

Abstract
Oxaliplatin is a third-generation platinum-based anticancer drug that is currently used in the treatment of metastatic colorectal cancer. Oxaliplatin, like other platinum-based anticancer drugs such as cisplatin and carboplatin, is known to induce apoptosis in tumor cells by binding to nuclear DNA, forming monoadducts, and intra- and interstrand diadducts. Previously, we reported an accelerator mass spectrometry (AMS) assay to measure the kinetics of oxaliplatin-induced DNA damage and repair (Hah, S. S.; Sumbad, R. A.; de Vere White, R. W.; Turteltaub, K. W.; Henderson, P. T. Chem. Res. Toxicol. 2007, 20, 1745). Here, we describe another application of AMS to the measurement of oxaliplatin–DNA adduct distribution in cultured platinum-sensitive testicular (833 K) and platinum-resistant breast (MDA-MB-231) cancer cells, which resulted in elucidation of cell-dependent differentiation of oxaliplatin–DNA adduct formation, implying that differential adduction and/or accumulation of the drug in cellular DNA may be responsible for the sensitivity of cancer cells to platinum treatment. Ultimately, we hope to use this method to measure the intrinsic platinated DNA adduct repair capacity in cancer patients for use as a biomarker for diagnostics or a predictor of patient outcome.
Keywords: Oxaliplatin, DNA adduct, Accelerator mass spectrometry
(trans-R,R)1,2-Diaminocyclohexaneoxalatoplatinum(II) (oxaliplatin) derives from the well-established platinum drug cis-diamminedichloroplatinum(II) (cisplatin) (structures shown in Fig. 1). It is the first platinum-based anticancer drug approved for the treatment of colorectal cancer, which is a major cause of cancer-related death in developed countries and is essentially equally distributed between men and women.1 Oxaliplatin has been licensed in the European Union since 1999, and in the United States since 2002.2 Oxaliplatin has a broad spectrum of anticancer activity, and importantly, preclinical studies showed that oxaliplatin in combination with 5-FU has greater in vitro and in vivo antiproliferative activity than either compound alone in several tumor models, including colon cancer.3
Figure 1.
Chemical structures of cisplatin, carboplatin, and oxaliplatin.
Oxaliplatin has diaminocyclohexane (DACH) as a carrier group and oxalato as a leaving group, and is known to exert the cytotoxicity effect by interaction with DNA to form monoadducts and intra- and interstrand diadducts (Fig. 2), like other platinum-based anticancer drugs such as cisplatin and cis-diammine(1,1-cyclobutyldicarboxylato)platinum(II) (carboplatin).3 Importantly, oxaliplatin shows a better safety profile than cisplatin in clinical studies, except for reversible damage to peripheral sensory nerves,2 and a lack of cross-resistance with cisplatin or carboplatin, presumably because DACH–Pt–DNA adducts formed by oxaliplatin are bulkier and more hydrophobic than cisplatin and carboplatin adducts, leading to different effects in cells.4 However, the clinical efficacy and toxicity resulting from oxaliplatin treatment vary from patient to patient, largely because of intrinsic and acquired resistance.2 Although resistance remains poorly understood, it is clearly influenced by variation in cellular rates of drug influx, export, and the formation and repair of platinum DNA adducts.
Figure 2.
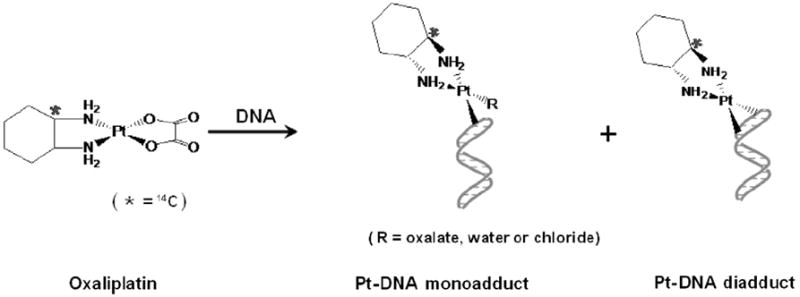
Simplified scheme for the reaction of oxaliplatin with DNA (the location of the 14C atom is asterisked). Because radiocarbon is located in the DACH carrier group, radiocarbon in oxaliplatin–DNA monoadducts and diadducts can be detected by AMS.
In spite of extensive analytical studies for Pt–DNA adduct formation/distribution under pharmacologically- or subpharmacologically-relevant conditions,3 measurement of the adducts in cells and in vivo with higher sensitivity and precision is still desirable, because it remains unclear which of the Pt–DNA adducts are responsible for the cytotoxic activity of platinum-based anticancer drugs. To address this issue, a sensitive and quantitative assay would be ideal to better understand mechanisms of resistance and to be predictive of patient outcome.
We recently reported the use of an extremely sensitive detection method of accelerator mass spectrometry (AMS),5 a highly sensitive technique with origins in geochemistry for radiocarbon dating, in order to study the kinetics of oxaliplatin–DNA adduct formation in DNA, both in vitro and in cultured cells, as well as to study rates of cellular drug influx, efflux, DNA damage and DNA repair in cultured platinum-sensitive testicular (833 K) and platinum resistant breast and bladder (MDA-MB-231 and T24, respectively) cancer cells incubated with a subpharmacological dose of oxaliplatin (0.2 μM) containing 2.16 pmol of [14C]oxaliplatin.6 Like other experiments using radioisotopes, AMS is specific only to the labeled compound in any chemical or biological medium, allowing the direct determination of the amount of isotope in a sample and thus the quantitative analysis of the fate of the radiolabeled probes under the given conditions.5 The lowest concentration of radiocarbon measured in our previous work was approximately 1 ± 0.1 amol/μg of DNA, when assaying 1 μg of DNA, of which the sensitivity for measuring oxaliplatin–DNA adducts is still the highest reported to date.6
In the present study, we report an application of our previous approach to those of oxaliplatin–DNA adduct distribution in cultured 833 K and MDA-MB-231 cancer cell lines of differing platinum sensitivity, so as to investigate oxaliplatin–DNA adduct formation and/or accumulation in these cells compared to that in T24 cells. We applied the same experimental conditions as reported in our previous oxaliplatin study,6,7 and determined oxaliplatin–DNA adduct distribution in 833 K and MDA-MB-231 cells which were incubated with a subpharmacological concentration of oxaliplatin (0.2 μM) for 24 h, respectively. This is a clinically relevant concentration for this assay, since plasma levels of oxaliplatin are reported to be a maximum of ~3.6 μM in patients undergoing therapy.8 This drug concentration allowed measurement of the accumulation of radiocarbon in cells and DNA without interference from acute toxicity.6
We were able to take advantage of radiocarbon located in the DACH carrier group. By following the DNA extraction, digestion and HPLC separation conditions in the literature,6,9 oxaliplatin–DNA adducts were characterized at the nucleoside level for mono- and diadduct formation using AMS because those adducts still have radiocarbon (Fig. 2). Genomic DNA extracted from 833 K and MDA-MB-231 cells was enzymatically digested by deoxyribonuclease and P1 nuclease to deoxyribonucleotides, and the resulting mixture was filtered and separated by HPLC. Fractions were taken at one minute intervals and then dried and converted to graphite for AMS measurement.
Figure 3 shows the retention times of the resulting radiocarbon-containing fractions, as compared to UV detection of authentic standards of the Pt(DACH)-adducted nucleotides and nucleosides which were prepared according to the literature.6,9 The resulting distribution of peaks could be assigned to several types of Pt(DACH) mono- and diadducts. The obtained data clearly show the presence of intrastrand and interstrand DNA adducts in addition to at least two products that were not observed in the previously shown experiments with naked DNA. The total radiocarbon contents in extracted DNA in MDA-MB-231 and 833 K cells were determined to be 3.1 ± 0.6 and 6.3 ± 0.4 amol 14C/μg of DNA, respectively, as previously reported,6 suggesting that approximately 1.2% of the cellular radiocarbon was found in nuclear DNA after one-day incubation, with the highest accumulation in the platinum-sensitive 833 K cells, which may be explained by DNA repair deficiencies in these cells as compared to the other cell types. The total radiocarbon recovered by HPLC relative to the undigested control was approximately 98% (Table 1). Because we had essentially quantitative recovery of the total injected radiocarbon from each HPLC injection, the data were indicative of the total DNA adduct distribution in the cellular DNA for the particular experimental conditions used in this study. Our observation also confirms previous Letters that there is a high correlation between decreased adduct accumulation and resistance to platinum.10 Interestingly, the mass distribution of the oxaliplatin–DNA adducts in MDA-MB-231 cells is also different from that of T24 cells. These results support that different cancer cell types may have contrasting abilities to repair DNA damage and tolerate platinum chemotherapy.
Figure 3.
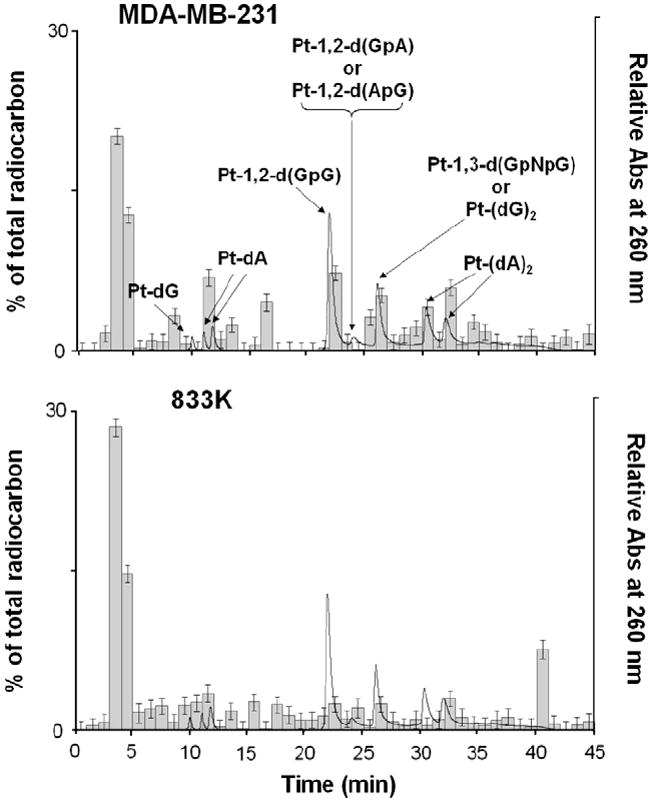
HPLC–AMS data of digested DNA resulting from the incubation of [14C]oxaliplatin with 833 K and MDA-MB-231 cells for 24 h. The radiocarbon due to oxaliplatin–DNA adducts was measured in triplicate by AMS as a fraction of the total radiocarbon (histogram). The retention times of synthetic authentic standards (solid line) were used to assign peak identity.
Table 1.
Percentage of the major oxaliplatin–DNA adducts obtained from HPLC–AMS data (Fig. 3), which summarizes the percentages of the radiocarbon-labeled oxaliplatin–DNA adducts from the HPLC fractions, to determine the cell-dependence manner of the individual adduct concentration (compositional pattern)
| MDA-MB-231 | 833K | T24 | |
|---|---|---|---|
| Monoadducts | 6.22 ± 1.43 | 8.10 ± 1.40 | 10.6 |
| Pt-1,2-d(GpG) | 3.79 ± 1.23 | 7.54 ± 1.19 | 3.63 |
| Pt-1,2-d(GpA) or Pt-1,2-d(ApG) | 3.12 ± 1.30 | 0.969 ± 0.59 | 0.977 |
| Pt-1,3-d(GpNpG) or Pt-(dG)2 | 3.83 ± 1.59 | 10.7 ± 1.53 | 8.89 |
| Pt-(dA)2 | 4.35 ± 1.42 | 11.1 ± 1.38 | 17.4 |
| Not determined | ~76.9 | ~59.5 | 54.9 |
| Σ | 98.2 | 97.9 | 96.4 |
Σ represents the percentage summation of the major oxaliplatin–DNA adducts listed and not determined row, compared to the total radiocarbon content in cellular DNA. The data for T24 cells were from our previously published paper (Ref. 6).
From a quantitative point of view, our data for oxaliplatin-adduct distribution in MDA-MB-231 cells suggest that after a 24-h incubation, monoadducts constituted approximately 6% of the total radioactivity from the HPLC fractions and oxaliplatin forms approximately 4% intrastrand Pt-1,2-d(GpG), 3% intrastrand Pt-1,2-d(GpA) or Pt-1,2-d(ApG), and 4% intrastrand Pt-1,3-d(GpNpG) or interstrand Pt-(dG)2 and 4% interstrand Pt-(dA)2 diadducts (Table 1), which shows slightly different compositional adduct distribution from a previous study6 reporting that oxaliplatin in T24 human bladder cancer cells forms approximately 10% monoadducts, 4% intrastrand Pt-1,2-d(GpG), 1% intrastrand Pt-1,2-d(GpA) or Pt-1,2-d(ApG), and 8% intrastrand Pt-1,3-d(GpNpG) or interstrand Pt-(dG)2 and 17% interstrand Pt-(dA)2 diadducts. The ratio of Pt-1,2-d(GpG) cross-links as compared to the other adduct standards is quite similar with the data from the cell-based experiment reported in our previous study, implying the similar repair efficiency of these intrastrand 1,2-cross-links in cells. The remaining HPLC fractions that eluted with elevated radiocarbon concentrations at 3, 13, 15, 17, and 40 min could not be assigned. These uncharacterized adducts, however, constituted approximately 77% of the total radioactivity from the HPLC fractions when including the fractions likely eluting with the solvent front. This result suggests that although the typical oxaliplatin–DNA adducts such as intrastrand Pt-1,2-d(GpG) and interstrand Pt-(dG)2 diadducts are the major products of oxaliplatin in MDA-MB-231 cells, other unidentified adducts also comprise of a significant portion and may play some roles in the cytotoxic activity of oxaliplatin in MDA-MB-231 cells.
More importantly, our AMS data for oxaliplatin-adduct distribution in platinum-sensitive 833 K cells are greatly different from the data for oxaliplatin-adduct distribution in platinum-resistant T24 and MDA-MB-231 cells. Our obtained data support that after a 24-h incubation, monoadducts constituted approximately 8% of the total radioactivity from the HPLC fractions and oxaliplatin forms approximately 7% intrastrand Pt-1,2-d(GpG), 1% intrastrand Pt-1,2-d(GpA) or Pt-1,2-d(ApG), and 10% intrastrand Pt-1,3-d(GpNpG) or interstrand Pt-(dG)2 and 11% interstrand Pt-(dA)2 diadducts (Table 1), which clearly demonstrates different compositional adduct distribution from the adduct distribution in MDA-MB-231 and T24 cells. It should be noted that 833 K cells, the most drug-sensitive cancers among three, showed the highest 14C accumulation in cells with approximately 2- and 2.5-fold higher AUC values as compared to the platinum-resistant MDA-MB-231 and T24 cells, respectively, and our data support that intrastrand Pt-1,2-d(GpG) in 833 K cells after one-day incubation with oxaliplatin (0.2 μM) constituted approximately 7% of the total radioactivity from the HPLC fractions. The remaining HPLC fractions that eluted with elevated radiocarbon concentrations at 3, 9, 13, 17, and 35 min could not be assigned. The ratio of Pt-1,2-d(GpG) crosslinks compared to the other adduct standards is approximately two-fold higher than the data from the T24 genomic DNA experiments, strongly implying that in addition to the cell-dependent drug uptake and efflux kinetics and drug–DNA adduct formation kinetics, the differential accumulation and/or repair of these intrastrand 1,2-crosslinks in these cells may be responsible for the sensitivity of cancer cells to platinum treatment.
To our knowledge, this is the first report describing the differential oxaliplatin–DNA adduct distribution from a cell-based study at a subpharmacological drug concentration. Identification of uncharacterized product retention times as well as elucidation of oxaliplatin–DNA adduct repair rates depending on the cell types are in progress with an ultimate aim to discover ‘biomarkers’ to correlate mass distribution of oxaliplatin adducts and/or relative amounts of specific cross-links to patient outcome, since employing our AMS-based assay, it is likely that mass distribution study for oxaliplatin–DNA adducts in small clinical samples such as tumor biopsies can be measured, which is a goal of our future work. In addition, we will investigate the adduct distribution close to the ‘real life’ clinical situation to complete the picture of the chemistry of oxaliplatin and the differences from cisplatin and carboplatin.
In summary, we used AMS to measure and characterize oxaliplatin–DNA adducts in cell-based experiments, and observed cell-specific differences in drug–DNA adduct distribution after one-day incubation. AMS was able to measure attomole per microgram concentrations of specific platinated DNA at several orders of magnitude lower concentrations in cells compared to other detection methods. The lowest concentration of radiocarbon measured by AMS was again approximately 1 amol/μg of DNA, when assaying 1 μg of DNA, demonstrating that the cell lines could be differentiated with respect to platinum sensitivity even though non-toxic sub-pharmacological concentrations of the drug were used. This observation opens the possibility of personalized medicine by extending the AMS assay to an in vivo setting with oxaliplatin and possibly other DNA-damaging drugs. Such an experiment has not been previously possible due to lack of sufficient analytical sensitivity.
Acknowledgments
Kurt Hack is acknowledged for preparing AMS samples. This work was performed at the Research Resource for Biomedical Accelerator Mass Spectrometry under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under contract DE-AC52-07NA27344, and was supported in part by DOE/LDRD Grant 06-LW-023 and by the Korea Science and Engineering Foundation (KOSEF) Grant funded by the Korea Government (MEST) (No. 2009-0064333).
References and notes
- 1.Saunders M, Iveson T. Br J Cancer. 2006;95:131. doi: 10.1038/sj.bjc.6603233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Graham J, Muhsin M, Kirkpatrick P. Nat Rev Drug Disc. 2004;3:11. doi: 10.1038/nrd1287. [DOI] [PubMed] [Google Scholar]
- 3.(a) Wang D, Lippard SJ. Nat Rev Drug Disc. 2005;4:307. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]; (b) Chaney SG, Campbell SL, Bassett E, Wu Y. Crit Rev Oncol Hematol. 2005;53:3. doi: 10.1016/j.critrevonc.2004.08.008. [DOI] [PubMed] [Google Scholar]; (c) Desoize B, Madoulet C. Crit Rev Oncol Hematol. 2002;42:317. doi: 10.1016/s1040-8428(01)00219-0. [DOI] [PubMed] [Google Scholar]; (d) Kartalou M, Essigmann JM. Mutat Res. 2001;478:1. doi: 10.1016/s0027-5107(01)00142-7. [DOI] [PubMed] [Google Scholar]; (e) Jamieson ER, Lippard SJ. Chem Rev. 1999;99:2467. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]; (f) Wong E, Giandomenico CM. Chem Rev. 1999;99:2451. doi: 10.1021/cr980420v. [DOI] [PubMed] [Google Scholar]; (g) Fuertes MA, Alonso C, Perez JM. Chem Rev. 2003;103:645. doi: 10.1021/cr020010d. [DOI] [PubMed] [Google Scholar]
- 4.Di Francesco AM, Ruggiero A, Riccardi R. Cell Mol Life Sci. 2002;59:1914. doi: 10.1007/PL00012514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Hah SS. J Biomed Sci. 2009;16:54. doi: 10.1186/1423-0127-16-54. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hah SS. Anal Sci. 2009;25:731. doi: 10.2116/analsci.25.731. [DOI] [PubMed] [Google Scholar]; (c) Mundt JM, Hah SS, Sumbad RA, Schramm V, Henderson PT. Nucleic Acids Res. 2008;36:228. doi: 10.1093/nar/gkm1032. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hah SS, Mundt JM, Ubick EA, Turteltaub KW, Gregg JP, Henderson PT. Nucl Instrum Methods Phys Res Sect B. 2007;259:763. [Google Scholar]; (e) Hah SS, Stivers KM, de Vere White RW, Henderson PT. Chem Res Toxicol. 2006;19:622. doi: 10.1021/tx060058c. [DOI] [PubMed] [Google Scholar]; (f) Hah SS, Kim HM, Sumbad RA, Henderson PT. Bioorg Med Chem Lett. 2005;15:3627. doi: 10.1016/j.bmcl.2005.05.113. [DOI] [PubMed] [Google Scholar]; (g) Vogel JS. Biotechniques. 2005;38:13. [Google Scholar]; (h) Lappin G, Garner RC. Anal Bioanal Chem. 2004;378:356. doi: 10.1007/s00216-003-2348-5. [DOI] [PubMed] [Google Scholar]
- 6.Hah SS, Sumbad RA, Vere White RW, Turteltaub KW, Henderson PT. Chem Res Toxicol. 2007;20:1745. doi: 10.1021/tx700376a. [DOI] [PubMed] [Google Scholar]
- 7.Unless otherwise noted, reagents were obtained from commercial suppliers and were used without further purification. Radiocarbon-labeled oxaliplatin ([14C]oxaliplatin) was purchased from GE Healthcare (formerly Amersham Biosciences, Piscataway, NJ), of which the specific activity was 58 mCi/mmol. MDA-MB-231 breast cancer cells were obtained from ATCC. 833K testicular cancer cells were generously provided by Drs. Robert Wood and Beate Koberle (University of Pittsburgh). Cells were maintained in tissue culture plates at 37 °C in an atmosphere of 5% CO2 in RPMI media supplemented with 10% fetal bovine serum, 100 IU/ml penicillin and 0.1 mg/ml streptomycin. Exponentially growing cells were harvested by means of trypsinization (0.5 ml of 0.25% trypsin solution for 5 min at 37 °C) and the resulting single cell suspension was plated into tissue culture plates. Cells were incubated for 24 h to allow attachment. Free cells were removed by washing three times with phosphate buffered saline (PBS). Cells were then dosed with oxaliplatin (0.2 μM) which contained 300 dpm (2.16 pmol) [14C]oxaliplatin (77.6 μCi/mmol) in 5 ml of media. After one-day incubation, the plates were washed three times with PBS, followed by trypsinization. The isolated cells were subjected to lysis, proteinase K treatment and DNA extraction. The total amounts of radiocarbon in the cells and extracted DNA measured by AMS were consistent with the values previously described (see Ref. 6).The resulting platinated DNA was enzymatically digested by deoxyribonuclease and P1 nuclease, which degraded the DNA to deoxyribonucleotides. Further digestion with alkaline phosphatase yielded deoxyribonucleosides, which were subjected to fractionation by HPLC. In brief, the eluent was 5–25% methanol in 0.4 M ammonium acetate (pH 7) over 40 min at a flow rate of 1 ml/min using a 250 × 4.6 mm, Hypersil ODS column (Thermo Electron Corporation, Bellefonte, PA). The HPLC retention times of the radiolabeled digested DNA adducts were compared to unmodified deoxyribonucleosides, dC, dG, dT and dA, and to those of authentic standards that were synthesized based upon literature protocols. The authentic standards include Pt(DACH) adducted to N1 and N7 of deoxyadenosine and N7 of deoxyguanosine, which are the major monoadduction products (Pt-dA and Pt-dG, respectively). The major intrastrand 1,2-crosslinks were represented in the standard collection by Pt(DACH) adducted to d(GpG), d(GpA), and d(ApG). The major interstrand crosslink products were Pt(DACH) adducted to (dG)2 and (dA)2, although the latter two products can be formed from enzymatic digestion of d(XpNpX) intrastrand 1,3-crosslinks (X = A or G and N = A, T, G or C). Measurement with AMS of the radiocarbon-containing HPLC fractions allowed quantitation of the resulting oxalipatin monoadducts and diadducts.The radiocarbon content in cells, extracted DNA, and HPLC franctions was measured by AMS as reported (see Ref. 6). Briefly, AMS samples were prepared in triplicate after addition of carrier carbon in the form of 1 μl of tributyrin, and the ratio of 14C to 12C in a biological sample that has been converted to solid carbon (graphite or fullerene) for analysis using a two-step oxidation–reduction process was measured. Graphite targets were measured at the Center for Accelerator Mass Spectrometry at Lawrence Livermore National Laboratory. Calculation of the number of attomole (amol) of 14C was performed using the conversion factor of 1 Modern equals 97.8 amol of 14C per mg of total carbon.Since AMS is specific only to the labeled compound in any chemical or biological medium, and DNA was extracted from the cultured cells, followed by enzymatic digestion to yield deoxyribonucleosides, AMS data in Figure 3 were from [14C]oxaliplatin-labeled deoxyribonucleosides, not from any other products.All experiments were performed in triplicate.
- 8.Ehrsson H, Wallin I, Yachnin J. Med Oncol. 2002;19:261. doi: 10.1385/MO:19:4:261. [DOI] [PubMed] [Google Scholar]
- 9.(a) Jennerwein MM, Eastman A, Khokhar A. Chem Biol Interact. 1989;70:39. doi: 10.1016/0009-2797(89)90061-6. [DOI] [PubMed] [Google Scholar]; (b) Eastman A. Biochemistry. 1982;21:6732. doi: 10.1021/bi00269a018. [DOI] [PubMed] [Google Scholar]
- 10.(a) Johnson SW, Swiggard PA, Handel LM, Brennan JM, Godwin AK, Ozols RF, Hamilton TC. Cancer Res. 1994;54:5911. [PubMed] [Google Scholar]; (b) Johnson SW, Laub PB, Beesley JS, Ozols RF, Hamilton TC. Cancer Res. 1997;57:850. [PubMed] [Google Scholar]