

Abstract
TGF-β can induce Foxp3+ inducible regulatory T cells (Treg) and also synergize with IL-6 and IL-4 to induce Th17 and Th9 cells. We now report that NO modulates TGF-β activity away from Treg but toward the Th1 lineage. NO potentiated Th1 differentiation in the presence of TGF-β in both IL-12–independent and –dependent fashions by augmenting IFN-γ–activated STAT-1 and T-bet. Differentiation into Treg, Th1, and Th17 lineages could be modulated by NO competing with other cofactors, such as IL-6 and retinoic acid. NO antagonized IL-6 to block TGF-β–directed Th17 differentiation, and together with IL-6, NO suppressed Treg development induced by TGF-β and retinoic acid. Furthermore, we show that physiologically produced NO from TNF and inducible NO synthase-producing dendritic cells can contribute to Th1 development predominating over Treg development through a synergistic activity induced when these cells cocluster with conventional dendritic cells presenting Ag to naive Th cells. This illustrates that NO is another cofactor allowing TGF-β to participate in development of multiple Th lineages and suggests a new mechanism by which NO, which is associated with protection against intracellular pathogens, might maintain effective Th1 immunity.
Thelper cells (CD4+) are crucial to immune function by producing distinct profiles of cytokines such as Th1, Th2, and Th17 that have been associated with individual responses against intracellular pathogens, parasites, allergens, and self-Ags linked to autoimmune disease (1-3). Naive CD4 T cells do not immediately express these phenotypes but are directed to differentiate into them by other cytokines thought largely to derive from innate cells that either present Ag to naive T cells or are activated to secrete directive cytokines early during an immune response. For example, dendritic cells (DC) producing IL-12 can support Th1 development (1), whereas mast cells or basophils producing IL-4 can support Th2 development (4). Additionally, naive CD4 T cells can differentiate into a further subset termed adaptive or inducible regulatory T cells (iTreg) that express Foxp3 and play important roles in suppressing immune responsiveness and antagonizing the activity of the Th1, Th2, and Th17 subsets (5-7). TGF-β is critical for promoting Foxp3 expression and directing iTreg differentiation. Although this action of TGF-β corresponds with the notion that it is an immunosuppressive cytokine, TGF-β can have proinflammatory activities. IL-6 promotes autoimmune predisposing Th17 cells and suppresses Treg development, but only in synergy with TGF-β (8, 9). Furthermore, recent data show that IL-4 can synergize with TGF-β to promote a novel subset of cells, termed Th9 (10, 11), that make IL-9 but not other classic Th2 cytokines and presumably participate in allergic-type reactions and protection against helminths. This has raised the question of whether other soluble mediators, produced by innate immune cells, might modulate Th differentiation and could act together with TGF-β to promote the development of subsets other than Th17 and Th9 cells.
NO, a product of l-arginine metabolism regulated by NO synthase (NOS), has been known to play a role in the immune system for ~20 y (12). It was first described to be a product of macrophages, made in response to microbes and cytokines such as IFN-γ, and functioned directly to kill or suppress replication of infectious pathogens such as bacteria, viruses, protozoa, and fungi. It is now clear that NO might have many modulatory actions on the immune system and can be produced by varying types of cells including neutrophils, eosinophils, and nonhematopoietic cells (13, 14). Interestingly, NO can also be made by subsets of DC (15, 16), yet its role in DC function is not understood. In particular, TNF and inducible NOS (iNOS)-producing DC (TipDC), or simply iNOS-producing inflammatory monocytes, have been identified as sources of NO in a variety of infections (16-18), suggesting that NO produced from TipDC is involved in both innate and adaptive immunity to pathogens. There are additionally reports suggesting that NO might be suppressive for certain T cell functions when present at high concentrations, such as blocking IL-2R signaling (14, 19), or might enhance IL-12–driven Th1 differentiation at lower concentrations by promoting expression of IL-12Rβ2 and enhancing IL-12 signaling in T cells (20, 21). Another more recent report has suggested that NO could enhance the generation of a type of Treg from naive CD4+ T cells that does not express Foxp3 but secretes IL-10 (22). This collectively implies that NO might display several modulatory activities, which can be positive or negative, depending on how much is made and the context in which it is available.
Because IFN-γ can promote the expression and activity of iNOS/NOS2 and is the hallmark of Th1 responses and clearance of intracellular pathogens, we questioned whether NO might help to promote Th1 responses in the presence of TGF-β. In this study, we show that NO strongly suppresses the induction of Foxp3+ Treg driven by TGF-β and instead results in T cells diverging into the Th1 lineage. Furthermore, IL-6 and NO compete in synergizing with TGF-β to determine differentiation into the Th17 versus Th1 lineages. Mechanistically, NO intensifies IFN-γR signals in Th cells by augmenting STAT-1 intracellular trafficking and increasing T-bet expression, resulting in a synergistic activity with IFN-γ that augments Th1 differentiation. Lastly, we show that TipDC potentiate Th1 differentiation by producing NO in collaboration with conventional DC (cDC). Thus, several soluble innate factors, two cytokines, IL-6 and IL-4, and a metabolite of l-arginine, NO, can determine whether T cell exposure to TGF-β results in Treg differentiation or T cell subsets associated with development of autoimmunity, development of allergy, or protection against extracellular and intracellular pathogens.
Materials and Methods
Mice and bacteria
OT-II mice on a C57BL/6 background were bred at La Jolla Institute for Allergy and Immunology. C57BL/6J (CD45.2+) and C57BL/6Pep3b/BoyJ (CD45.1+) mice were purchased from The Jackson Laboratory. Listeria monocytogenes were grown and purified as previously reported (16). All experiments were in compliance with the regulations of the La Jolla Institute for Allergy and Immunology animal care committee in accordance with guidelines of the Association for the Assessment and Accreditation of Laboratory Animal Care.
Cells
DC from spleen were isolated by MACS with CD11c-microbeads as described previously (23). TipDC (CD11bhiLy-6ChiCD11c−/loMHC class II+) and cDC (CD11bmedLy-6Clo/−CD11chiMHC class II+) were from spleens of mice that were i.v. infected with L. monocytogenes (5,000–10,000 CFU) purified by cell sorting with FACSAria (BD Biosciences) 2 d postinfection. OT-II or polyclonal CD4 T cells were enriched over MACS LS columns by negative isolation with a CD4+ T cell kit (Miltenyi Biotec). Naive CD4 T cells were further purified by cell sorting with FACSAria or FACSDiva (BD Biosciences) by gating cells as CD4+CD25−CD44loCD62Lhi.
In vitro lineage differentiation of CD4+ T cells
A total of 1 × 105 naive OT-II T cells and 1–2 × 104 DC were cultured in 96-well plates in 250 ml in complete IMDM media (Invitrogen) in the presence of OVA323–339 peptides (1 μM) for 4 d. Exogenous cytokines and reagents were used as follows: recombinant human TGF-β1 (5–20 ng/ml), IL-6 (1–20 ng/ml) (both from R&D Systems); all-trans retinoic acid (RA; 0.1–10 nM; Sigma-Aldrich); (Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl) amino]diazen-1-ium-1,2-diolate (DETA-NONOate; 10–100 μM), S-nitroso-N-acetyl-d-l-penicillamine (SNAP; 10–100 μM), 8-Br-cGMP (0.1–1 μM), N6-(1-iminoethyl)-l-lysine dihydrochloride (L-NIL; 0.1–1 mM) (all from Alexis Biochemicals); anti–IFN-γ (XMG1.2), anti–IL-12/23p40 (C17.8; 5–20 μg/ml) (BD Biosciences); and rat IgG (KLH/G1-2-2; Southern Biotechnology Associates). Th cell lineages were determined by intracellular staining of IFN-γ (Th1), IL-17A (Th17), and Foxp3 (Treg) after restimulating cells with PMA and ionomycin (Sigma-Aldrich) for 5 h in the presence of GolgiPlug (BD Biosciences). The following Abs were used for flow cytometry: biotin-conjugated rat IgG1 (RTK2071; Biolegend); FITC-conjugated anti-CD44 (1M7), anti–IFN-γ (XMG1.2) (both from BD Biosciences); anti–Ly-6C (HK1.4; Biolegend); PE-conjugated anti-CD25 (PC61), anti–IFN-γ (XMG1.2) (BD Biosciences); anti–IL-17A (TC11-18H10.1; Biolegend); anti-NOS2 (N-20; Santa Cruz Biotechnology), streptavidin (Invitrogen); allophycocyanin-conjugated anti-CD11c (HL3; BD Biosciences); anti-CD62L (MEL-14), anti-Foxp3 (FJK-16s; eBioscience); PerCP-conjugated anti-CD4 (RM4.5; BD Biosciences); PerCP-Cy5.5–conjugated anti-CD11b (M1/70; BD Biosciences); and Pacific Blue-conjugated anti–I-A/E (M5/114.15.2; Biolegend). For measuring mRNA expression of transcription factors, CD4 T cells were harvested, washed with PBS, and then RNA was isolated using TRIzol (Invitrogen), and cDNA was synthesized from total RNA using the Superscript III system (Invitrogen) following the instructions provided by the manufacturer. Subsequently, the cDNA was subject to real-time PCR using SYBR Green (Roche) and the RT-PCR–specific primers. The sequences of primers were as follows: Foxp3 forward, 5′-GGC CCT TCT CCA GGA CAG A-3′; Foxp3 reverse, 5′-GCT GAT CAT GGC TGG GTT GT-3′; RA-related orphan receptor γt (RORγt) forward, 5′-GGA CAG GGA GCC AAG TTC TCA G-3′; RORγt reverse, 5′-CAC AGG TGATAA CCC CGT AGT GG-3′; T-bet forward, 5′-CAA CAA CCC CTT TGC CAA AG-3′; T-bet reverse, 5′-TCC CCC AAG CAG TTG ACA GT-3′; IFN-γ forward, 5′-AAC GCT ACA CAC TGC ATC TTG G-3′; IFN-γ reverse, 5′-GCC GTG GCA GTA ACA GCC-3′; iNOS forward, 5′-GAA GAA AAC CCC TTG TGC TG-3′; iNOS reverse, 5′-GTC GAT GTC ACA TGC AGC TT-3′; L32 forward, 5′-GAA ACT GGC GGA AAC CCA-3′; L32 reverse, 5′-GGA TCT GGC CCT TGA ACC TT-3′; β-actin forward, 5′-ACA GCT TCT TTG CAG CTC CT-3′; and β-actin reverse, 5′-AGT CCT TCT GAC CCA TTC C-3′. Data were collected and analyzed on a Roche LightCycler 480 (Roche).
Immunofluorescent staining
T cells (1× 105) were loaded onto a poly-l-lysine–coated coverslip (BD Biosciences), fixed in 4% paraformaldehyde solution (USB), permeabilized in 0.3% saponin (Sigma-Aldrich)/PBS, and treated with 5% BSA/PBS. Cells were incubated with mouse anti–STAT-1 Ab (BD Transduction Laboratory) followed by additional incubation with goat anti-mouse IgG (H+L) Cy3 (Molecular Probes). Cells were finally stained with DAPI to visualize the nuclei. Coverslips were mounted with FluorSave (Calbiochem) reagent, and immunofluorescence was analyzed under the Zeiss Axiovert 200M scope (Carl Zeiss), integrated with 3i’s Slidebook 4.2 software (Intelligent Imaging Innovations). Cells that displayed nuclear STAT-1 colocalized with DAPI were quantified and expressed as the percentage of the total cells. The data shown are representative of one of three independent double-blinded experiments. For immunofluorescent staining of spleens, frozen sections were cut and permeabilized with 0.2% Triton X-100 in PBS. After blocking with goat IgG (Calbiochem), sections were stained with anti-iNOS (rabbit polyclonal IgG; Millipore) followed by incubation with HRP-conjugated anti-rabbit IgG (Pierce). Cell-surface markers CD3e, CD4, and CD11c were further stained, and the nucleus was stained with DAPI. Sections were then mounted with a mounting medium (Fluoromount G; Southern Biotechnology Associates) and analyzed using a Marianas fluorescence microscope (Intelligent Imaging Innovations). Images were processed and analyzed with ImageJ software (National Institutes of Health).
Western blots
Cellular extracts were prepared by lysing cells in cell lysis buffer (Cell Signaling Technology; 20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1.0% Triton, and 1 mM EGTA) supplemented with a protease and phosphatase inhibitor mixture. Equal amounts of protein were separated by electrophoresis in SDS-polyacrylamide gel and electrotransferred onto a polyvinylidene difluoride membrane (Invitrogen). After blocking, blots were incubated at 4°C overnight with Abs to phospho-Smad3 and actin (Cell Signaling Technology) followed by HRP-conjugated anti-rabbit IgG. Signals were developed with ECL Western blotting Substrate (Thermo Fisher Scientific), and the membranes were exposed to x-ray films.
Results
NO modulates TGF-β differentiation signals
TGF-β has been found to induce differentiation of adaptive Treg by upregulating Foxp3 expression in responding naive CD4 T cells (5, 6). To investigate whether NO might modulate TGF-β activity and alter Th differentiation, naive CD4 (CD25−Foxp3−CD44loCD62Lhi) T cells specific for OVA were exposed to cognate Ag presented on DC in the presence of TGF-β. As an exogenous source of NO, two NO donors were added, DETA-NONOate and SNAP. As expected, TGF-β strongly promoted a population of T cells that expressed Foxp3. In contrast, the NO donors dose-dependently suppressed this activity (Fig. 1A; 32 to ~3% Foxp3+). Significantly, whereas few IFN-γ– or IL-17–producing effector T cells resulted with TGF-β in isolation, the NO donors strongly promoted the Th1 subset when combined with TGF-β (Fig. 1A; ~4 to 40–50% IFN-γ+), but had no effect on induction of Th17 cells. Thus, NO has the capacity to reprogram TGF-β differentiation signals into the Th1 lineage.
FIGURE 1.
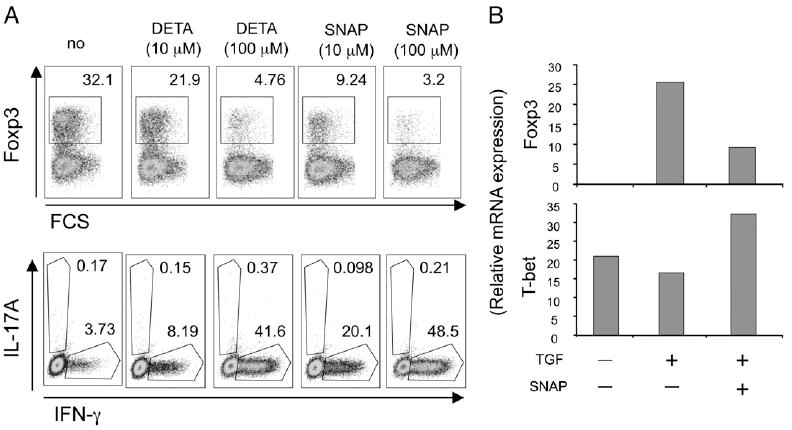
NO blocks Foxp3 induction driven by TGF-β and skews naive CD4 T cell differentiation to Th1. A and B, Naive (CD4+CD25−CD44loCD62Lhi) OT-II CD4 T cells were stimulated with splenic DC and OVA peptide (1 µM) in the presence of TGF-β. NO donors (DETA-NONOate or SNAP) were added at the start of culture at varying concentrations. A, Foxp3 expression versus forward scatter (top panel) or IL-17A versus IFN-γ intracellular cytokine expression (bottom panel) assessed after 4 d. Cytokines were detected after restimulation of cells for 5 h with PMA/ionomycin. B, mRNA levels of Foxp3 and T-bet were evaluated at day 4 by real-time PCR without restimulation. Results are representative of three experiments.
In our cultures with splenic DC, differentiation under neutral conditions was toward Th1 in that peptide presentation led to upregulation of T-bet but not RORγt or Foxp3 mRNA expression (Fig. 1B, Supplemental Fig. 1). Of note, NO alone in the absence of exogenous cytokines was able to enhance T-bet expression as well as Th1 differentiation (Supplemental Fig. 1). Exogenous TGF-β did not markedly affect T-bet expression, but strongly promoted Foxp3, and NO suppressed this Foxp3 expression while augmenting T-bet (Fig. 1B). This suggests that NO modulates the Treg/Th1 balance in the presence of TGF-β by regulating the expression of Th subset-specific transcription factors.
NO competes with IL-6 and RA to determine the Th1/Th17/Treg balance
Treg differentiation induced by TGF-β can be antagonized by IL-6 that reprograms differentiation into the Th17 subset (8, 9). NO also suppressed Treg development, but its action was separable from that of IL-6 in that NO had no ability to promote Th17 cells and IL-6 no ability to promote Th1 cells (Figs. 1A, 2A). To further address this, we titrated IL-6 in the presence of NO to determine how varying the levels of IL-6R signals would influence the activity of NO and modulate differentiation into alternate Th subsets. Addition of the NO donor SNAP in the presence of TGF-β and a low dose of IL-6 (0.1 ng/ml) preferentially promoted Th1 differentiation (Fig. 2A). Increasing doses of IL-6 (1–10 ng/ml) suppressed NO-induced Th1 differentiation and promoted Th17 cells. Another NO donor, DETA, showed a similar competitive activity with varying doses of IL-6 (Fig. 2A). As expected, IL-6 together with TGF-β greatly induced mRNA for RORγt while suppressing T-bet (Fig. 2B), essentially opposite to the action of NO, which elevated T-bet with no induction of RORγt. In contrast, IL-6, when titrated into cultures with the NO donor and TGF-β, blocked T-bet first and then at higher concentrations promoted RORγt (Fig. 2B). However, RORγt and IL-17 production was significantly lower when the NO donor was active in the presence of IL-6 and TGF-β. Interestingly, a medium dose of IL-6 in combination with NO resulted in almost no T-bet or RORγt mRNA. These results directly show that NO and IL-6 compete to determine the Th1/Th17 balance when naive T cells are exposed to the directive influence of TGF-β.
FIGURE 2.
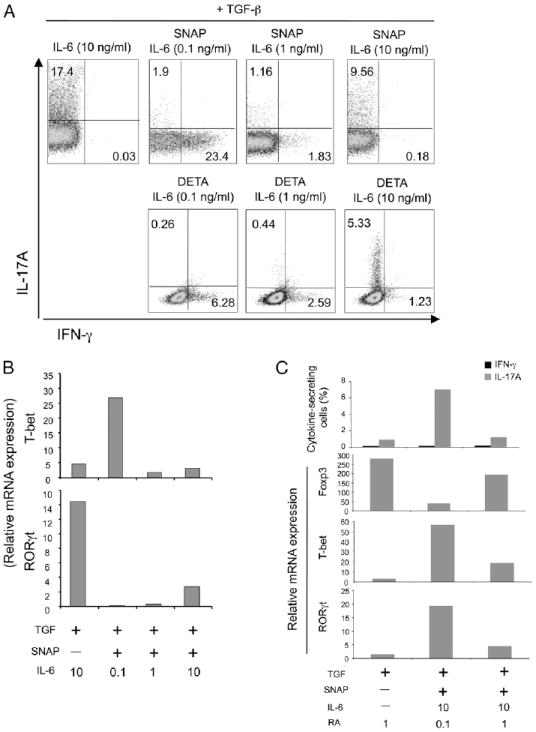
NO competes with IL-6 and RA to determine the balance of Th1/Th17/Treg differentiation. Naive OT-II CD4 T cells were stimulated with splenic DC and OVA peptide in the presence of TGF-β with NO donors, SNAP, and DETA (100 µM). Where indicated, various doses of IL-6 (ng/ml) and RA (nM) were added at the start of culture. Expression of intracellular IL-17A versus IFN-γ was determined at day 4 after restimulation (A, C). mRNA levels of Foxp3, T-bet, and RORγt were evaluated by realtime PCR without restimulation (B, C). Results are representative of three experiments.
To further assess competition between differentiation signals, we assessed the effects of NO when RA was present. RA strongly enhanced expression of Foxp3 in the presence of TGF-β (Fig. 2C) as reported previously (24-27). Moreover, a high level of RA (1 nM) allowed TGF-β signals to maintain differentiation into the Treg subset by still promoting strong Foxp3 expression even when high concentrations of IL-6 and the NO donor were used separately or together (Fig. 2C and data not shown). When we used a lower concentration of RA (0.1 nM), NO together with IL-6 then repressed the activity of RA for promoting Foxp3 expression. These data suggest that both NO and IL-6 have the capacity to modulate TGF-β differentiation signals away from Foxp3 and Treg, but if there is a source of RA, such as a gut-resident DC, the activity of RA will prevail over that of both NO and IL-6 and lead to a preponderance of Foxp3+ Treg rather than Th1 or Th17 cells.
IFN-γ–dependent and –independent activity of NO
IFN-γ has been linked to NO production and activity of iNOS (12, 13), but IFN-γ has also been shown to antagonize Foxp3 expression as well as itself promote Th1 differentiation (3, 28-30). We then questioned whether another feedback link between these molecules might exist. Significantly, blocking IFN-γ during the priming of naive T cells largely prevented NO in combination with TGF-β from antagonizing induction of Foxp3 and enhancing the appearance of IFN-γ–secreting effector T cells (Fig. 3A). Prior results showed that NO in the absence of TGF-β potentiated exogenous IL-12–driven Th1 differentiation (20, 21). However, the splenic DC obtained from naive mice that were used in our cultures were unlikely to be high IL-12 producers, suggesting that IL-12 might not be active in this system. Confirming this, blocking IL-12 did not prevent NO from downregulating Foxp3 and up-regulating IFN-γ in the presence of TGF-β (Fig. 3A). Furthermore, when IFN-γ was neutralized in cultures with TGF-β plus the NO donor and IL-6, no difference in Foxp3 expression was observed but NO still reduced differentiation into the Th17 lineage (Fig. 3B). Therefore, NO can exert both IFN-γ–dependent and–independent activity and IL-12–independent activity in modulating the Treg/Th1/Th17 balance when TGF-β is present.
FIGURE 3.
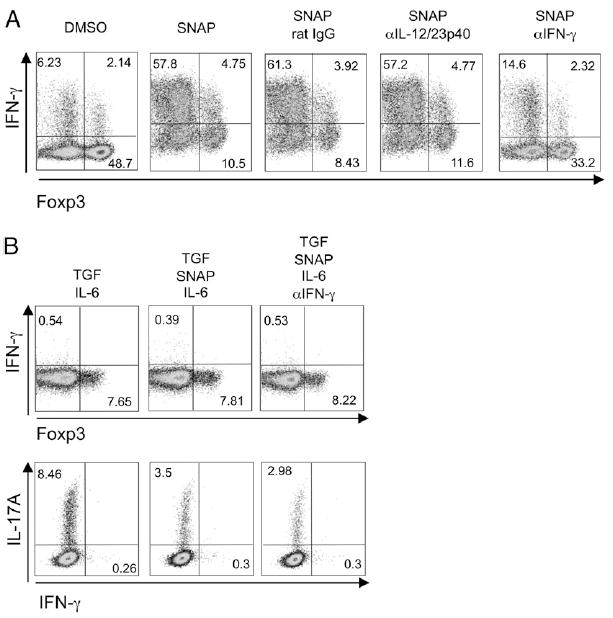
IFN-γ–dependent and –independent activity of NO in suppressing Foxp3 and Th17, and promoting Th1 differentiation. Naive OT-II CD4 T cells were stimulated with splenic DC and OVA peptide in the presence of TGF-β (5 ng/ml). Where indicated, SNAP (100 µM) and neutralizing Abs (20 µg/ml) to IL-12 or IFN-γ (A) or SNAP (100 μM), IL-6 (1 ng/ml), and anti–IFN-γ (20 μg/ml) (B) were added at the start of culture. Intracellular Foxp3, IFN-γ, and IL-17 were determined at day 4 after restimulation. Results are representative of three experiments.
Next, we assessed whether targeting one of the major signaling pathways activated by NO could mimic its activity in modulating Th1 and Treg differentiation. NO binds to and activates guanylyl cyclase, which catalyzes the dephosphorylation of GTP to cyclic GMP (cGMP) (31). cGMP has been described to be responsible for a number of activities of NO on various cell types (19, 21). To determine if cGMP might account for the differentiative capacity of NO on T cells, we supplemented TGF-β cultures with a cGMP analog. 8-Br-cGMP dose-dependently suppressed Foxp3 induction in response to TGF-β and concomitantly promoted some T cells to differentiate into IFN-γ producers (Supplemental Fig. 2). The action of cGMP then partially replicated that of the NO donor, although levels of conversion into Th1 cells were lower. These data therefore suggest that NO functions at least in part by inducing cGMP production and that altering cGMP levels has the potential to dictate the Treg/Th1 balance.
Synergy between NO and IFN-γ for Th1 differentiation
To address whether NO directly targeted T cells, we stimulated naive Th cells with plate-bound anti-CD3 and soluble anti-CD28 in the presence of TGF-β. In these cultures, 50–60% of T cells became Foxp3+, and ~2% of T cells gained expression of IFN-γ (Fig. 4A, DMSO control). NO increased Th1 differentiation (~6%) and T-bet expression, whereas it modestly reduced Treg differentiation in these APC-free cultures (Fig. 4A, 4B). As Foxp3 expression inversely correlated with IFN-γ expression, and neutralizing IFN-γ largely abolished NO activity in cultures with DC (Fig. 3), we also blocked endogenous IFN-γ in the APC-free cultures. This completely inhibited Th1 differentiation induced by NO, shown by minimal expression of IFN-γ and T-bet (Fig. 4A, 4B). Interestingly, we still found the same modest reduction in Treg development with blockade of IFN-γ (Fig. 4A), indicating that NO can suppress Foxp3 expression in part independently of IFN-γ similar to the IFN-γ –independent action on suppressing Th17 differentiation (Fig. 3).
FIGURE 4.
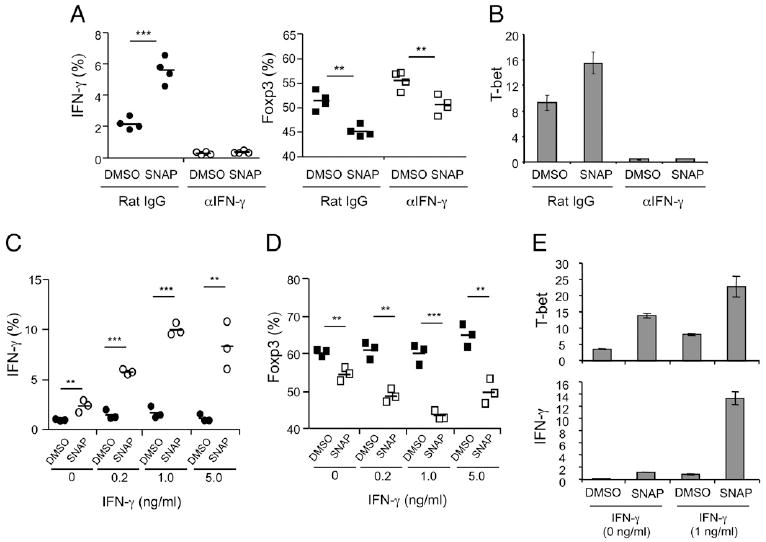
Direct synergy between NO and IFN-γ on CD4 T cells. Naive OT-II CD4 T cells were stimulated with anti-CD3 (plate-coated, 10 mg/ml), anti-CD28 (soluble, 1 mg/ml), and IL-2 (5 ng/ml) in the presence of TGF-β (1 ng/ml). Where indicated, SNAP (10 µM) or DMSO was added at the start of culture. A and B, Endogenous IFN-γ was neutralized by the addition of anti–IFN-γ (10 μg/ml). Expression of intracellular Foxp3 and IFN-γ was determined at day 4 after restimulation, and mRNA levels of T-bet were measured without restimulation. C–E, Naive Th cells were stimulated as in A and B in the presence of various doses of exogenous IFN-γ. Intracellular staining of IFN-γ and Foxp3 and mRNA levels of T-bet and IFN-γ were measured as above. Results are representative of three experiments. **p > 0.01, ***p > 0.001 (two-tailed Student t test).
As these data suggested a cooperative action of the low level of endogenous IFN-γ with NO, we asked if increasing concentrations of IFN-γ would potentiate NO activity with TGF-β. Th1 differentiation was strongly enhanced when exogenous IFN-γ was combined with NO at the start of culture, and, at the same time, Treg differentiation was inhibited (Fig. 4C, 4D). The addition of IFN-γ in the absence of NO had no effect on induction of Treg, revealing a synergistic action of NO with IFN-γ, and showing that IFN-γ alone was not sufficient to counteract the suppressive action of TGF-β on Th1 differentiation. NO enhanced T-bet expression and IFN-γ mRNA expression when added to T cells in the presence of TGF-β, and T-bet and IFN-γ expression were amplified when exogenous IFN-γ was combined with NO (Fig. 4E). Collectively, these results suggest that NO regulates Th1 differentiation through a direct action on the T cell and that this activity of NO is mediated through upregulating T-bet expression in a feedback loop driven by a source of IFN-γ.
To address how NO modulated TGF-β signaling and potentiated Th1 differentiation, we first tested the possibility that NO directly inhibited TGF-βR signaling in Th cells. A prior study with endothelial cell lines reported that NO blocked the phosphorylation and nuclear translocation of Smad2/3 induced by TGF-β (32). In contrast, NO addition to Th cells revealed no inhibitory effect on the phosphorylation or the nuclear accumulation of Smad2/3 (Supplemental Fig. 3A–D). Because of the synergy with IFN-γ revealed in our APC-free cultures, we then hypothesized that NO might augment IFN-γ R signaling and determined the effect on nuclear translocation of STAT-1 that is a crucial part of IFN-γR action. STAT-1 was localized in the cytoplasm in naive Th cells (Fig. 5A), and, as reported in other studies, exposure to high-dose IFN-γ could promote strong STAT-1 accumulation in the nucleus (Fig. 5B, 5E). In contrast, simultaneous exposure of anti-CD3/CD28–activated Th cells to TGF-β with exogenous IFN-γ resulted in only a low level of STAT-1 translocated into the nucleus, showing inefficient activation of IFN-γR signaling in the presence of suppressive signals from TGF-βR. Most significantly, nuclear STAT-1 was strongly increased by additional exposure of T cells to NO in these cultures (Fig. 5C–E). We found no activation of STAT-4 when Th cells were cultured with TGF-β and NO (Supplemental Fig. 3E, 3F) in line with our prior data showing no role for IL-12 in mediating the activity of NO in the cultures with splenic DC from naive animals. This also showed that NO did not exert any IL-12–independent effect on STAT-4. These data suggest that NO can potentiate IFN-γR signaling in Th cells through regulating the mobilization of STAT-1.
FIGURE 5.
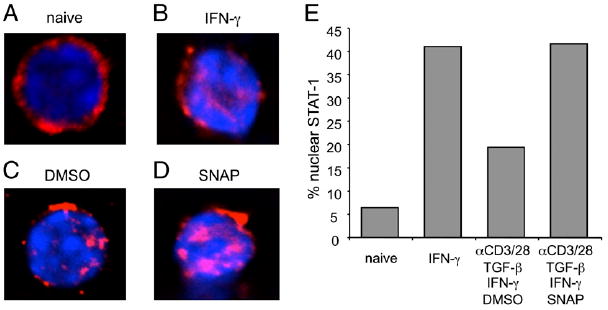
NO augments IFN-γR signaling in Th cells. Naive OT-II CD4 T cells were treated with PBS (A) or as a control IFN-γ (B, 20 ng/ml). Other T cells were stimulated with anti-CD3/CD28, IL-2, TGF-β (1 ng/ml), and IFN-γ (1 ng/ml) in the presence of DMSO (C) and SNAP (D, 10 μM). Cells were fixed and stained with anti–STAT-1 (red) and DAPI (blue). Original magnification ×60. The percentages of cells having nuclear STAT-1 proteins were analyzed (E). Results are representative of three experiments.
TipDC as physiological sources of NO
We then assessed whether endogenous NO production could play a role in the Treg/Th1 balance in physiological situations in which naive Th cells encountered Ag. TipDC have previously been identified as a subset of DC involved in immunity against L. monocytogenes, and these specialized cells were proposed to play a major role in clearance of the intracellular bacteria (16). A recent study by Kang et al. (33) found that TipDC developed from monocytes recruited from the blood, and these cells coclustered with other innate cells, such as NK cells and granulocytes, in the T cell zone of the spleen. This led us to hypothesize that TipDC might also regulate Th differentiation through NO production in this location. As shown previously (33), inflammatory monocytes can be identified as CD11b+Ly-6ChiCD11c−/lo, separable from cDC that are CD11bmedLy-6C−/loCD11chi (Supplemental Fig. 4A). Intracellular staining (Supplemental Fig. 4B) and mRNA expression (Supplemental Fig. 4C) for iNOS indicated that the majority of monocytes had matured into TipDC after Listeria infection, whereas cDC minimally expressed iNOS (Supplemental Fig. 4C). Thus, we isolated TipDC to test whether NO endogenously produced from these cells played a role in Th differentiation. We also isolated cDC as a control APC population.
Surprisingly, in isolation, TipDC had less Ag-presenting activity in promoting Th1 induction in cultures with naive T cells when compared with cDC, and also resulted in ~2% of cells gaining Foxp3 (Fig. 6A, left panel). This effect was further amplified if exogenous TGF-β was added (Fig. 6A, right panel). Blockade of iNOS with a specific inhibitor, L-NIL, did not reveal any role for NO in these cultures (Fig. 6A). Although some effector Th cells were generated in the TipDC cultures, further assays based on measuring proliferation of T cells showed that TipDC were very poor at activating naive Th cells compared with cDC, and this was not explained by the production of NO (Fig. 6B). Additionally, presentation of Ag by TipDC failed to maintain survival of T cells in culture, particularly at later times, leading to weak accumulation of Th cells over time (data not shown). Interestingly, TipDC expressed MHC class II and many cell-surface molecules that have T cell costimulatory or coinhibitory activity at levels similar to or higher than cDC (not shown). These results suggest that TipDC, albeit expressing a mature phenotype, are intrinsically poor at stimulating naive T cells and therefore might not represent a primary APC for initiating the T cell response.
FIGURE 6.
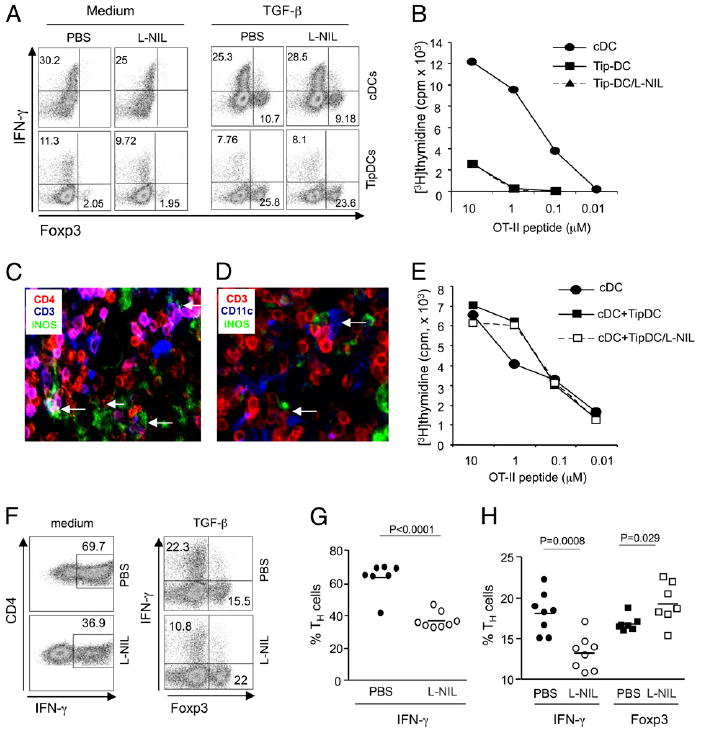
Physiological NO produced by TipDC regulates Th1/Treg differentiation. cDC and TipDC were isolated as described in Materials and Methods from mice infected for 2 d with Listeria. Naive OT-II CD4 T cells were stimulated with cDC or TipDC in isolation (A, B) or in coculture of both cDC and TipDC (E–H) in the presence of OVA peptide with or without TGF-β (5 ng/ml). Where indicated, L-NIL (100 µM) was added. Expression of Foxp3 and IFN-γ was determined at day 4 after restimulation (A, F–H). Th cells were stimulated with DC mixtures in the absence (G) or presence (H) of TGF-β. The proliferation of Th cells was measured by [3H]thymidine incorporation at day 3 (B, E). C and D, Immunohistochemistry of spleen sections from Listeria-infected mice stained for CD4, CD3, iNOS, and CD11c as indicated. Relevant cell conjugates/clusters were marked with arrows. Original magnification ×20. Results are representative of two (C, D) to three (A, B, E–H) experiments.
As TipDC were found to locate to the T-zone during infection and coclustered with other innate cells (33), this raised the possibility that TipDC might also cluster with cDC, and any activity they exert on T cells could be secondary to cDC or in cooperation with cDC. Indeed, analyses of splenic tissue sections by immunohistochemistry after Listeria infection clearly indicated that iNOS-producing TipDC interacted with Th cells (Fig. 6C), and, moreover, three-cell conjugates comprising TipDC, T cells, and cDC could be identified (Fig. 6D). To further explore this, we cocultured naive Th cells with a mixture of TipDC and cDC. We found strong proliferation of Th cells and no apparent suppressive activity of TipDC (Fig. 6E). Moreover, in this coculture system, TipDC cooperated with cDC to induce greater Th1 differentiation when compared with cDC alone (Fig. 6F versus 6A). Most significantly, inhibition of iNOS now blocked Th1 differentiation and resulted in proportions of IFN-γ–secreting cells similar to those seen with cDC alone (Fig. 6F, 6G). Furthermore, iNOS blockade resulted in the generation of more Treg when exogenous TGF-β was added into culture (Fig. 6F, 6H).
Although we found IL-12 had no role in our earlier systems, IL-12 production was far higher in DC isolated from Listeria-infected mice than that in DC from naive animals (data not shown). We therefore tested whether IL-12 could also cooperate with NO in this scenario. In contrast to the previous results (Fig. 3A), neutralization of IL-12 reduced Th1 development while upregulating Treg differentiation in the presence of TGF-β, mimicking the effect observed through iNOS blockade (Fig. 7). Simultaneous inhibition of iNOS and IL-12 showed no further effect in modulating the ratio of Treg to Th1 cells. This suggests that both NO and IL-12R signaling can be equally important and function interdependently in Th cell differentiation in inflammatory situations. Collectively, this shows that NO physiologically produced from TipDC can modulate the differentiation of Th cells and could contribute to determining the Treg/Th1 balance.
FIGURE 7.
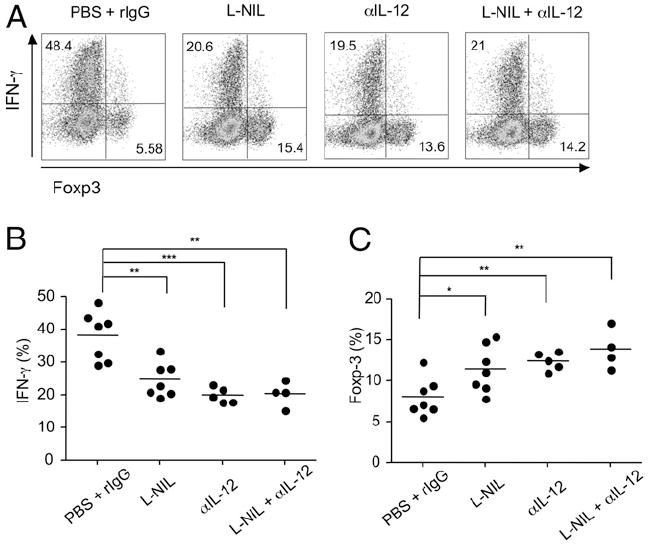
NO and IL-12 interdependently regulate Th1/Treg differentiation in coculture with cDC and TipDC. (A–C) Naive OT-II CD4 T cells were stimulated in coculture with both cDC and TipDC in the presence of OVA peptide and TGF-β (5 ng/ml). Where indicated, L-NIL (100 μM) or anti–IL-12 (10 μg/ml) was added. Expression of Foxp3 and IFN-γ were determined at day 4 after restimulation. Results are representative of two experiments.
Discussion
TGF-β has recently taken center stage in immunology due to its ability to promote Foxp3+ iTreg, but for many years, it was known to have both pro- and anti-inflammatory activities (34, 35). The demonstration that TGF-βR signals to T cells can be modified by the concomitant provision of signals through the IL-6R and IL-4R to enhance differentiation into proinflammatory subsets of Th17 and Th9 cells has provided some rationale for the divergent activities of TGF-β (8-11). We now show that another soluble mediator, NO, can also modulate the action of TGF-β, again away from induction of Treg and in this case toward the expansion or development of another major subset of Th cells, namely Th1. These data further illustrate the proinflammatory activity of TGF-β and provide a new mechanism by which NO, which has been associated with protection against intracellular pathogens, could be a directive influence to allow the efficient development or maintenance of Th1 immunity.
NO is an important effector molecule involved in protection against bacteria, parasites, and some viruses (12-14). It has also been reported to be active in the pathological manifestations of a number of autoimmune diseases such as diabetes, rheumatoid arthritis, and systemic lupus erythematosus (13). Generally, the immune response associated with these infections and diseases has been classified as Th1-type, although roles for Th17 cells are also becoming known. This has led to the suggestion that NO might be active in determining the quality of the T cell response, as well as being a product and effector of IFN-γ action when produced by a T cell. Our data now show a novel activity of NO, interacting with other cofactors such as TGF-β, IL-6, RA, and IL-12, that not only allows greater development of Th1 cells, but also antagonizes Foxp3+ iTreg and Th17 differentiation. Especially important is the concept that as TGF-β is quite ubiquitous and can be produced by many cell types, the default pathway of differentiation for a naive CD4 T cell might be to upregulate Foxp3 and become a Treg. However, with the finding that NO can reprogram TGF-β action toward Th1, and the previous knowledge that IL-6 and IL-4 can also synergize with TGF-β to direct expansion of other lineages, places TGF-β as a central modulator for revealing the activity of other directive soluble factors in dictating the nature of much of T cell immunity. This implies that NO has the ability to promote Th1 responses in several ways that might vary depending on the inflammatory environment and whether other cofactors are produced.
NO regulated Th subset differentiation in an IL-12–dependent fashion when Th cells were cultured with inflammatory DC that secreted high levels of IL-12. This might agree with a previous report that showed that exposure of T cells to a source of NO, when they were cultured under standard Th1-type conditions with rIL-12 and anti–IL-4, potentiated the effects of IL-12 and promoted greater development of Th1 populations (20). In addition, this was also seen with CD8 T cells cultured under similar conditions and was found to be a cGMP-dependent activity mediated by upregulation of IL-12Rβ2 on the T cells (21). In contrast, our results show that NO is also able to regulate the Th1/Treg balance in an IL-12–independent manner when Th cells were cultured with cDC from naive spleen or stimulated in APC-free systems in which IL-12 was limited or absent. Therefore, NO might augment Th1 responses independent of IL-12 when strong innate signals do not drive APC to make this cytokine, whereas in inflammatory situations, an IL-12–dependent activity might dominate. Furthermore, our results indicated that the relative levels of NO and IL-6 can determine Th1 versus Th17 differentiation in the presence of TGF-β. This coincides with a recent report showing that an NO donor, S-nitrosoglutathione, inhibited Th17 responses in an experimental autoimmune encephalomyelitis model (36).
Our results indicated that NO can directly regulate differentiative signals in Th cells. Unlike the inhibitory action shown in endothelial cells (32), NO did not block TGF-βR signaling in Th cells. In contrast, NO functioned at least in part by reinforcing IFN-γR signaling through mobilizing STAT-1. We found that when a naive Th cell was exposed to TGF-β and IFN-γ simultaneously, TGF-βR signals dominated over IFN-γR signals, resulting in Th cells diverging into the Treg lineage. The provision of NO may then allow Th cells to receive quantitatively more IFN-γR/STAT-1–directed signals, resulting in either a direct or indirect activity in blocking Foxp3 induction. As IFN-γ was previously shown to block Foxp3 upregulation through the induction of T-bet (28, 29), this likely explains much of the direct suppression of Foxp3 by NO when IL-12 was limiting. The early IFN-γR signal allows Th cells to express T-bet and leads to further IFN-γ production, which in turn will act to intensify a positive-feedback circuit involving IFN-γR signaling, T-bet induction, and again IFN-γ production, eventually leading to Th1 development in the presence of TGF-β. IL-12 may potentiate this activity by simply promoting initial production of IFN-γ by responding T cells that then synergize with direct signals from NO, and, as described above, NO may also enhance these feedback circuits by upregulating IL-12Rβ2 on the T cells (21).
Interestingly, some NO activities were IFN-independent. NO partially inhibited Treg differentiation in situations in which IFN-γ was neutralized and T-bet was downregulated. Furthermore, the NO activity in blocking Th17 differentiation was also independent of the expression of IFN-γ. Prior reports have suggested that NO derived from myeloid suppressor cells might antagonize IL-2R signaling in T cells (14). As IL-2 is required for the induction of Foxp3 in naive CD4 T cells (37, 38), this could be a separate action by which NO blocks Treg development. Of interest for future studies is how NO modulates multiple signals during T cell priming. NO has been shown in some systems to potentiate TCR signals, promoting proximal phosphorylation of CD3ζ and ZAP70 followed by activation of Ras/ERK pathways (39, 40). It remains to be addressed whether this type of activity on TCR signals by NO might play a role in the IFN-γ–independent activities of NO in suppressing Treg or Th17 differentiation and in enhancing STAT-1 mobilization initiated by IFN-γR signaling. Moreover, it might be possible that the activities of many proteins that are involved in Th differentiation are modulated by direct protein modification with S-nitrosylation (41).
A central question was the contribution of NO produced from physiological sources in modulating Th differentiation. We showed that NO from iNOS-expressing TipDC induced during Listeria infection could affect cDC-regulated naive T cell priming. This subpopulation of inflammatory monocytes was designated as one subset of DC (16), because they express CD11c, albeit at a much lower level than cDC, as well as express MHC class II and many costimulatory molecules. However, we found that TipDC could not support optimal proliferation and accumulation of Th cells stimulated by cognate peptide, which is in contrast to a previous report that TipDC induced robust T cell proliferation in an MLR (16). We believe that this discrepancy is likely due to the allogeneic system involving a mixed population of unseparated T cells as opposed to the peptide-specific stimulation of naive Th cells in our study. Regardless of the weak stimulatory capacity of TipDC for naive CD4 T cells, our data argue that they may actively participate in modulating Th cell responses as third-party accessory cells by coclustering with Th cells and cDC. Through the secretion of NO and IL-12 in close proximity to primed Th cells, TipDC have the ability to potentiate Th1 differentiation. Of note, TipDC are also surrounded by other innate cells, such as NK and NKT cells, that are strong IFN-γ producers (33). Thus, it is tempting to speculate that these cells, by providing IFN-γ, will additionally potentiate the effects of TipDC-derived NO in vivo in driving Th1 differentiation, particularly during infections in which type 1 innate responses are triggered.
In summary, we report a new activity of NO in modulating and synergizing with TGF-β and IFN-γ but antagonizing IL-6 to alter the balance among Treg, Th17, and Th1 cells. These data have significant implications regarding how Th immunity is sustained and perpetuated.
Supplementary Material
Acknowledgments
We thank Y. Park, D. Mucida, and R. Arens for technical assistance and discussion.
This work was supported by National Institutes of Health Grants CA91837 and AI42944 (to M.C.). This is manuscript No. 1111 from the La Jolla Institute for Allergy and Immunology.
Abbreviations used in this article
- cDC
conventional dendritic cell
- cGMP
cyclic GMP
- DC
dendritic cell
- DETA-NONOate
(Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate
- iNOS
inducible NO synthase
- iTreg
inducible regulatory T cell
- L-NIL
N6-(1-iminoethyl)-l-lysine dihydrochloride
- NAP
S-nitroso-N-acetyl-d-l-penicillamine
- NOS
NO synthase
- RA
retinoic acid
- RORγt
retinoic acid-related orphan receptor γt
- TipDC
TNF and inducible NO synthase-producing dendritic cell
- Treg
regulatory T cell
Footnotes
The online version of this article contains supplemental material.
Disclosures The authors have no financial conflicts of interest.
References
- 1.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 2.Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature. 2008;453:1051–1057. doi: 10.1038/nature07036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 4.Sokol CL, Barton GM, Farr AG, Medzhitov R. A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat Immunol. 2008;9:310–318. doi: 10.1038/ni1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bluestone JA, Abbas AK. Natural versus adaptive regulatory T cells. Nat Rev Immunol. 2003;3:253–257. doi: 10.1038/nri1032. [DOI] [PubMed] [Google Scholar]
- 6.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+ CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 8.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 9.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, Martin B, Wilhelm C, Stockinger B. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. 2008;9:1341–1346. doi: 10.1038/ni.1659. [DOI] [PubMed] [Google Scholar]
- 11.Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, Mitsdoerffer M, Strom TB, Elyaman W, Ho IC, et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat Immunol. 2008;9:1347–1355. doi: 10.1038/ni.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 13.Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 14.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 15.Lu L, Bonham CA, Chambers FG, Watkins SC, Hoffman RA, Simmons RL, Thomson AW. Induction of nitric oxide synthase in mouse dendritic cells by IFN-gamma, endotoxin, and interaction with allogeneic T cells: nitric oxide production is associated with dendritic cell apoptosis. J Immunol. 1996;157:3577–3586. [PubMed] [Google Scholar]
- 16.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 17.Aldridge JR, Jr, Moseley CE, Boltz DA, Negovetich NJ, Reynolds C, Franks J, Brown SA, Doherty PC, Webster RG, Thomas PG. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc Natl Acad Sci USA. 2009;106:5306–5311. doi: 10.1073/pnas.0900655106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Trez C, Magez S, Akira S, Ryffel B, Carlier Y, Muraille E. iNOS-producing inflammatory dendritic cells constitute the major infected cell type during the chronic Leishmania major infection phase of C57BL/6 resistant mice. PLoS Pathog. 2009;5:e1000494. doi: 10.1371/journal.ppat.1000494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fischer TA, Palmetshofer A, Gambaryan S, Butt E, Jassoy C, Walter U, Sopper S, Lohmann SM. Activation of cGMP-dependent protein kinase Ibeta inhibits interleukin 2 release and proliferation of T cell receptor-stimulated human peripheral T cells. J Biol Chem. 2001;276:5967–5974. doi: 10.1074/jbc.M009781200. [DOI] [PubMed] [Google Scholar]
- 20.Niedbala W, Wei XQ, Piedrafita D, Xu D, Liew FY. Effects of nitric oxide on the induction and differentiation of Th1 cells. Eur J Immunol. 1999;29:2498–2505. doi: 10.1002/(SICI)1521-4141(199908)29:08<2498::AID-IMMU2498>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 21.Niedbala W, Wei XQ, Campbell C, Thomson D, Komai-Koma M, Liew FY. Nitric oxide preferentially induces type 1 T cell differentiation by selectively up-regulating IL-12 receptor beta 2 expression via cGMP. Proc Natl Acad Sci USA. 2002;99:16186–16191. doi: 10.1073/pnas.252464599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niedbala W, Cai B, Liu H, Pitman N, Chang L, Liew FY. Nitric oxide induces CD4+CD25+ Foxp3 regulatory T cells from CD4+CD25 T cells via p53, IL-2, and OX40. Proc Natl Acad Sci USA. 2007;104:15478–15483. doi: 10.1073/pnas.0703725104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee SW, Park Y, So T, Kwon BS, Cheroutre H, Mittler RS, Croft M. Identification of regulatory functions for 4-1BB and 4-1BBL in myelopoiesis and the development of dendritic cells. Nat Immunol. 2008;9:917–926. doi: 10.1038/ni.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 25.Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007;204:1765–1774. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coombes JL, Siddiqui KR, Arancibia-Cárcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei J, Duramad O, Perng OA, Reiner SL, Liu YJ, Qin FX. Antagonistic nature of T helper 1/2 developmental programs in opposing peripheral induction of Foxp3+ regulatory T cells. Proc Natl Acad Sci USA. 2007;104:18169–18174. doi: 10.1073/pnas.0703642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hall JA, Bouladoux N, Sun CM, Wohlfert EA, Blank RB, Zhu Q, Grigg ME, Berzofsky JA, Belkaid Y. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity. 2008;29:637–649. doi: 10.1016/j.immuni.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bradley LM, Dalton DK, Croft M. A direct role for IFN-gamma in regulation of Th1 cell development. J Immunol. 1996;157:1350–1358. [PubMed] [Google Scholar]
- 31.Stamler JS, Lamas S, Fang FC. Nitrosylation. the prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 32.Saura M, Zaragoza C, Herranz B, Griera M, Diez-Marqués L, Rodriguez-Puyol D, Rodriguez-Puyol M. Nitric oxide regulates transforming growth factor-beta signaling in endothelial cells. Circ Res. 2005;97:1115–1123. doi: 10.1161/01.RES.0000191538.76771.66. [DOI] [PubMed] [Google Scholar]
- 33.Kang SJ, Liang HE, Reizis B, Locksley RM. Regulation of hierarchical clustering and activation of innate immune cells by dendritic cells. Immunity. 2008;29:819–833. doi: 10.1016/j.immuni.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang LC, Tsao LT, Chang CS, Chen CJ, Huang LJ, Kuo SC, Lin RH, Wang JP. Inhibition of nitric oxide production by the carbazole compound LCY-2-CHO via blockade of activator protein-1 and CCAAT/enhancer-binding protein activation in microglia. Biochem Pharmacol. 2008;76:507–519. doi: 10.1016/j.bcp.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 35.Li MO, Flavell RA. Contextual regulation of inflammation: a duet by transforming growth factor-beta and interleukin-10. Immunity. 2008;28:468–476. doi: 10.1016/j.immuni.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 36.Nath N, Morinaga O, Singh I. S-nitrosoglutathione a physiologic nitric oxide carrier attenuates experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2010;5:240–251. doi: 10.1007/s11481-009-9187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL-2 is essential for TGF-beta to convert naive CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol. 2007;178:2018–2027. doi: 10.4049/jimmunol.178.4.2018. [DOI] [PubMed] [Google Scholar]
- 38.Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting Edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J Immunol. 2007;178:4022–4026. doi: 10.4049/jimmunol.178.7.4022. [DOI] [PubMed] [Google Scholar]
- 39.Ibiza S, Pérez-Rodríguez A, Ortega A, Martínez-Ruiz A, Barreiro O, García-Domínguez CA, Víctor VM, Esplugues JV, Rojas JM, Sánchez-Madrid F, Serrador JM. Endothelial nitric oxide synthase regulates N-Ras activation on the Golgi complex of antigen-stimulated T cells. Proc Natl Acad Sci USA. 2008;105:10507–10512. doi: 10.1073/pnas.0711062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ibiza S, Víctor VM, Boscá I, Ortega A, Urzainqui A, O’Connor JE, Sánchez-Madrid F, Esplugues JV, Serrador JM. Endothelial nitric oxide synthase regulates T cell receptor signaling at the immunological synapse. Immunity. 2006;24:753–765. doi: 10.1016/j.immuni.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 41.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.