

SUMMARY
Objectives
To determine the role of Smad1 in bone development and postnatal bone formation.
Methods
Col2a1-Cre transgenic mice were bred with Smad1fx/fx mice to produce chondrocyte-specific Smad1 conditional knockout (cKO) mice. Embryonic skeletal preparation and staining were performed, alkaline phosphatase activity (ALP) and relative gene expression were examined in isolated primary cells. Smad1fx/fx mice were also bred with Col1a1-Cre transgenic mice to produce osteoblast-specific Smad1 cKO mice. Postnatal bone formation was assessed by micro-computed tomography (μCT) and histological analyses in 2-month-old mice. Mineralized bone nodule formation assay, 5-bromo-2′-deoxy-uridine (BrdU) labeling and gene expression analysis were performed.
Results
Mice with chondrocyte- and osteoblast-specific deletion of the Smad1 gene are viable and fertile. Calvarial bone development was delayed in chondrocyte-specific Smad1 cKO mice. In osteoblast-specific Smad1 cKO mice, BMP signaling was partially inhibited and mice developed an osteopenic phenotype. Osteoblast proliferation and differentiation were impaired in osteoblast-specific Smad1 cKO mice.
Conclusions
Smad1 plays an essential role in bone development and postnatal bone formation.
Keywords: Smad1, Conditional knockout, BMP signaling, Chondrocyte, Osteoblast
Introduction
The vertebrate skeletal system is composed of two distinct tissues: cartilage and bone. The variant cartilage types are connective tissues important in numerous and varied roles during prenatal and postnatal skeletal development1. Bones are rigid mineralized organs formed in a variety of shapes. The cell type located in cartilage is the chondrocyte and in bone the osteoblast and osteoclast. Chondrocytes and osteoblasts are both originally derived from a common mesenchymal progenitor2, while osteoclasts have a hematopoietic origin. There are two different mechanisms of bone formation:membranous bone formation and endochondral ossification3–5 and both of these mechanisms start with condensation of mesenchymal cells. During endochondral ossification most mesenchymal cells will differentiate into chondrocytes and eventually these chondrocytes will be replaced by osteoblasts and most bone tissues in the body are developed through this process. In contrast, bones of the skull vault, or cranium, and the clavicle form directly from mesenchymal progenitors in a process termed membranous ossification.
Prenatal and postnatal bone formation is a complicated process that is regulated by multiple growth factors, signaling molecules, and transcription factors. Sox9, Runx2 and β-catenin are important transcription factors and a signaling protein, respectively, which affect cell fate during early prenatal skeletal development. Sox9 inactivation in the cranial neural crest causes cranial neural crest-derived cells to deviate from their chondrogenic differentiation into an osteoblastic lineage6. Runx2-deficient calvarial cells differentiate into chondrocytes instead of osteoblasts when cultured with BMP-27. Conditional inactivation of the β-catenin gene in mouse mesenchymal cells results in loss of osteoblasts and ectopic chondrocyte formation in bone tissues through membranous and endochondral bone formation processes8. During the postnatal stage, Ihh and β-catenin play an important role in normal cartilage and bone development. Postnatal conditional ablation of Ihh leads to condylar disorganization and growth retardation9, while late stage postnatal conditional activation of the β-catenin gene causes articular cartilage degradation10.
BMPs, especially BMP-2, and their signaling proteins are required in both chondrogenesis and bone formation at embryonic and postnatal stages11–13. Smad1 is an immediate downstream transducing molecule of the BMP receptor and plays a central role in mediating BMP signaling14. Activation of Smad1 is achieved through phosphorylation by the BMP type I receptor in a ligand-dependent manner. Phosphorylated Smad1 thereafter forms a heteromeric complex with Smad4. This heteromeric Smad complex translocates and accumulates in the nucleus participating in gene transcription in conjunction with other transcription factors. Smad1 is critical in embryonic development. Targeted deletion of the Smad1 gene results in early embryonic lethality due to failure of the allantois to fuse to the chorion and animals die around embryonic day 10 (E10)15,16. Since bone development starts at a later stage of embryonic development and the first ossification center forms around E14.5 in mice17, it is impossible to study the role of Smad1 in bone development and subsequent bone formation postnatally using Smad1 knockout (KO) mice.
Although an accumulating body of evidence shows that BMP signaling is critical for chondrogenesis and osteoblast differentiation, the physiological role of bone morphogenetic protein (BMP) signaling in prenatal and postnatal bone formation in vivo remains to be defined. Recently, Retting et al. found that Smads 1 and 5 are required for chondrogenesis by analyzing cartilage-specific Smad1 and Smad5 double KO mice18. However, the precise role of Smad1 in prenatal bone development and postnatal bone formation remains undefined. In this study, we generated chondrocyte- and osteoblast-specific Smad1 cKO mice using the Cre-loxP system19–21 and investigated the specific role of Smad1 in bone development and postnatal bone formation. We found that chondrocyte-specific deletion of the Smad1 gene causes ectopic cartilage formation in calvaria at the embryonic stage, and further leads to delayed calvarial bone ossification. Moreover, osteoblast-specific Smad1 cKO mice have impaired postnatal bone formation. Our findings provide direct evidence that Smad1 can regulate prenatal bone development and postnatal bone formation.
Materials and methods
Generation of the Smad1 cKO Mice
To generate Smad1fx/fx mice, the Smad1 locus was targeted by pLoxpneo-loxP-Smad1 in which exon 2 was flanked with loxP sites by insertion of pLoxpneo in intron 1 and a third loxP site in intron 222. Viable homozygous Smad1fx/fx mice were obtained, indicating that the insertion of pLoxpneo in intron1 does not interfere with expression of the Smad1 gene. To generate chondrocyte-specific Smad1 cKO mice, Smad1fx/fx mice were bred with Col2a1-Cre transgenic mice (Table I). In these Smad1 cKO mice (hereafter referred to as Smad1Col2a1), the Smad1 gene was deleted by Cre recombinase which was driven by the 3 kb murine type II collagen promoter (Col2a1-Cre)23. To determine the role of Smad1 in postnatal bone formation, we also generated osteoblast-specific Smad1 cKO mice. To facilitate the excision activity of Cre, we bred Smad1fx/fx mice with CMV-Cre mice first24 (Table II). The strain of CMV-Cre mice mediates deletion of loxP-flanked genes in all tissues, including germ cells25. In this mouse line every single cell would have only one floxed Smad1 allele, removal of which would result in compound heterozygosity for the deleted-null allele (CMV-Cre; Smad1fx/wt). After that, we crossed Smad1del/wt mice with the Col1a1-Cre transgenic mice in which Cre recombinase was driven by the 2.3 kb murine osteoblast-specific type I collagen promoter26–28 (Table II). The 2.3 kb type I collagen promoter is mainly active in mature osteoblasts29,30. Mating of these animals with mice homozygous for the floxed allele (Smad1fx/fx) generated the progeny with desired genotype, Col1a1-Cre;Smad1del/fx mice (osteoblast-specific Smad1 cKO) (hereafter referred to as Smad1Col1a1). The Cre-dependent recombination was detected by polymerase chain reaction (PCR) [Fig. 2(B), Lane 1, 4 and 5].
Table I.
Breeding and production of Smad1Col2a1 mice
| Breeding | Desired progeny |
|---|---|
| a) Col2a1-Cre+/− ×Smad1fx/fx | a) Col2a1-Cre+/−; Smad1fx/wt |
| b) Col2a1-Cre+/−; Smad1fx/wt× Smad1fx/fx | b) Col2a1-Cre+/−; Smad1fx/fx |
Table II.
Breeding and production of Smad1Col1a1 mice
| Breeding | Desired progeny |
|---|---|
| a) CMV-Cre+/−× Smad1fx/fx | a) Smad1del/wt |
| b) Col1a1-Cre+/−×Smad1del/wt | b) Col1a1-Cre+/−; Smad1del/wt |
| c) Col1a1-Cre+/−; Smad1del/wt ×Smad1fx/fx | c) Col1a1-Cre+/−; Smad1del/fx |
The sequences of PCR primers for genotyping and detecting Cre transgene are Cre-5′, 5′-CCT ggA AAA TgC TTC TgT CCg TTT gCC-3′; Cre-3′, 5′-gAg TTg ATA gCT ggC Tgg Tgg Cag ATg-3′, and the size of the PCR product is 600-bp. The sequences of PCR primers for genotyping Smad1fx/fx mice are SIF400, 5′-gTT CCC ATT Tgg TTC CAA gC-3′; LoxP2, 5′-gAg CTC TgC TCC gCC ACT CA-3′. The 360-bp PCR product was detected in wild-type mice and the 410-bp PCR product was detected in Smad1fx/fx mice [Fig. 1(A)]. The sequences of PCR primers for genotyping Smad1 cKO mice are S1F400, 5′-gTT CCC ATT Tgg TTC CAA gC-3′; LoxP2, 5′-gAg CTC TgC TCC gCC ACT CA-3′; Rec-Smad1, 5′-CAC CTg TgC CCC CTC CAA gT-3′. The 360-bp PCR product was detected in wild-type mice, the 410-bp PCR product was detected in Smad1fx/fx mice, and the 300-bp product was detected after Smad1 recombination [Fig. 2(A)].
Fig. 1.


Generation and analysis of Smad1Col2a1 mice. (A) Heterozygous Smad1 floxed mice (Smad1fx/wt) were bred with each other (Table I) and the production of homozygous Smad1 floxed mice (Smad1fx/fx) were identified by PCR using primers SIF400/LoxP2 (lower panel, Lane 1, 410-bp band). Smad1fx/fx mice were then bred with Co21a1-Cre mice (Table I, step a and b) and Smad1Col2a1 mice were identified by PCR using Cre-5′/Cre-3′ primers (600-bp PCR product) and SIF400/LoxP2 primers (410-bp PCR product). (B–E) Total RNAs were extracted from calvarial tissues of E16.5 Cre-negative and Smad1Col2a1 embryos. Expression of Col2a1 mRNA was detected in both Cre-negative and Smad1Col2a1 embryos and expression of Cre mRNA was only detected in Cre-positive Smad1Col2a1 embryos. Expression of Smad1 mRNA was significantly reduced in E16.5 Smad1Col2a1 embryos. (F) Chondrocyte-specific Smad1Col2a1 embryos and littermate control embryos are indistinguishable in size at embryonic day 16.5 (E16.5). Alizarin red/Alcian blue staining was performed on E16.5 embryos. E16.5 Smad1Col2a1 embryos had delayed mineralization and ectopic cartilage formation of frontal [F] and parietal [P] bones compared to the same aged control embryos. (G) Calvarial Alizarin red/Alcian blue staining showed delayed mineralization of supraoccipital [S] bone in E16.5 Smad1Col2a1 embryo. (H) The formation of ossification center of proximal phalangeal bone was also delayed in E16.5 Smad1Col2a1 embryos. (I and J) μ-CT analysis showed reduced calvarial BV and mineralization in E16.5 Smad1Col2a1 embryos. Unpaired Student’s t-test, P = 0.045 (n = 3). (K) E18.5 Smad1Col2a1 embryos and littermate control embryos are indistinguishable in size. No delayed mineralization and ectopic cartilage formation were observed in E18.5 Smad1Col2a1 embryos.
Fig. 2.

Reduction in marker gene expression in sternal chondrocytes and calvarial cells derived from Smad1Col2a1 mice. (A) Primary sternal chondrocytes were isolated from 3-day-old Cre-negative and Smad1Col2a1 mice and total RNAs were extracted from these cells. Expression of genes related to chondrocyte differentiation and was measured by real-time PCR. Significant reduction in expression of these genes was observed in Smad1-deficient cells. Unpaired Student’s t-test, Col2a1, P = 0.033; Col10a1, P = 0.005; Runx2, P = 0.012; ALP, P = 0.048; Osterix, P = 0.004; OC, P = 0.080; Mmp13, P = 0.022 (Cre-negative, n = 6; Smad1Col2a1, n = 4). (B) Primary calvarial cells were isolated from 3-day-old Cre-negative and Smad1Col2a1 mice and total RNAs were extracted from these cells. Expression of genes related to osteoblast differentiation and mineralization was measured by real-time PCR. Unpaired Student’s t-test, Col2a1, P = 0.005; Runx2, P = 0.001; Alp, P = 0.452; Osterix, P < 0.001; OC, P = 0.002; Mmp13, P = 0.032 (Cre-negative, n = 6; Smad1Col2a1, n = 4). (C) Primary calvarial cells were isolated from 3-day-old Smad1Col2a1 and their littermate control mice. Cells were treated with vehicle or BMP-2 (100 ng/ml) for 24 h, followed by ALP staining. Smad1-deficient cells had decreased basal ALP activity. BMP-2 stimulated ALP activity in control cells but had no effect on Smad1-deficient cells.
The Smad1-loxP, Col1a1-Cre and Col2a1-Cre mice were produced with mixed backgrounds22,23,26 and were then converted into C57 background in our lab after breeding with C57 mice for over 15 generations.
Isolation of calvarial mRNA from E16.5 embryos
E16.5 embryos were sacrificed and tail tissues were obtained for genotyping. Calvarial tissues of E16.5 Smad1Col2a1 or Cre-negative control embryos were harvested to obtain total mRNA. After washing with sterile phosphate buffered saline (PBS) twice, the calvariae were collected in 1.5 ml micro-centrifuge tubes. 1 ml Trizol reagent (Qiagen) and one 5 mm stainless steal bead (Qiagen) were added into each tube. This was followed by vibrating the tube at 4°C for 1 min using Tissue Lyser (Qiagen) to breakdown the tissue. Total mRNA was harvested following Trizol manufacture’s instructions. Expression of Col2a1, Cre and Smad1 mRNA was analyzed by PCR or real-time PCR.
Embryonic skeletal alizarin red/alcian blue staining
Embryos were sacrificed at E16.5 and E18.5 and tail tissues were obtained for genotyping. Embryo skin was removed before placing them in 95% ethanol for 1–2 days to dehydrate. Embryos were then transferred in acetone (J. T. Baker) for 1 day to remove fat. The alcian blue/alizarin red staining was performed using a solution containing 5 ml 0.3% alcian blue 8GS (Sigma) dissolved in 70% ethanol, 5 ml 0.1% Alizerin red S (Sigma) dissolved in 95% ethanol, 5 ml glacial acetic acid (J. T. Baker), and 85 ml 70% ethanol in 37°C incubator for 3 days. The embryos were then transferred in 2% KOH (Sigma) for 1–2 days until skeletons are clearly visible through the surrounding skin. The embryos were transferred in 20% glycerin (Sigma) containing 1% KOH, and finally store in 100% glycerin.
Assessment of three-dimensional (3D) trabecular microstructure
The calvariae of chondrocyte-specific Smad1 cKO embryos (E16.5) and proximal tibiae of osteoblast-specific Smad1Col1a1 mice (2-month-old) were scanned using a micro-computed tomography (μCT) scanner (μCT 40, Scanco Medical AG, Bassersdorf, Switzerland) and Cre-negative littermates were used as controls. A small X-ray tube with a μ focal spot is used as a source. The detector consists of a linear charged coupled device (CCD)-array. A scout view scan was obtained first for selection of the examination volume of the specimens, followed by automatic positioning, measurement, and offline reconstruction32. The field of view was 30 mm×30 mm, and matrix size was 2048 × 2048. Images with isotropic resolution of 15 μm3 were obtained.
Alkaline phosphatase staining
Twenty-four hours after isolated from 3-day-old Cre-negative and Smad1Col2a1 mice, primary calvarial osteoblasts were treated with vehicle or BMP-2 (100 ng/ml) for 48 h. Prior to staining, cells were washed with PBS and fixed with 10% Neutral buffered formalin (NBF) at 4°C for 15 min. Cells were washed twice with milliQ H2O after removal of NBF. Alkaline phosphatase (ALP) staining was performed using a solution containing 66.7 μl 25 mg/ml naphthol AS-MS-PO4 (Sigma) dissolved in N-N-Dimethyl formamide (Sigma), 8.33 ml 0.2 M Tris–HCl (pH= 8.3), 8.33 ml milliQ H2O, and 10 mg Red Violet LB Salt in 37°C incubator for 45 min.
Isolation of primary sternal chondrocytes
Three-day-old neonatal pups were sacrificed and tail tissues were obtained for genotyping. The anterior rib cage and sternum of Smad1Col2a1 and Cre-negative control mice were harvested to obtain sternal chondrocytes. This was followed by washing with sterile PBS twice, and digesting with 2mg/ml pronase (Roche) dissolved in PBS in a 37°C water bath with continuous shaking for 60 min. The rib cage and sternum were then incubated in 3 mg/ml collagenase D (Roche) solution dissolved in serum-free Dulbecco’s modified Eagle’s medium (DMEM)(GIBCO) at 37°C for 90 min. The soft tissue debris was carefully removed. This was followed by further digestion in fresh 3 mg/ml collagenase D solution in petri dishes at 37°C for 5 h with intermittent shaking. The digestion solution was filtered using a 40 μm cell strainer to collect the sternal chondrocytes. Cells were counted and plated in complete DMEM with 50 μg/ml ascorbic acid (Sigma).
Isolation of primary calvarial osteoblasts
Three-day-old neonatal pups were sacrificed and tail tissues were obtained for genotyping. Calvariae of 3-day-old Cre-negative and Smad1Col2a1 mice were harvested to obtain calvariae. Calvariae were washed with PBS twice, serum-free α-Minimum Essential Medium (α-MEM) (GIBCO)twice, and then digested with 1 mg/ml Collagenase A (Roche) dissolved in serum-free α MEM in a 37°C water bath with continuous shaking. Keep changing fresh 1 mg/ml Collagenase A every 15–20 minutes for four times. On the 4th time digestion, a 40 μm cell strainer was used to filter the digestion solution. Cells were centrifuged and resuspended in complete α MEM (GIBCO) with 50 μg/ml Ascorbic Acid (Sigma). The remaining calvariae were further digested twice with Collagenase A for 15–20 minutes each time to collect digestion solution. Cells were centrifuged and resuspended under the same condition.
Western blot analysis
The cells were lysed using Golden Lysis Buffer supplemented with protease inhibitor cocktail tablets (Sigma). The Bio-Rad Protein Assay (Bio-Rad) kit was used to detect protein concentration. The protein was detected with mouse monoclonal antibody against Smad1 (Santa Cruz) or phosphorylated Smad1/5/8. A monoclonal antibody against β-actin (Sigma) at a dilution of 1:8000 was used as loading control. The immune complexes were detected using West Pico chemiluminescent agents (Pierce).
Histological and histomorphometric analyses
Histomorphometric analyses were performed on proximal tibial metaphyses in 2-month-old osteoblast-specific Smad1Col1a1 mice and control littermates that had been injected intraperitoneally with calcein 7 and 2 days before animals were sacrificed28,31. Half of the bone samples from each group were decalcified in 14% ethylenediaminetetraacetic acid (EDTA), embedded in paraffin, and sections stained with H&E or for tartrate-resistant acid phosphatase (TRAP). Histological changes in trabecular bones were analyzed and changes in osteoblast and osteoclast numbers were quantified as previously described28,31. The other half of the bone samples were embedded in methylmethacrylate and sections used for measurements of bone formation rates (BFR) or stained with von Kossa to determine changes in the trabecular bone mineralization and changes in bone volume (BV). Trabecular BV was quantified in the bone marrow cavity 0.5–2.5 mm from the growth plates and expressed as a percentage of total tissue volume (TV). Values were expressed as the mean ± standard error calculated from three non-consecutive sections per mouse from each of 10 Smad1Col1a1 mice and littermate control mice. The calcein labeled bone surface or mineralizing surface (MS) and the distance between two fluorescent labels was measured and BFR/bone surface (BS), (μm3/μm2/day), were calculated.
5-bromo-2′-deoxy-uridine (BrdU) labeling
BrdU labeling was performed using Zymed BrdU labeling reagent and BrdU staining kit according to the manufacture’s protocols (Zymed). Briefly, BrdU was injected intraperitoneally into mice 24 h before sacrifice. Bone tissues were fixed in 10% NBF and embedded in paraffin. 3–4 μm thick sections were cut and placed on polylysine coated slides. Slides were dried in a 60°C oven overnight, deparaffinized in two changes of xylene for 5 min each, and then rehydrated in a graded series of alcohol. Slides were stained for BrdU according to manufacture’s protocols. Streptavidin-peroxidase was used as a signal generator for the BrdU system and deaminobenzidine in the presence of hydrogen peroxide was used as a chromogen. BrdU-incorporated nuclei were stained in a dark brown color34.
Mineralized bone nodule formation assay
Primary osteoblasts isolated from the calvariae of neonatal Smad1Col1a1 mice and littermate control mice were used for mineralized bone nodule formation assays as described previously28,31,33. Cells were plated in 24-well culture plates at a density of 2 ×104 cells/well and cultured with αMEM supplemented with 10% fetal calf serum (FCS). When cells reached confluence (day 0), the medium was changed to αMEM containing 5% FCS, 100 μg/ml ascorbic acid and 5 mM β-glycerol phosphate with or without addition of 100 ng/ml of BMP-2. The medium was changed every other day and fresh reagents were added. After 10-day incubation, von Kossa staining was performed to analyze the formation of mineralized bone nodules.
Luciferase assay
Primary osteoblasts isolated from Smad1Col1a1 and Cre-negative control mice were transiently transfected with BMP signaling reporter construct 12× SBE-OC-Luc followed by treatment with 4, 20 and 100 ng/ml of BMP-2 using Superfect (Qiagen, CA, USA) according to the manufacturer’s instruction. The DNA to transfection reagent ratio was 1:3 (w/w) in all experiments. β-gal plasmid was cotransfected as an internal control. Cells were lysed using passive lysis buffer (Promega, WI, USA) and extracts were prepared to detect the luciferase activity using the Dual Luciferase Assay System (Promega, WI, USA) following manufacturer’s directions.
Statistical analysis
The data were presented as mean ± 95% confidence limit (CL). For comparison of two groups of data, unpaired student’s t-test was performed. For comparison of multiple groups of data, one-way analysis of variance (ANOVA) was performed followed by Dunnett’s test. *P< 0.05 was considered as significant difference between two experimental groups.
Results
To determine the role of Smad1 in skeletal development, E16.5 and E18.5 chondrocyte-specific Smad1Col2a1 embryos were analyzed. We first extracted RNA from calvarial tissues of E16.5 Cre-negative and Smad1Col2a1 embryos to determine if Col2a1 gene is expressed and Cre-recombination occurs in calvarial tissues of Smad1Col2a1 embryos. We detected Col2a1 and Cre mRNA expression in calvariae of Cre-negative and Smad1Col2a1embryos [Fig. 1(B and C)]. We then analyzed Smad1 expression and found that Smad1 expression was significantly reduced (about 70% reduction) in the calvaria of Smad1Col2a1 mice [Fig. 1(D and E)]. These findings indicate that mouse calvarial tissues indeed express the Col2a1 gene at early developmental stage. Embryos were placed in 95% ethanol for 2 days for dehydration and then placed in acetone for 1 day to remove fat. Alizarin red/Alcian blue staining was performed. Smad1Col2a1 embryos and their littermate Cre-negative embryos are similar in size [Fig. 1(F)]. Delayed ossification of frontal, parietal and supraoccipital bones was observed in chondrocyte-specific Smad1Col2a1 embryos at E16.5 stage [Fig. 1(F and G)]. In addition, formation of the ossification center of proximal phalangeal bones was also delayed in E16.5 Smad1Col2a1 embryos [Fig. 1(H)]. We also analyzed calvarial bone formation and mineralization of E16.5 Smad1Col2a1 embryos by μ-CT and found that calvarial bone formation and mineralization were reduced in Smad1Col2a1 embryos [Fig. 1(I and J)]. However, no obvious difference was observed in E18.5 Smad1Col2a1 embryos compared to same aged Cre-negative embryos [Fig. 1(K)], indicating that the delayed calvarial mineralization and ectopic cartilage formation observed at E16.5 Smad1Col2a1 embryos are stage-specific phenomenon.
To further characterize these mice, we isolated primary sternal chondrocytes and primary calvarial cells from 3-day-old chondrocyte-specific Smad1Col2a1 mice and Cre-negative control mice. We analyzed the expression of genes related to calvarial bone formation and mineralization and ALP activity. We found that several of chondrocyte and osteoblast differentiation-related genes were down-regulated in Smad1Col2a1 mice [Fig. 2(A and B)] and ALP activity in calvarial cells was reduced [Fig. 2(C)]. It seems that transition from cartilage to bone phase at the specific developmental stage is delayed in Smad1Col2a1 mice and Smad1-mediated BMP signaling plays an important role in this process.
To determine the role of Smad1 in postnatal bone formation, we have generated osteoblast-specific Smad1Col1a1 mice. The Smad1Col1a1 mice are viable, fertile, and survive into adulthood. To determine the expression of Smad1 mRNA and protein, we isolated bone marrow cells from 2-month-old Smad1Col1a1 mice and control littermates. Expression of Smad1 mRNA was reduced about 80% in Smad1Col1a1 cells [Fig. 3(C)]. Smad1 protein expression was detected by Western blotting using an anti-Smad1 mouse monoclonal antibody (Santa Cruz, CA, USA) in osteoblasts derived from control mice. In Smad1-Col1a1 cells, expression of Smad1 protein was significantly reduced [Fig. 3(D)]. These results demonstrate that expression of the Smad1 gene is disrupted in bone marrow cells in osteoblast-specific Smad1Col1a1mice. To determine changes intrabecular bone formation between Smad1Col1a1 mice and littermate controls, 2-month-old mice were analyzed using histology and μCT methods on proximal tibial metaphyses32. BV was measured on von Kossa-stained tibia sections. Compared to the Cre-negative control mice, the trabecular BV of 2-month-old osteoblast-specific Smad1Col1a1 mice was reduced 30% [Fig. 3(E and F)]. To investigate the dynamic changes in bone formation in Smad1Col1a1 mice, we measured BFR by performing calcein/calcein labeling experiments. Fluorescent labels were administered by intraperitoneal injection of calcein 7 and 2 days before mice were sacrificed28,31. Un decalcified bone tissues were processed by methylmethacrylate embedding. A significant reduction (36%) in BFR was found in osteoblast-specific Smad1Col1a1 mice [Fig. 3(G and H)]. Since trabecular BV and BFR represent the percentage of bone in a well-defined area of bone marrow cavities these results suggest that Smad1Col1a1 mice developed osteopenia. We also analyzed bone mass in the heterozygous Smad1Col1a1 mice and found that there is no significant reduction in bone mass in those heterozygous Smad1Col1a1 mice (data not shown).
Fig. 3.

Generation and analysis of Smad1Col1a1 mice. (A and B) Smad1del/wt mice were bred with Col1a1-Cre mice and Col1a1-Cre;Smad1del/wt mice were identified by PCR using Cre-5′/Cre-3′ primers (lower panel, Lane 1, 4, 5, 600-bp PCR product) and Rec-Smad1/LoxP2 primers (300-bp PCR product). Heterozygous Smad1 null allele mediated by CMV-Cre was detected by Rec-Smad1/LoxP2 primers. To generate osteoblast-specific Smad1Col1a1 mice, the Col1a1-Cre;Smad1del/wt mice were bred with Smad1fx/fx mice. The desired progeny, Col1a1-Cre;Smad1del/fx mice (Smad1Col1a1 mice), were identified by PCR using SIF400/LoxP2 primers (tail tissue: 410-bp PCR product), Cre-5′/Cre-3′ primers (600-bp PCR product) and Rec-Smad1/LoxP2 primers (300-bp PCR product). (C and D) Expression of Smad1 mRNA and protein was measured by real-time PCR and Western blotting in bone marrow cells. Expression of Smad1 mRNA was reduced about 80%. Unpaired Student’s t-test, P < 0.001 (n = 3). Expression of Smad1 protein was detected by Western blotting using an anti-Smad1 monoclonal antibody in bone marrow cells derived from control mice but significantly reduced in those derived from Smad1Col1a1 mice. (E and F) BV was analyzed in a defined area 0.5–2.5 mm from the growth plate in proximal tibiae of 2-month-old Smad1Col1a1 mice and littermate control mice. The BV was normalized to TV. A 30% decrease in BV was observed in Smad1Col1a1 mice compared with littermate controls. Unpaired Student’s t-test, P = 0.006 (n = 6). (G and H) The animals were labeled with calcein/calcein. In Smad1Col1a1 mice, the distance between the two labels was significantly reduced as indicated by white arrows. The BFR/BS were measured in the area of the marrow cavity 0.5–2.5 mm from the growth plates of proximal tibiae and expressed as mm3/mm2/day (F). Unpaired Student’s t-test, P = 0.004 (n = 6). (I) Osteoblast numbers in trabecular bone in the same area were quantitated and a 19% decrease in osteoblast numbers was found in Smad1Col1a1 mice. Unpaired Student’s t-test, P < 0.001 (n = 6).
μCT analysis confirmed trabecular BV per unit of metaphyses to be significantly decreased in osteoblast-specific Smad1Col1a1 mice [Fig. 4(A)]. Similarly, trabecular number (Th.N), trabecular thickness (Tb.Th), and connectivity density (CD) were significantly decreased in Smad1 cKO mice compared with Cre-negative controls [Fig. 4(B, C and E)]. Trabecular separation (Tb.Sp) and structure model index (SMI) were significantly higher in Smad1Col1a1 mice [Fig. 4(D and F)]. These results suggest that trabecular BV and mechanical property of the bone are reduced in Smad1Col1a1 mice.
Fig. 4.
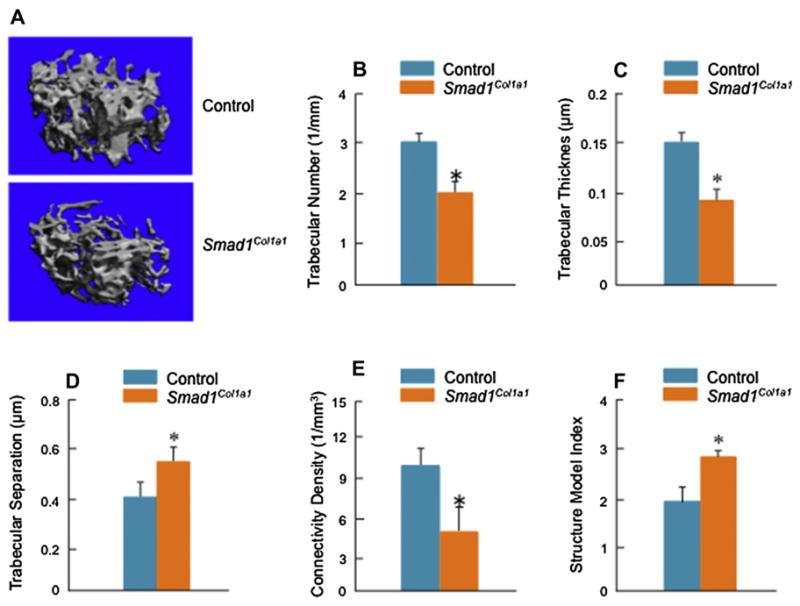
μ-CT analysis of Smad1Col1a1 mice. μ-CT images showed a significant reduction in bone mass in Smad1Col1a1 mice. (A) Trabecular structural parameters in the secondary spongiosa of the proximal tibia were measured using μCT analysis. Samples were examined with an isotropic resolution of 5-μm voxel size. Tb.N (B), Tb.Th (C) and trabecular connectivity density (Conn D) (E) were significantly decreased in Smad1Col1a1 mice compared with littermate controls. Trabecular separation (Tb.Sp) (D) and SMI (F) were significantly increased in Smad1Col1a1 mice. Unpaired Student’s t-test; Tb.N., P = 0.047; Tb.Th., P = 0.001; Tb.Sp., P = 0.007; Conn D, P = 0.012; SMI, P < 0.001 (n = 6).
To determine if there were changes in osteoblast number, we quantified osteoblast number in the trabecular bone area of mouse tibiae and found a 19% decrease in Smad1 cKO mice compared with Cre-negative control mice [Fig. 3(I)]. No obvious changes in osteoclast numbers were observed in Smad1Col1a1 mice. To determine whether osteoblast proliferation was affected, BrdU labeling experiments were performed28. BrdU was injected intraperitoneally into mice 24 h before sacrifice. Bone tissues were then processed and stained. A significant decrease (22%) in BrdU-positive osteoblasts was found on calvarial bone surfaces in Smad1Col1a1 mice [Fig. 5(A and B)]. These results demonstrate that defects in bone formation in osteoblast-specific Smad1Col1a1 mice may be partially due to the impairment of osteoblast proliferation.
Fig. 5.

Inhibition of osteoblast proliferation and differentiation in Smad1Col1a1 mice. (A and B) Paraffin-embedded sections of calvariae were stained with BrdU. The BrdU-positive osteoblasts were indicated by red arrows and a 22% reduction of BrdU-positive osteoblasts was found in Smad1Col1a1 mice. Unpaired Student’s t-test, P = 0.002 (n = 4). (C) Primary osteoblasts isolated from Smad1Col1a1 mice and littermate control mice were cultured for 10 days in the absence or presence of BMP-2 (100 ng/ml). von Kossa staining was performed after the cell culture was ended and the formation of mineralized bone nodules was analyzed. In osteoblasts derived from Smad1Col1a1 mice, the formation of mineralized bone nodules was inhibited. (D) Primary osteoblasts isolated from Smad1Col1a1 mice and littermate control mice were transfected with BMP signaling reporter construct 12× SBE-OC-Luc and treated with 4, 20 and 100 ng/ml of BMP-2. The luciferase activity was measured and normalized to β-gal activity. In osteoblasts derived from Smad1 cKO mice, BMP signaling was significantly inhibited compared with cells isolated from littermate control mice. One-way ANOVA followed by Dunnett’s test, BMP-2 (4 ng/ml), P = 0.003; BMP-2 (20 ng/ml), P = 0.014; BMP-2 (100 ng/ml), P < 0.001 (n = 3) between control and Smad1Col1a1 cells.
To determine changes in osteoblast differentiation, primary osteoblasts were isolated from the calvariae of neonatal Smad1Col1a1 mice and Cre-negative control mice and were used to examine mineralized bone nodule formation8,31,33. The mineralized bone nodules were formed at day 10 and treatment with BMP-2 (100 ng/ml) stimulated the nodule formation in osteoblast cells derived from control mice [Fig. 5(C, left panels)]. However, either at basal levels or with BMP-2 treatment, nodule formation was inhibited in osteoblasts derived from Smad1Col1a1 mice [Fig. 5(C, right panels)]. To determine if BMP signaling is blocked in osteoblasts derived from Smad1Col1a1 mice, we transfected the BMP signaling reporter construct 12×SBE-OC-Luc8 into primary osteoblasts of Smad1Col1a1 mice and Cre-negative control mice followed by BMP-2 treatment. In osteoblasts derived from Smad1Col1a1 mice, BMP signaling was significantly inhibited compared with that in osteoblasts from Cre-negative control mice [Fig. 5(D)]. We also analyzed osteoblast maker gene expression34 and found that expression of osteoblast marker genes such as Runx2, osterix, bone sialoprotein (BSP) and osteocalcin was significantly reduced in osteoblasts derived from Smad1Col1a1 mice compared with those from Cre-negative control mice [Fig. 6(A–D)]. These results demonstrate that BMP signaling and osteoblast differentiation are impaired in osteoblast-specific Smad1Col1a1 mice. To determine if Smad5/8 phosphorylation is impaired in Smad1Col1a1 mice, we performed Smad5/8 phosphorylation assay. We found that BMP-2-induced Smad5/8 phosphorylation remains intact in Smad1-Col1a1 cells although the levels were reduced compared to control cells [Fig. 6(E)]. Our previous study demonstrates that β-catenin could act upstream of BMP signaling35. To determine if deletion of the Smad1 gene could inhibit β-catenin signaling in osteoblasts, we treated osteoblasts derived Cre-negative and Smad1Col1a1 mice with BIO, a GSK-3β inhibitor, which mimics the activation of β-catenin signaling. We found that BIO significantly up-regulated ALP activity in control cells. In contrast, its stimulatory effect was significantly reduced in Smad1Col1a1 cells [Fig. 6(F)]. These results suggest that BMP-Smad1 could serve as downstream signaling molecules of β-catenin signaling in osteoblasts. Since BMP signaling was not completely blocked in osteoblasts derived from Smad1Col1a1 mice, the results suggest that BMP signaling through Smad5 or Smad8 remains intact in these osteoblasts and Smad5/8 may partially compensate the loss of Smad1 function. Generation of Smad1/5 double KO mice or Smad1/5/8 triple KO mice would be necessary to examine this possibility and to clarify the individual contributions of Smad1, 5 and 8 to BMP signaling in postnatal bone formation and adult bone remodeling.
Fig. 6.

Reduction in expression of osteoblast marker genes in Smad1Col1a1 mice. (A–D) mRNA expression of osteoblast marker genes was examined by real-time PCR. The expression of Runx2, Osterix (Osx), BSP and osteocalcin (D) genes was significantly decreased in osteoblasts derived from Smad1Col1a1 mice. Unpaired Student’s t-test; Runx2, P = 0.002; Osterix, P = 0.022; BSP, P = 0.050; OC, P = 0.002 (n = 6). (E) Primary osteoblasts were starved for 12 h and then treated with BMP-2 (100 ng/ml) for 6 h. Smad1/5/8 phosphorylation was measured by Western blotting using an anti-p-Smad1/5/8 antibody. BMP-2-induced Smad1/5/8 phosphorylation was reduced in Smad1-deficient cells. (F) Primary osteoblasts were treated with BIO (1 μM) for 48 h and ALP activity in cell lysates was measured. Significant inhibition of ALP activity was found in Smad1-deficient cells. One-way ANOVA followed by Dunnett’s test, basal ALP, P = 0.127; BIO, P < 0.001 (n = 3), between control and Smad1-deficient cells.
Discussion
To determine the specific contribution of Smad1 in bone development and postnatal bone formation, we have generated chondrocyte- and osteoblast-specific Smad1 cKO mice using Col2a1-Cre (the Cre transgene is driven by the 3 kb Col2a1 transgene) and Col1a1-Cre (the Cre transgene is driven by the 2.3 kb Col1a1 promoter) transgenic mice. Our findings demonstrate a delay in calvarial bone mineralization and reduction of postnatal bone formation in these Smad1 cKO mice. Abzhanov et al. showed that type II collagen is expressed in mouse calvaria during the embryonic stage and large population of calvarial cells are derived from Col2a1-positive chondrocyte-like cells36. In the paper published by Ovchinnikov et al.23, the authors analyzed β-gal activity at E15 embryos of Col2a1-Cre transgenic mice and they found Col2a1 promoter activity is active in the axial and appendicular skeleton, calvarial bones (temporal and basioccipital bones), and the other elements of the skull. Our results show that chondrocyte-specific deletion of the Smad1 gene causes a transient ectopic cartilage phase and delayed ossification of the calvarial bones during the prenatal stage. Our previous studies demonstrate that hypertrophic marker genes such as ColX and Alp are upregulated by Wnt3a in primary chondrocytes. Activation of Wnt/β-catenin signaling up-regulated Bmp-2 and Bmp4 expression and addition of noggin, a BMP antagonist, inhibited Wnt3a-induced chondrocyte differentiation37. In the present study, we further demonstrated that BIO-induced ALP activity was inhibited in Smad1-deficient cells. These findings suggest that BMP/Smad1 signaling is downstream of Wnt/β-catenin signaling in osteoblasts. Interestingly, ectopic cartilage formation in calvariae was also observed when the β-catenin gene was inactivated8, and these results suggest that the disruption of the β-catenin-BMP/Smad1 signaling pathway may be responsible for the inhibition of osteoblast differentiation in calvariae.
Cumulative evidence shows that intact BMP signaling is necessary for postnatal bone formation. Our previous studies demonstrate that disruption of normal BMP signaling by expressing dominant-negative type I BMP receptor causes osteopenic phenotype28. Tob is a member of anti-proliferative protein family and inhibits BMP-induced and Smad-dependent transcription in osteoblasts by direct inhibition of Smad1 or 538. Tob KO mice show increased BMP signaling and enhanced responsiveness to BMP-2-induced osteoblast proliferation and differentiation. Bone mass and BMP-2-induced local bone formation are increased in Tob KO mice38,39. These observations demonstrate that Smad1 and other BMP-activated Smad proteins play an important role in postnatal bone formation. The importance of BMP signaling in postnatal bone formation is reinforced by studies using transgenic mice in which noggin and sclerostin transgenes were overexpressed in mature osteoblasts. In these mice, osteopenic and osteoporotic phenotypes were observed40–42. These findings provide evidence that activation of endogenous BMP signaling can enhance bone formation and that regulation of the amount of BMP signaling in postnatal life is required for normal bone formation. Our current findings are consistent with previous observations in Bmpr1a cKO mice. Deletion of the Bmpr1a gene in mature osteoblasts targeted by the Og2-Cre transgenic mice showed reduced BV and BFR in 3-month-old cKO mice43. However, when the Bmpr1a gene is deleted in early progenitor cells using the 3.2Col1-CreER transgenic mice, bone mass was increased in the cKO mice44. The different phenotypes of these mice may be due to targeting different populations of cells at different stages. In our study, we used 2.3 Col1-Cre transgenic mice which target mature osteoblasts population similar to Og2-Cre transgenic mice and found similar osteopenic phenotype. Although both Smad1 and Smad5 are expressed in primary osteoblasts and multiple osteoblast-like cell lines, such as 2T3 and MC3T3 cells (data not shown), our present findings suggest that Smad1 is one of the critical molecules mediating BMP signaling and regulating postnatal bone formation. To fully understand the BMP signaling in postnatal bone formation, osteoblast-specific deletion of the Smad1/5 genes or Smad1/5/8 genes will be required.
Acknowledgments
Role of the funding source
This work was supported by Grants R01 AR055915 and R01 AR054465 to DC from National Institute of Health and by Contract C024320 to DC from New York State Department of Health and Empire State Stem Cell Board.
We thank Ryan Smith and Michael Thullen for their expert technical support.
Footnotes
Author contributions
MeinaWang: (1) acquisition of data, (2) analysis of data, and (3) drafting the manuscript.
Hongting Jin: (1) acquisition of data.
Dezhi Tang: (1) acquisition of data.
Shixia Huang: (1) acquisition of data.
Michael J. Zuscik: (1) analysis and interpretation of data.
Di Chen: (1) design of study, (2) analysis and interpretation of data, (3) revision of the manuscript, (4) final approval of the submitted manuscript.
Conflict of interest
The authors have no conflict of interest to disclose.
References
- 1.Shum L, Coleman CM, Hatakeyama Y, Rocky TS. Morphogenesis and dysmorphogenesis of the appendicular skeleton. Birth Defects Res C Embryo Today. 2003;69:102–22. doi: 10.1002/bdrc.10012. [DOI] [PubMed] [Google Scholar]
- 2.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89(5):747–54. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 3.Zelzer E, Olsen BR. The genetic basis for skeletal diseases. Nature. 2003;423:343–8. doi: 10.1038/nature01659. [DOI] [PubMed] [Google Scholar]
- 4.Yang X, Karsenty G. Transcription factors in bone: developmental and pathological aspects. Trends Mol Med. 2002;8:340–5. doi: 10.1016/s1471-4914(02)02340-7. [DOI] [PubMed] [Google Scholar]
- 5.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–6. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 6.Mori-Akiyama Y, Akiyama H, Rowitch DH, de Crombrugghe B. Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc Natl Acad Sci USA. 2003;100:9360–5. doi: 10.1073/pnas.1631288100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi H, Gao Y-H, Ueta C, Yamaguchi A, Komori T. Multilineage differentiation of Cbfa1-deficient calvarial cells in vitro. Biochem Biophys Res Commun. 2000;273:630–6. doi: 10.1006/bbrc.2000.2981. [DOI] [PubMed] [Google Scholar]
- 8.Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8:739–50. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 9.Ochiai T, Shibukawa Y, Nagayama M, Mundy C, Yasuda T, Okabe T, et al. Indian hedgehog roles in post-natal TMJ development and organization. J Dent Res. 2010;89(4):349–54. doi: 10.1177/0022034510363078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu M, Tang D, Wu Q, Hao S, Chen M, Xie C, et al. Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J Bone Miner Res. 2009;24(1):12–21. doi: 10.1359/JBMR.080901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoon BS, Pogue R, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, et al. BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development. 2006;133:4667–78. doi: 10.1242/dev.02680. [DOI] [PubMed] [Google Scholar]
- 12.Haas AR, Tuan RS. Chondrogenic differentiation of murine C3H10T1/2 multipotential mesenchymal cells: II. Stimulation by bone morphogenetic protein-2 requires modulation of N-cadherin expression and function. Differentiation. 1999;64:77–89. doi: 10.1046/j.1432-0436.1999.6420077.x. [DOI] [PubMed] [Google Scholar]
- 13.Cao X, Chen D. The BMP signaling and in vivo bone formation. Gene. 2005;357:1–8. doi: 10.1016/j.gene.2005.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoodless PA, Haerry T, Abdollah S, Stapleton M, O’Connor MB, Attisano L, et al. MADR1, a MAD-related protein that functions in BMP2 signaling pathways. Cell. 1996;85(4):489–500. doi: 10.1016/s0092-8674(00)81250-7. [DOI] [PubMed] [Google Scholar]
- 15.Tremblay KD, Dunn NR, Robertson E. Mouse embryos lacking Smad1 signals display defects in extra-embryonic tissues and germ cell formation. Development. 2001;128:3609–21. doi: 10.1242/dev.128.18.3609. [DOI] [PubMed] [Google Scholar]
- 16.Lechleider RJ, Ryan JL, Garrett L, Eng C, Deng C, Wynshaw-Boris A, et al. Targeted mutagenesis of Smad1 reveals an essential role in chorioallantoic fusion. Dev Biol. 2001;240:157–67. doi: 10.1006/dbio.2001.0469. [DOI] [PubMed] [Google Scholar]
- 17.Kaufman MH. The Atlas of Mouse Development. London, UK: Academic Press; 1995. pp. 1–8. [Google Scholar]
- 18.Retting KN, Song B, Yoon BS, Lyons KM. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development. 2009;136(7):1093–104. doi: 10.1242/dev.029926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoess RH, Abremski K. Mechanism of strand cleavage and exchange in the Cre-lox site-specific recombination system. J Mol Biol. 1985;181:351–62. doi: 10.1016/0022-2836(85)90224-4. [DOI] [PubMed] [Google Scholar]
- 20.Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000;26:99–109. [PubMed] [Google Scholar]
- 21.Kwan KM. Conditional alleles in mice: practical considerations for tissue-specific knockouts. Genesis. 2002;32:49–62. doi: 10.1002/gene.10068. [DOI] [PubMed] [Google Scholar]
- 22.Huang S, Tang B, Usoskin D, Lechleider RJ, Jamin SP, Li C, et al. Conditional knockout of theSmad1 gene. Genesis. 2002;32:76–9. doi: 10.1002/gene.10059. [DOI] [PubMed] [Google Scholar]
- 23.Ovchinnikov DA, Deng JM, Ogunrinu G, Behringer RR. Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis. 2000;26:145–6. [PubMed] [Google Scholar]
- 24.Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, et al. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–64. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 25.Schwenk F, Baron U, Rajewsky K. A Cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–1. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dacquin R, Starbuck M, Schinke T, Karsenty G. Mouse alpha 1(I)-collagen promoter is the best known promoter to drive efficient Cre recombinase expression in osteoblast. Dev Dyn. 2002;224:245–51. doi: 10.1002/dvdy.10100. [DOI] [PubMed] [Google Scholar]
- 27.Rossert JA, Eberspaecher H, de Crombrugghe B. Separate cis-acting DNA elements of the mouse pro-alpha 1(I) collagen promoter direct expression of reporter genes to different type I collagen-producing cells in transgenic mice. J Cell Biol. 1995;129:1421–32. doi: 10.1083/jcb.129.5.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao M, Harris SE, Horn D, Geng Z, Nishimura R, Mundy GR, et al. Bone morphogenetic protein receptor signaling is necessary for normal murine postnatal bone formation. J Cell Biol. 2002;157:1049–60. doi: 10.1083/jcb.200109012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalajzic I, Kalajzic Z, Kaliterna M, Gronowicz G, Clark SH, Lichtler AC, et al. Use of type I collagen green fluorescent protein transgenes to identify subpopulations of cells at different stages of the osteoblast lineage. J Bone Miner Res. 2002;17:15–25. doi: 10.1359/jbmr.2002.17.1.15. [DOI] [PubMed] [Google Scholar]
- 30.Kalajzic Z, Liu P, Kalajzic I, Du Z, Braut A, Mina M, et al. Directing the expression of a green fluorescent protein transgene in differentiated osteoblasts: comparison between rat type I collagen and rat osteocalcin promoters. Bone. 2002;31:654–60. doi: 10.1016/s8756-3282(02)00912-2. [DOI] [PubMed] [Google Scholar]
- 31.Zhao M, Qiao M, Harris SE, Oyajobi BO, Mundy GR, Chen D. Smurf1 inhibits osteoblast differentiation and bone formation in vitro and in vivo. J Biol Chem. 2004;279:12854–9. doi: 10.1074/jbc.M313294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang Y, Zhao J, Mitlak BH, Wang O, Genant HK, Eriksen EF. Recombinant human parathyroid hormone (1–34) [teriparatide] improves both cortical and cancellous bone structure. J Bone Miner Res. 2003;18:1932–41. doi: 10.1359/jbmr.2003.18.11.1932. [DOI] [PubMed] [Google Scholar]
- 33.Feng JQ, Xing L, Zhang J, Zhao M, Horn D, Chan J, et al. NF-kappaB specifically activates BMP-2 gene expression in growth plate chondrocytes in vivo and in a chondrocyte cell line in vitro. J Biol Chem. 2003;278:29130–5. doi: 10.1074/jbc.M212296200. [DOI] [PubMed] [Google Scholar]
- 34.Chen D, Ji X, Harris MA, Feng JQ, Karsenty G, Celeste AJ, et al. Differential roles for bone morphogenetic protein (BMP) receptor type IB and IA in differentiation and specification of mesenchymal precursor cells to osteoblast and adipocyte lineages. J Cell Biol. 1998;142:295–305. doi: 10.1083/jcb.142.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan Y, Tang D, Chen M, Huang J, Xie R, Jonason JH, et al. Axin2 controls bone remodeling through the beta-catenin-BMP signaling pathway in adult mice. J Cell Sci. 2009;122(Pt 19):3566–78. doi: 10.1242/jcs.051904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abzhanov A, Rodda SJ, McMahon AP, Tabin CJ. Regulation of skeletogenic differentiation in cranial dermal bone. Development. 2007;134:3133–44. doi: 10.1242/dev.002709. [DOI] [PubMed] [Google Scholar]
- 37.Chen M, Zhu M, Awad H, Li TF, Sheu TJ, Boyce BF, et al. Inhibition of beta-catenin signaling causes defects in postnatal cartilage development. J Cell Sci. 2008;121(Pt 9):1455–65. doi: 10.1242/jcs.020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshida Y, Tanaka S, Umemori H, Minowa O, Usui M, Ikematsu N, et al. Negative regulation of BMP/Smad signaling by Tob in osteoblasts. Cell. 2000;103:1085–97. doi: 10.1016/s0092-8674(00)00211-7. [DOI] [PubMed] [Google Scholar]
- 39.Usui M, Yoshida Y, Yamashita T, Tsuji K, Isao I, Yamamoto T, et al. Enhancing effect of Tob deficiency on bone formation is specific to bone morphogenetic protein-induced osteogenesis. J Bone Miner Res. 2002;17:1026–33. doi: 10.1359/jbmr.2002.17.6.1026. [DOI] [PubMed] [Google Scholar]
- 40.Devlin RD, Du Z, Pereira RC, Kimble RB, Economides N, Jorgetti V, et al. Skeletal overexpression of noggin results in osteopenia and reduced bone formation. Endocrinology. 2003;144:1972–8. doi: 10.1210/en.2002-220918. [DOI] [PubMed] [Google Scholar]
- 41.Wu XB, Li Y, Schneider A, Yu W, Rajendren G, Iqbal J, et al. Impaired osteoblastic differentiation, reduced bone formation, and severe osteoporosis in noggin-overexpressing mice. J Clin Invest. 2003;112:924–34. doi: 10.1172/JCI15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22:6267–76. doi: 10.1093/emboj/cdg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mishina Y, Starbuck MW, Gentile MA, Fukuda T, Kasparcova V, Seedor JG, et al. Bone morphogenetic protein type IA receptor signaling regulates postnatal osteoblast function and bone remodeling. J Biol Chem. 2004;279:27560–6. doi: 10.1074/jbc.M404222200. [DOI] [PubMed] [Google Scholar]
- 44.Kamiya N, Ye L, Kobayashi T, Lucas DJ, Mochida Y, Yamauchi M, et al. Disruption of BMP signaling in osteoblasts through type IA receptor (BMPRIA) increases bone mass. J Bone Miner Res. 2008;23:2007–17. doi: 10.1359/JBMR.080809. [DOI] [PMC free article] [PubMed] [Google Scholar]