild-type, dyW−/− and dyW−/−;itga7+ mice were placed in a clean cage by themselves and monitored for 5 minutes. Periods of moving about the cage, standing up and digging were considered times of activity. Additionally, during this time period the number of times the mouse stood up was recorded. Stand-up testing was only performed on animals who were physically able to stand up. Some mice were excluded from these samples owing to the extent of their peripheral neuropathy.
Hematoxylin and eosin staining
Cryosections from 4-week-old TA and diaphragm muscle were stained using hematoxylin and eosin, as previously described (Rooney et al., 2009a), and were used to determine the percentage of myofibers that contained CLN using a Zeiss Axioskop 2 Plus fluorescence microscope. A minimum of 1000 fibers per animal (five animals per group) were counted and the percentage of myofibers with CLN calculated. Images were captured using a Zeiss AxioCam HRc digital camera and Axiovision 4.1 software.
Myofiber area determination
Cryosections from TA and diaphragm muscles from 4-week-old mice were fixed for 5 minutes in 4% PFA and rehydrated in PBS. Myofibers were outlined with 2 μg/ml Oregon-Green-488-conjugated WGA (Molecular Bioprobes) for 30 minutes. Sections were then washed with PBS for 15 minutes and mounted in Vectashield. A minimum of 1000 fibers per animal with five animals per group were assessed for the TA muscle. For diaphragm muscle, a minimum of 500 fibers per animal with five animals per genotyped were used. Myofiber cross-sectional area was determined with a Zeiss Axioskop 2 Plus fluorescence microscope and images were captured with a Zeiss AxioCam HRc digital camera with Axiovision 4.1 software.
Quantitative real-time PCR analysis
Total RNA was purified from five 4-week-old male wild-type, dyW−/−, and dyW−/−;itga7+ mice gastrocnemius muscles using TRIzol (Invitrogen). After the concentration was determined, mRNA was pooled equally by genotype for cDNA production. The cDNA was prepared from 4 μg of pooled total RNA with random hexamers and Superscript III (Invitrogen) using standard procedures. Quantitative real-time PCR was conducted with 50 pg total cDNA using SYBR Green Jumpstart (Sigma–Aldrich). Primer sequences for the mouse extracellular matrix genes are listed in supplementary Table S1 and levels were normalized to that of Gapdh. The fold change over wild-type was calculated using the ΔΔCt method after normalization and the average fold change in transcript (±s.e.m.) were calculated.
Statistics
Data is reported as the mean±s.d. One-way analysis of variance (ANOVA) was used to compare animals across groups. A Kaplan–Meier log-rank test was used to determine significance of lifespan changes. Myofiber cross-sectional area was analyzed using the GLIMMIX statistical analysis package in SAS. P<0.05 was considered significant.
Supplementary Material
Acknowledgments
The authors thank Paul Martin and Eva Engvall for the dyW mice and Stephen Kaufman (University of Illinois, Urbana-Champaign, IL) for the anti-α7 integrin antibodies. The authors thank Rebecca Evans and Honglin Tian for technical assistance, Heather Burkin for critically reading the manuscript and George Fernandez for statistical analysis. This study was supported by NIH/NIAMS R01AR053697 to D.J.B. and NIH-NCRR 5P20RR015581. Deposited in PMC for release after 12 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/13/2287/DC1
References
- Bentzinger C. F., Barzaghi P., Lin S., Ruegg M. A. (2005). Overexpression of mini-agrin in skeletal muscle increases muscle integrity and regenerative capacity in laminin-alpha2-deficient mice. FASEB J. 19, 934-942 [DOI] [PubMed] [Google Scholar]
- Boppart M. D., Volker S. E., Alexander N., Burkin D. J., Kaufman S. J. (2008). Exercise promotes alpha7 integrin gene transcription and protection of skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R1623-R1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkin D. J., Kaufman S. J. (1999). The alpha7beta1 integrin in muscle development and disease. Cell Tissue Res. 296, 183-190 [DOI] [PubMed] [Google Scholar]
- Burkin D. J., Gu M., Hodges B. L., Campanelli J. T., Kaufman S. J. (1998). A functional role for specific spliced variants of the alpha7beta1 integrin in acetylcholine receptor clustering. J. Cell Biol. 143, 1067-1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkin D. J., Kim J. E., Gu M., Kaufman S. J. (2000). Laminin and alpha7beta1 integrin regulate agrin-induced clustering of acetylcholine receptors. J. Cell Sci. 113, 2877-2886 [DOI] [PubMed] [Google Scholar]
- Burkin D. J., Wallace G. Q., Nicol K. J., Kaufman D. J., Kaufman S. J. (2001). Enhanced expression of the alpha 7 beta 1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J. Cell Biol. 152, 1207-1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkin D. J., Wallace G. Q., Milner D. J., Chaney E. J., Mulligan J. A., Kaufman S. J. (2005). Transgenic expression of {alpha}7{beta}1 integrin maintains muscle integrity, increases regenerative capacity, promotes hypertrophy, and reduces cardiomyopathy in dystrophic mice. Am. J. Pathol. 166, 253-263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Li Y. (2009). Role of matrix metalloproteinases in skeletal muscle: migration, differentiation, regeneration and fibrosis. Cell Adh. Migr. 3, 337-341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn R. D., Campbell K. P. (2000). Molecular basis of muscular dystrophies. Muscle Nerve 23, 1456-1471 [DOI] [PubMed] [Google Scholar]
- Collo G., Starr L., Quaranta V. (1993). A new isoform of the laminin receptor integrin alpha 7 beta 1 is developmentally regulated in skeletal muscle. J. Biol. Chem. 268, 19019-19024 [PubMed] [Google Scholar]
- Colognato H., Winkelmann D. A., Yurchenco P. D. (1999). Laminin polymerization induces a receptor-cytoskeleton network. J. Cell Biol. 145, 619-631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumic J., Dabelic S., Flogel M. (2006). Galectin-3: an open-ended story. Biochim. Biophys. Acta 1760, 616-635 [DOI] [PubMed] [Google Scholar]
- Erb M., Meinen S., Barzaghi P., Sumanovski L. T., Courdier-Fruh I., Ruegg M. A., Meier T. (2009). Omigapil ameliorates the pathology of muscle dystrophy caused by laminin-alpha2 deficiency. J. Pharmacol. Exp. Ther. 331, 787-795 [DOI] [PubMed] [Google Scholar]
- Flintoff-Dye N. L., Welser J., Rooney J., Scowen P., Tamowski S., Hatton W., Burkin D. J. (2005). Role for the alpha7beta1 integrin in vascular development and integrity. Dev. Dyn. 234, 11-21 [DOI] [PubMed] [Google Scholar]
- Furtak V., Hatcher F., Ochieng J. (2001). Galectin-3 mediates the endocytosis of beta-1 integrins by breast carcinoma cells. Biochem. Biophys. Res. Commun. 289, 845-850 [DOI] [PubMed] [Google Scholar]
- Gawlik K., Miyagoe-Suzuki Y., Ekblom P., Takeda S., Durbeej M. (2004). Laminin alpha1 chain reduces muscular dystrophy in laminin alpha2 chain deficient mice. Hum. Mol. Genet. 13, 1775-1784 [DOI] [PubMed] [Google Scholar]
- Gawlik K. I., Mayer U., Blomberg K., Sonnenberg A., Ekblom P., Durbeej M. (2006). Laminin alpha1 chain mediated reduction of laminin alpha2 chain deficient muscular dystrophy involves integrin alpha7beta1 and dystroglycan. FEBS Lett. 580, 1759-1765 [DOI] [PubMed] [Google Scholar]
- Girgenrath M., Dominov J. A., Kostek C. A., Miller J. B. (2004). Inhibition of apoptosis improves outcome in a model of congenital muscular dystrophy. J. Clin. Invest. 114, 1635-1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girgenrath M., Beermann M. L., Vishnudas V. K., Homma S., Miller J. B. (2009). Pathology is alleviated by doxycycline in a laminin-alpha2-null model of congenital muscular dystrophy. Ann. Neurol. 65, 47-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M., Wang W., Song W. K., Cooper D. N., Kaufman S. J. (1994). Selective modulation of the interaction of alpha 7 beta 1 integrin with fibronectin and laminin by L-14 lectin during skeletal muscle differentiation. J. Cell Sci. 107, 175-181 [DOI] [PubMed] [Google Scholar]
- Guo L. T., Zhang X. U., Kuang W., Xu H., Liu L. A., Vilquin J. T., Miyagoe-Suzuki Y., Takeda S., Ruegg M. A., Wewer U. M., et al. (2003). Laminin alpha2 deficiency and muscular dystrophy; genotype-phenotype correlation in mutant mice. Neuromuscul. Disord. 13, 207-215 [DOI] [PubMed] [Google Scholar]
- Hayashi Y. K., Chou F. L., Engvall E., Ogawa M., Matsuda C., Hirabayashi S., Yokochi K., Ziober B. L., Kramer R. H., Kaufman S. J., et al. (1998). Mutations in the integrin alpha7 gene cause congenital myopathy. Nat. Genet. 19, 94-97 [DOI] [PubMed] [Google Scholar]
- Helbling-Leclerc A., Zhang X., Topaloglu H., Cruaud C., Tesson F., Weissenbach J., Tome F. M., Schwartz K., Fardeau M., Tryggvason K., et al. (1995). Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat. Genet. 11, 216-218 [DOI] [PubMed] [Google Scholar]
- Hodges B. L., Hayashi Y. K., Nonaka I., Wang W., Arahata K., Kaufman S. J. (1997). Altered expression of the alpha7beta1 integrin in human and murine muscular dystrophies. J. Cell Sci. 110, 2873-2881 [DOI] [PubMed] [Google Scholar]
- Jones K. J., Morgan G., Johnston H., Tobias V., Ouvrier R. A., Wilkinson I., North K. N. (2001). The expanding phenotype of laminin alpha2 chain (merosin) abnormalities: case series and review. J. Med. Genet. 38, 649-657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang W., Xu H., Vachon P. H., Liu L., Loechel F., Wewer U. M., Engvall E. (1998). Merosin-deficient congenital muscular dystrophy. Partial genetic correction in two mouse models. J. Clin. Invest. 102, 844-852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang W., Xu H., Vilquin J. T., Engvall E. (1999). Activation of the lama2 gene in muscle regeneration: abortive regeneration in laminin alpha2-deficiency. Lab. Invest. 79, 1601-1613 [PubMed] [Google Scholar]
- Martin P. T., Ettinger A. J., Sanes J. R. (1995). A synaptic localization domain in the synaptic cleft protein laminin beta 2 (s-laminin). Science 269, 413-416 [DOI] [PubMed] [Google Scholar]
- Martin P. T., Kaufman S. J., Kramer R. H., Sanes J. R. (1996). Synaptic integrins in developing, adult, and mutant muscle: selective association of alpha1, alpha7A, and alpha7B integrins with the neuromuscular junction. Dev. Biol. 174, 125-139 [DOI] [PubMed] [Google Scholar]
- Mayer U., Saher G., Fassler R., Bornemann A., Echtermeyer F., von der Mark H., Miosge N., Poschl E., von der Mark K. (1997). Absence of integrin alpha 7 causes a novel form of muscular dystrophy. Nat. Genet. 17, 318-323 [DOI] [PubMed] [Google Scholar]
- Mendell J. R., Boue D. R., Martin P. T. (2006). The congenital muscular dystrophies: recent advances and molecular insights. Pediatr. Dev. Pathol. 9, 427-443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagoe-Suzuki Y., Nakagawa M., Takeda S. (2000). Merosin and congenital muscular dystrophy. Microsc. Res. Tech. 48, 181-191 [DOI] [PubMed] [Google Scholar]
- Muntoni F., Voit T. (2004). The congenital muscular dystrophies in 2004, a century of exciting progress. Neuromuscul. Disord. 14, 635-649 [DOI] [PubMed] [Google Scholar]
- Naom I. S., D'Alessandro M., Topaloglu H., Sewry C., Ferlini A., Helbling-Leclerc A., Guicheney P., Weissenbach J., Schwartz K., Bushby K., et al. (1997). Refinement of the laminin alpha2 chain locus to human chromosome 6q2 in severe and mild merosin deficient congenital muscular dystrophy. J. Med. Genet. 34, 99-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrotzki R., Willem M., Miosge N., Brinkmeier H., Mayer U. (2003). Defective integrin switch and matrix composition at alpha 7-deficient myotendinous junctions precede the onset of muscular dystrophy in mice. Hum. Mol. Genet. 12, 483-495 [DOI] [PubMed] [Google Scholar]
- Occhi S., Zambroni D., Del C. U., Amadio S., Sirkowski E. E., Scherer S. S., Campbell K. P., Moore S. A., Chen Z. L., Strickland S., et al. (2005). Both laminin and Schwann cell dystroglycan are necessary for proper clustering of sodium channels at nodes of Ranvier. J. Neurosci. 25, 9418-9427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patarroyo M., Tryggvason K., Virtanen I. (2002). Laminin isoforms in tumor invasion, angiogenesis and metastasis. Semin. Cancer Biol. 12, 197-207 [DOI] [PubMed] [Google Scholar]
- Patton B. L., Miner J. H., Chiu A. Y., Sanes J. R. (1997). Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J. Cell Biol. 139, 1507-1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peat R. A., Smith J. M., Compton A. G., Baker N. L., Pace R. A., Burkin D. J., Kaufman S. J., Lamande S. R., North K. N. (2008). Diagnosis and etiology of congenital muscular dystrophy. Neurology 71, 312-321 [DOI] [PubMed] [Google Scholar]
- Pegoraro E., Mancias P., Swerdlow S. H., Raikow R. B., Garcia C., Marks H., Crawford T., Carver V., Di C. B., Hoffman E. P. (1996). Congenital muscular dystrophy with primary laminin alpha2 (merosin) deficiency presenting as inflammatory myopathy. Ann. Neurol. 40, 782-791 [DOI] [PubMed] [Google Scholar]
- Philpot J., Bagnall A., King C., Dubowitz V., Muntoni F. (1999a). Feeding problems in merosin deficient congenital muscular dystrophy. Arch. Dis. Child 80, 542-547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot J., Cowan F., Pennock J., Sewry C., Dubowitz V., Bydder G., Muntoni F. (1999b). Merosin-deficient congenital muscular dystrophy: the spectrum of brain involvement on magnetic resonance imaging. Neuromuscul. Disord. 9, 81-85 [DOI] [PubMed] [Google Scholar]
- Ringelmann B., Roder C., Hallmann R., Maley M., Davies M., Grounds M., Sorokin L. (1999). Expression of laminin alpha1, alpha2, alpha4, and alpha5 chains, fibronectin, and tenascin-C in skeletal muscle of dystrophic 129ReJ dy/dy mice. Exp. Cell Res. 246, 165-182 [DOI] [PubMed] [Google Scholar]
- Rooney J. E., Gurpur P. B., Burkin D. J. (2009a). Laminin-111 protein therapy prevents muscle disease in the mdx mouse model for Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 106, 7991-7996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney J. E., Gurpur P. B., Yablonka-Reuveni Z., Burkin D. J. (2009b). Laminin-111 restores regenerative capacity in a mouse model for alpha7 integrin congenital myopathy. Am. J. Pathol. 174, 256-264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T., Giltay R., Talts U., Timpl R., Talts J. F. (2002). Expression and distribution of laminin alpha1 and alpha2 chains in embryonic and adult mouse tissues: an immunochemical approach. Exp. Cell Res. 275, 185-199 [DOI] [PubMed] [Google Scholar]
- Song W. K., Wang W., Sato H., Bielser D. A., Kaufman S. J. (1993). Expression of alpha 7 integrin cytoplasmic domains during skeletal muscle development: alternate forms, conformational change, and homologies with serine/threonine kinases and tyrosine phosphatases. J. Cell Sci. 106, 1139-1152 [DOI] [PubMed] [Google Scholar]
- Vachon P. H., Xu H., Liu L., Loechel F., Hayashi Y., Arahata K., Reed J. C., Wewer U. M., Engvall E. (1997). Integrins (alpha7beta1) in muscle function and survival. Disrupted expression in merosin-deficient congenital muscular dystrophy. J. Clin. Invest. 100, 1870-1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Mark H., Williams I., Wendler O., Sorokin L., von der Mark K., Poschl E. (2002). Alternative splice variants of alpha 7 beta 1 integrin selectively recognize different laminin isoforms. J. Biol. Chem. 277, 6012-6016 [DOI] [PubMed] [Google Scholar]
- Xu R., Chandrasekharan K., Yoon J. H., Camboni M., Martin P. T. (2007). Overexpression of the cytotoxic T cell (CT) carbohydrate inhibits muscular dystrophy in the dyW mouse model of congenital muscular dystrophy 1A. Am. J. Pathol. 171, 181-199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziober B. L., Vu M. P., Waleh N., Crawford J., Lin C. S., Kramer R. H. (1993). Alternative extracellular and cytoplasmic domains of the integrin alpha 7 subunit are differentially expressed during development. J. Biol. Chem. 268, 26773-26783 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}