

Abstract
Structural plasticity of dendritic spines and synapses is a fundamental mechanism governing neuronal circuits and may form an enduring basis for information storage in the brain. We find that the p65 subunit of the nuclear factor-κB (NF-κB) transcription factor, which is required for learning and memory, controls excitatory synapse and dendritic spine formation and morphology in murine hippocampal neurons. Endogenous NF-κB activity is elevated by excitatory transmission during periods of rapid spine and synapse development. During in vitro synaptogenesis, NF-κB enhances dendritic spine and excitatory synapse density and loss of endogenous p65 decreases spine density and spine head volume. Cell-autonomous function of NF-κB within the postsynaptic neuron is sufficient to regulate the formation of both presynaptic and postsynaptic elements. During synapse development in vivo, loss of NF-κB similarly reduces spine density and also diminishes the amplitude of synaptic responses. In contrast, after developmental synaptogenesis has plateaued, endogenous NF-κB activity is low and p65 deficiency no longer attenuates basal spine density. Instead, NF-κB in mature neurons is activated by stimuli that induce demand for new synapses, including estrogen and short-term bicuculline, and is essential for upregulating spine density in response to these stimuli. p65 is enriched in dendritic spines making local protein–protein interactions possible; however, the effects of NF-κB on spine density require transcription and the NF-κB-dependent regulation of PSD-95, a critical postsynaptic component. Collectively, our data define a distinct role for NF-κB in imparting transcriptional regulation required for the induction of changes to, but not maintenance of, excitatory synapse and spine density.
Introduction
Changes in the strength and connectivity of synapses within neural circuits underlie developmental and experience-dependent plasticity. Structural change at the level of synapses and dendritic spines, the primary postsynaptic sites for excitatory input, can impart endurance to these forms of plasticity (Bonhoeffer and Yuste, 2002; Bhatt et al., 2009). Spine morphology and the formation or retraction of dendritic spines is activity responsive to potentiating or depressing stimuli, respectively (Matsuzaki et al., 2004; Nägerl et al., 2004; Zhou et al., 2004; Kopec et al., 2007). Recent in vivo work indicates that changes in dendritic spine density outlast experience and may provide a structural basis for long-term information storage (Hofer et al., 2009; Restivo et al., 2009). In addition, diseases associated with developmental disorders of cognitive function, such as autism, trisomy 21, and fragile X syndrome, are characterized by altered dendritic spine density and morphology (Nimchinsky et al., 2002).
Endogenous transcriptional pathways capable of coordinating the essential upregulation of dendritic spine and excitatory synapse density during development and plasticity are not well characterized. The nuclear factor-κB (NF-κB) transcription factor family is required for synaptic plasticity and learning in multiple organisms including crabs (Freudenthal and Romano, 2000), Drosophila (Heckscher et al., 2007), and rodents (Meffert et al., 2003; Kaltschmidt et al., 2006; O'Riordan et al., 2006; Ahn et al., 2008), and genes containing NF-κB regulatory elements are selectively enhanced by learning (Levenson et al., 2004; O'Sullivan et al., 2007), further indicating the significance of transcriptional regulation by NF-κB during plasticity. Nonetheless, relatively little is known regarding cellular pathways that could underlie this function of NF-κB.
NF-κB transcription factors function as dimers containing one or more of five mammalian subunits and can be activated by degradation of the inhibitor of NF-κB, IκB. Three NF-κB family members, including p50 (Kassed et al., 2002), c-Rel (Levenson et al., 2004; O'Riordan et al., 2006; Ahn et al., 2008), and RelA or p65 (Meffert et al., 2003), are implicated in learning and memory. The p65:p50 heterodimer of NF-κB is localized in the cytoplasm and synapses of hippocampal neurons where it can be activated by excitatory stimulation (Kaltschmidt et al., 1993; Suzuki et al., 1997; Wellmann et al., 2001; Meffert et al., 2003). p65-deficient mice lack synaptic NF-κB and demonstrate significant deficits in hippocampal-dependent learning (Meffert et al., 2003).
We now report a requirement for the p65 subunit of NF-κB in regulating dendritic spine and synapse density in hippocampal neurons both during development and in response to stimuli that increase the demand for new synapses in mature neurons. These findings demonstrate a conserved endogenous regulatory pathway used for both initial spinogenesis and later structural plasticity. Our data also indicate that NF-κB positively regulates dendritic spine head size and the amplitude of synaptic responses. The specialized scaffold protein, postsynaptic density protein-95 (PSD-95), is a critical NF-κB target gene. These findings expand our understanding of NF-κB functions in learning and other forms of adaptive plasticity and reveal a novel pathway driving transcriptional regulation of spine and excitatory synapse density.
Materials and Methods
The care and use of animals met all guidelines of the local Institutional Animal Care and Use Committee (The Johns Hopkins University School of Medicine).
Expression constructs and lentiviral preparation
Wild-type and mutant human p65 expression constructs were cloned by in-frame insertion into the Clontech C1 vector at the C terminus of either enhanced green fluorescent protein (GFP) or mCherry (gift from R. Tsien, University of California at San Diego, La Jolla, CA). The p65ΔTAD construct encodes p65 residues 2–442 fused to GFP; the p65 DNA binding domain mutant (p65R33,35A) was made using site-directed mutagenesis of full-length p65 by PCR amplification with primers (F-GCAGCGGGGCATGGCCTTCGCCTACAAGTGCGAGG; R-CCTCGCACTTGTAGGCGAAGGCCATGCCCCGCTGC). EBFP2 (Ai et al., 2007) followed by an F2A sequence (Szymczak et al., 2004) and an inducible Cre recombinase fused to a mutant estrogen receptor fragment (CreERT2) responding specifically to 4-hydroxy tamoxifen (OHT) (Feil et al., 1997) were inserted into a replication-incompetent lentiviral vector to produce EBFP2-F2A-CreERT2 (supplemental Fig. S1, available at www.jneurosci.org as supplemental material); the F2A sequence causes the ribosome to skip the formation of a peptide bond producing a 1:1 ratio of EBFP2 and CreERT2. Lentiviral stocks were prepared as previously described (Lois et al., 2002). Cre-IRES-dsRed plasmid was a gift from A. Kolodkin (Johns Hopkins University School of Medicine, Baltimore, MD), and GFP-PSD-95 was a gift from R. Huganir (Johns Hopkins University School of Medicine, Baltimore, MD).
Small interfering RNA (siRNA) oligos (Dharmacon) targeting PSD-95 (Elias et al., 2006) used the sense sequence: 5′-TCACGATCATCGCTCAGTATA-3′ or control oligo with every fourth base replaced with adenosine: TCAAGATAATCACTCAGTAAA.
Hippocampal cultures and stimulation
Dissociated hippocampal cultures were prepared from postnatal day 0 (P0) male and female mice as previously described (Meffert et al., 2003) and plated onto poly-l-lysine-coated glass-bottomed dishes (MatTek) at a density of 100,000 cell/cm2 (low density) or 280,000 cell/cm2 (high density). Cultures were maintained in Neurobasal A medium (Invitrogen; 10888) with B27 Supplement (Invitrogen; 17504-44). Neurons were transiently transfected with Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen) 16–48 h before experimentation. Cultures from RelAF/F mice were infected at a multiplicity of infection of 5–10 at 2 d in vitro (DIV 2) with EBFP2-F2A-CreERT2-expressing lentivirus. CreERT2 was activated by OHT (400 nm; Sigma-Aldrich; H6278) addition at DIV 5 or as required. This interval ensured that neurons did not experience toxicity because of concomitant viral infection and p65 loss.
To induce demand for new synapses, neurons were incubated for 24 h with 25 μm bicuculline methobromide (Enzo Life Sciences; 550-040-M050) in growth media or 48–72 h with 0.3–1.7 μm 17β-estradiol (E2) (Sigma-Aldrich; E4389) in growth media with reduced B27. Neurons were imaged in Tyrode's buffer (in mm: 119 NaCl, 4.5 KCl, 2 CaCl2, 0.5 MgCl2, 25 glucose, 10 HEPES, 0.01 glycine, pH 7.33) with or without bicuculline and E2. NF-κB reporter assays were conducted as previously described (Pomerantz et al., 2002; Meffert et al., 2003), and neurons were stimulated with bicuculline (25, 50 μm) or 17β-estradiol (0.3, 1.7 μm).
Imaging and quantification
Confocal images of pyramidal neurons (determined by morphology) in 0.4–1.0 μm Z sections were acquired using a 63×, 1.4 numerical aperture, oil-immersion lens and 0.7 or 4× optical zoom on a LSM5 Pascal system (Carl Zeiss) at 37°C in Tyrode's buffer (as above with the addition of 2 mm EGTA). GFP was excited at 488 nm and emissions collected at 505–530 nm; mCherry was excited at 543 nm and emissions collected above 560 nm. Laser power and gain were adjusted to minimize phototoxicity and avoid saturation. All experiments were from a minimum of three independent cultures, with at least two dishes per condition per day and no more than five neurons per dish; the experimenter was blinded to conditions during acquisition and analysis.
Z-stacks containing the entire neuron or process of interest were analyzed using Pascal and ImageJ software. Sholl analysis from Z-compressed projections began with a circle of 45 μm diameter with subsequent dendritic intersections counted using circles of increasing 30-μm-diameter increments. Spines were defined as protrusions 0.4–2.5 μm in length with or without a head. Secondary or tertiary dendritic branch segments were chosen to ensure a uniform population. Spine-associated synapses were defined as Bassoon puncta that colocalized in x, y, and z dimensions with a dendritic spine from the transfected neuron. Soma area was measured by tracing the outline of the soma from a Z-compressed stack. Automated analysis of dendritic spine morphology (Imaris 6.3; Bitplane) determined spine head size by measuring a terminal-fitted sphere. A 0.15 μm threshold was used for the lower limit of spine diameter to avoid artifactual measurements.
Neurons expressing Cre recombinase constructs were binned by fluorescent intensity from the associated fluorophore. Neurons infected with EBFP2-F2A-CreERT2 were only included in analysis if a cytoplasmic or nuclear region of interest (ROI) returned a blue fluorescence value of 50 or greater. Neurons transfected with Cre-IRES-dsRed were included in the analysis if they had a red fluorescence value >300. Imaging conditions for fluorophores coexpressed with Cre were kept constant to permit comparison of Cre expression levels across experiments.
Analysis of spine density in p65 rescue experiments was only conducted on neurons expressing low (nontoxic) levels of p65 or p65 mutants as monitored by ROI and by lack of concentration in the nucleus relative to the cytoplasm.
Analysis of p65 concentration in dendritic spines was conducted on low expressing neurons with fluorescence intensity determined using ROI measurement in confocal images of the spine head and adjacent dendritic segment. Enrichment was determined using the ratio of GFP (alone or fused to p65) and mCherry fluorescent intensities according to the following equations:
 |
 |
Reporter assays
NF-κB-dependent transcription was assayed by transient transfection of an NF-κB luciferase reporter (Igκ2-IFN-LUC) containing two or three copies of the Ig light chain κB site, or a mutant reporter with four residues of the NF-κB consensus binding sites mutated as previously described. Cotransfection of the pCSK-lacZ vector, which constitutively expresses β-galactosidase and is not regulated by NF-κB, served to normalize transfection efficiency and extract recovery for each sample in all reporter assays. Each reporter experiment included extracts from cells transfected with pcDNA3 alone as a reference control. Cultures were lysed using 1× lysis buffer (reporter lysis buffer; Promega), and luciferase (Promega) and chemiluminescent β-gal (Roche) reporter assays were conducted 46 h after transfection and 3–4 h after stimulation according to manufacturer's instructions using a plate-reading luminometer (PerkinElmer). Samples were compared by subtracting the background activity of the reference control, and then normalizing the luciferase activity of each sample to its β-gal activity. When required, fold induction was calculated by dividing normalized stimulated samples by normalized unstimulated samples.
Immunocytochemistry and immunoblotting
Immunoblotting.
Primary neuronal cultures were lysed in buffer containing 1% Triton X-100, 0.5% sodium deoxycholic acid, and 0.1% SDS, subjected to Bradford protein assay, and equivalent total protein amounts resolved on SDS-PAGE gels, transferred to PVDF (polyvinylidene difluoride) membrane and probed using the following primary antibodies: anti-p65 (Santa Cruz Biotechnology; sc372; 1:4000), anti-PSD-95 (NeuroMab; K28/43; 1:500), anti-IκBα (Santa Cruz Biotechnology; sc374; 1:3000), anti-EIF4E (Cell Signaling; 9742; 1:1000), anti-GFP (NeuroMab; N86/8; 1:500), anti-GAPDH (Calbiochem; CB1001; 1:50,000), and anti-β-tubulin (University of Iowa Developmental Studies Hybridoma Bank; E-7; 1:500).
Immunocytochemistry.
DIV 16 neurons were fixed with 4% paraformaldehyde with 4% sucrose for 30 min, permeabilized with 0.2% Triton X-100 for 30 min, blocked for 1 h in 10% BSA, and incubated with primary antibodies: anti-GFP (Invitrogen; A11120, A11122; 1:500), anti-dsRED (Clontech; 632496; 1:600), anti-Bassoon (Nventa Biopharmaceuticals; VAM-PS003; 1:600), and anti-vesicular GABA transporter (VGAT) (Synaptic Systems; 151003; 1:1000). Alexa Fluor 488 and 568 secondary antibodies (Invitrogen; 1:1000) were incubated for 1 h at room temperature in 10% BSA and mounted with 0.1 m n-propyl gallate in 50% glycerol.
In vivo spine analysis and electrophysiology
The RelAF/F mouse line was crossed to the SLICK-V line, which exhibits sparse neuronal labeling of yellow fluorescent protein (YFP) and CreERT2 from bicistronic Thy1 promoters (Young et al., 2008). Mice were maintained as homozygous for the RelAF/F locus and heterozygous for SLICK-V. OHT (0.25 mg) was delivered to mice by subcutaneous injection for 4 consecutive days starting at P7 or P8. Control mice for spine analysis received vehicle (sunflower seed oil) alone.
Immunofluorescence.
At P12, brains were removed and fixed with 4% paraformaldehyde with 4% sucrose in PBS for 4 h at 4°C. Brains were then cryoprotected in 4% sucrose in PBS at 4°C overnight or until brains sank and then were embedded in Tissue-Tek OCT (Sakura; 4583) and frozen. Forty micrometer coronal hippocampal cryostat sections were mounted on slides and postfixed with 4% paraformaldehyde with 4% sucrose for 5 min, followed by blocking and overnight incubation at 4°C with chicken anti-GFP antibody (Aves; GFP-1020). Secondary detection was by a goat anti-chicken Alexa Fluor 488 antibody (Invitrogen; A11039; 1:500), and slides were then mounted with 2.5% DABCO (diazabicyclo[2.2.2]octane) (Sigma-Aldrich; D2522) in Fluoromount G (Beckman Coulter; 731604). The experimenter was blinded during data analysis.
Electrophysiology.
Coronal forebrain slices were prepared from P11–P13 OHT-treated mice as previously described (De Biase et al., 2010). Whole-cell recordings from YFP-positive or control hippocampal pyramidal neurons were performed using a 2.5–3.3 MΩ glass electrodes filled with the following (in mm): 100 CsMeS, 20 TEA (tetraethylammonium)-chloride, 20 HEPES, 1 MgCl2, 10 EGTA, 2 sodium-ATP, and 0.2 sodium-GTP, pH 7.3, 295 mOsm. Miniature EPSC (mEPSC) recordings were made without series resistance compensation and were performed in the presence of the following: TTX (1 μm), gabazine (5 μm), and (RS)-3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (RS-CPP) (NMDA receptor antagonist; 5 μm). Responses were recorded using a MultiClamp 700A amplifier (Molecular Devices), filtered at 3 kHz, digitized at 50 kHz, and recorded to disk using pClamp9.2 software (Molecular Devices). Data were analyzed off-line using Clampfit (Molecular Devices), Origin (OriginLab), and Mini analysis (Synaptosoft) software. Input resistance and membrane capacitance were calculated from a 2 mV hyperpolarizing step from a holding potential of −70 mV. mEPSC frequency (>5 pA amplitude, <1 ms rise time) was quantified by monitoring activity during continuous recording for at least 8 min. The experimenter was blinded to the condition during data analysis.
RNA analysis
Total RNA was extracted from murine hippocampi or hippocampal cultures throughout development using Tri-Reagent (Molecular Research Center). RNase protection assay was performed with 5 μg of RNA using a custom Riboquant MultiProbe set (BD Pharmingen) according to the manufacturer's instructions.
Statistical and bioinformatics analysis
Neuron dishes were randomly assigned to treatment conditions and the experimenter was blinded during data acquisition and analysis. Graphs illustrate the arithmetic mean and error bars are SEM. For statistical analyses, two-tailed t tests were used (paired or unpaired) with α = 0.05, unless otherwise noted. Data for spine density are not normally distributed (Fmax test); therefore, analyses were conducted with correction for unequal variance. To maintain an αexperiment-wise = 0.05 with multiple pairwise comparisons for spine density experiments, α = 0.01 was set for each t test. The α level was calculated by , where c is the number of pairwise comparisons. Statistical significance for mEPSC amplitudes was determined using the Kolmogorov–Smirnov (K-S) test.
Putative consensus NF-κB binding sites in the promoter region of Dlg4 were determined using TESS: Transcriptional Element Search System (http://www.cbil.upenn.edu/tess/) and analyzed for conservation between Mus musculus, Rattus norvegicus, and Homo sapiens.
Results
NF-κB regulates dendritic spine and excitatory synapse number
The potential for NF-κB-dependent regulation of dendritic spines and synapses in the mammalian brain was initially evaluated by testing the effect of exogenous p65 expression in hippocampal neurons. Enhanced GFP-tagged p65 (GFPp65) or GFP was expressed in low-density hippocampal cultures at DIV 16 for 24 h before immunostaining for GFP and the presynaptic marker Bassoon, and confocal imaging of dendritic spines and synapses. Expression of exogenous p65 or GFPp65 activates NF-κB and induces NF-κB-dependent gene expression (Meffert et al., 2003) (see Fig. 5A), presumably by overwhelming endogenous production of the inhibitor of NF-κB, IκB. GFPp65 expression in pyramidal neurons increased the density of dendritic spines by 46% compared with GFP (GFPp65: 4.1 ± 0.15 spines/10 μm, n = 95 dendrites; GFP: 2.8 ± 0.09, n = 183 dendrites; p = 1.3 × 10−11). Spine-associated synapses, which are predominantly glutamatergic and excitatory, were increased 106% by GFPp65 expression compared with GFP (GFPp65: 3.3 ± 0.21 synapses/10 μm; GFP: 1.6 ± 0.16; p = 1.8 × 10−9) (Fig. 1A,C). Although p65 expression increased both spine and excitatory synapse density, the proportionally larger increase in synapses was accounted for by an increase in the average number of presynaptic puncta on spines participating in synapses (GFPp65, 1.32 puncta/spine; GFP, 1.17 puncta/spine; p = 0.001) as well as an increase in the percentage of dendritic spines opposed by presynaptic puncta (GFPp65: 66.1 ± 2.6%; GFP: 49.1 ± 3.75%; p = 6.8 × 10−5).
Figure 5.
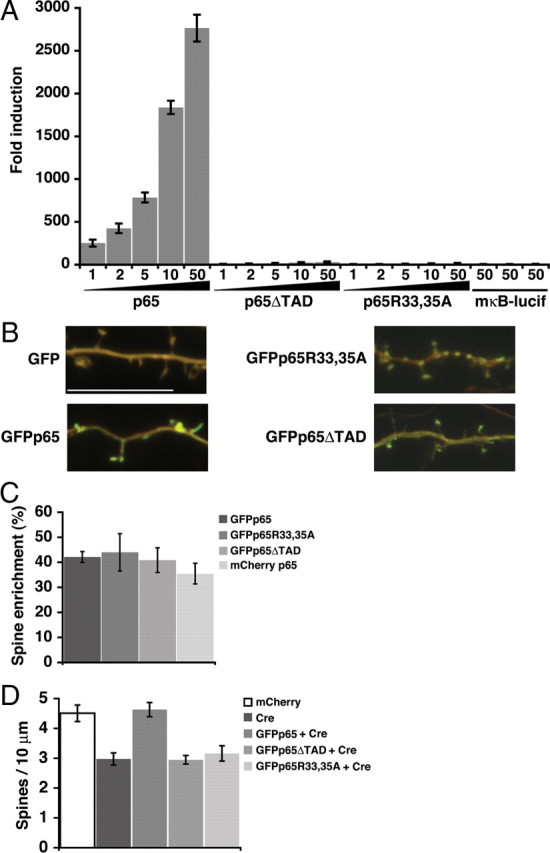
Requirement for NF-κB-dependent gene expression. A, Fold induction in an NF-κB luciferase reporter assay of 293T cells transfected with the indicated amount (in nanograms) of GFP-tagged wild-type p65 or transcriptionally inactive mutants (p65ΔTAD, p65R33,35A). A reporter containing a mutant NF-κB binding sequence (mκB-lucif) is not activated by transfection with 50 ng of p65, p65ΔTAD, or p65R33,35A. B, Representative confocal projections of dendrites from pyramidal neurons coexpressing mCherry and the indicated GFP or GFP-tagged p65 construct; red and green channels are overlaid to permit visualization of spine enrichment. Scale bar, 10 μm. C, Regions of interest in dendritic spines and shafts were quantitated to calculate normalized dendritic spine enrichment values and percentage enrichment relative to GFP (see Materials and Methods); wild-type p65 and mutant constructs did not significantly differ in spine enrichment. D, Expression of Cre (Cre-IRES-dsRed) in DIV 16 hippocampal pyramidal neurons resulted in a decrease in dendritic spine density that could be rescued by coexpression of GFPp65 but not by the transcriptionally inactive mutants of p65, GFPp65ΔTAD, or GFPp65R33,35A. Dendritic spine density in rescued neurons expressing GFPp65 plus Cre did not significantly differ from control neurons expressing mCherry alone (see text). Data are means ± SEM.
Figure 1.

Exogenous expression of NF-κB increases spine and excitatory synapse density. A, B, Confocal projections of dendrites and dendritic spines from DIV 16 murine hippocampal pyramidal neurons plated at low-density and immunostained for GFP and either a presynaptic marker Bassoon (A) or a marker of inhibitory presynaptic terminals VGAT (B). Neurons express either GFP or GFPp65 as indicated. Scale bar, 20 μm. C, Dendritic spine density and spine-associated predominantly excitatory synapses (colocalized Bassoon puncta) are both significantly increased (*p < 0.001) (see text) in DIV 16 pyramidal neurons expressing GFPp65. Inhibitory synapse density (VGAT staining) is not significantly different. Error bars indicate SEM.
In contrast to the marked effects on dendritic spines and associated excitatory synapses, p65 did not regulate inhibitory synapse density. Quantification of inhibitory synapse number by immunostaining for the VGAT revealed no significant differences in hippocampal pyramidal neurons expressing either GFP or GFPp65 (Fig. 1B,C).
Endogenous NF-κB regulates dendritic spine development
To determine whether NF-κB was required at endogenous levels for regulating dendritic spine and synapse growth, we used a previously characterized mouse strain carrying a loss-of-function allele of the RelA gene (encoding p65) in which exons 7–10 are flanked by loxP sites (Geisler et al., 2007) (RelAF/F). Effective recombination at the RelA site was achieved by infecting hippocampal cultures with lentivirus encoding OHT-inducible Cre recombinase (CreERT2) coupled to the expression of equivalent levels of enhanced blue fluorescent protein 2 (EBFP2) (Ai et al., 2007) by a picornavirus-derived F2A sequence (Szymczak et al., 2004) (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Increasing titers of viral infection resulted in dose-dependent loss of p65 protein in the presence, but not in the absence, of OHT (Fig. 2A). CreERT2-mediated loss of endogenous p65 significantly diminished the induction of NF-κB-dependent gene expression in hippocampal cultures stimulated with bicuculline (50 μm) to enhance excitatory activity; NF-κB reporter activity in stimulated cultures lacking p65 was not significantly different from unstimulated p65-deficient conditions (Fig. 2B) (n = 4; p = 0.707).
Figure 2.

Endogenous NF-κB regulates spine density and morphology during spinogenesis. A, Dose-dependent excision of p65 by CreERT2 in the presence of OHT. The indicated microliters of lentivirus coexpressing CreERT2 and EBFP2 were added to DIV 9 cultured RelAF/F hippocampal neurons, followed by 72 h of OHT exposure beginning at DIV 12; lysates were immunoblotted for p65 protein and EBFP2. The asterisk indicates p65 peptide remaining after excision. B, Loss of p65 (OHT) decreases NF-κB transcriptional activity in unstimulated cultures and prevents an increase in NF-κB-dependent transcription in stimulated conditions [bicuculline (Bic), 50 μm] as determined by luciferase reporter assay. C, D, p65 deficiency did not significantly affect dendritic arbor complexity as determined by Sholl analysis, or soma size of hippocampal pyramidal neurons at any tested DIV (ANOVA). E, Representative confocal projections of dendrites from CreERT2-expressing pyramidal neurons during maturation. Neurons were either exposed to OHT at DIV 5 to activate CreERT2 or untreated. Scale bar, 10 μm. F, Quantitation of dendritic spine density from pyramidal neurons treated as in E. Spine density was significantly decreased by loss of p65 (DIV 12, 14, 16, *p ≤ 0.005) (see text) except in mature cultures (DIV 19–21). Neurons were grown in high-density cultures. G, Delaying the time of p65 excision by exposing cultures to OHT at DIV 13–16 (OHT late), compared with DIV 5–8 (OHT early), did not uncover a role for NF-κB in regulating dendritic spines in mature cultures. H, Culture-wide p65 deficiency by viral CreERT2 introduction (Population) or focal p65-deficiency by transfected Cre (Individual) both similarly reduced dendritic spine density in DIV 16 pyramidal neurons. I, J, Cumulative distribution curves for total protrusion length (p = 4.2 × 10−9) (I) and spine diameter (J). K, Loss of p65 (OHT) significantly decreased average spine head volume in DIV 16 hippocampal pyramidal neurons (*p < 0.001) (see text) using the data in J. Error bars indicate SEM. See also supplemental Figure S1 (available at www.jneurosci.org as supplemental material).
NF-κB transcription factors regulate aspects of cell growth and proliferation in many tissues. To examine potential roles for NF-κB in growth and metabolism of hippocampal neurons, the effect of p65 deficiency on cell soma size and dendrite arbor complexity was evaluated at distinct time points that span the early stages of rapid dendritic spine and synapse formation, concluding with relatively mature neurons in which changes in spine and synapse density have stabilized. Cultures were infected at DIV 2 with CreERT2 lentivirus, treated with or without OHT at DIV 5, and transfected with mCherry fluorophore for morphological visualization 24 h before confocal imaging. Loss of p65 did not significantly affect cell soma size or dendrite complexity of pyramidal neurons at any tested culture DIV (Fig. 2C,D). In contrast, analysis of dendritic spine density showed significantly fewer spines in neurons lacking p65 at all tested time points until ≥DIV 19 (Fig. 2E,F) [DIV 12: n = 68 dendrites (−OHT), 38 (+OHT), p = 7.3 × 10−4; DIV 14: n = 82 (−OHT), 82 (+OHT), p = 2.0 × 10−5; DIV 16: n = 67 (−OHT), 44 (+OHT), p = 0.006; DIV 19–21: n = 117 (−OHT), 121 (+OHT), p = 0.57]. Although spine density did increase during DIV 12–19 in p65-deficient neurons, it lagged behind the spine density of p65 wild-type neurons with ∼32% fewer spines/10 μm at DIV 14 and 16 (Fig. 2E, F).
The possibility that the lack of p65 effect on spine density in older cultures could be attributable to a longer period available for compensation was evaluated by comparison of spine density in cultures made p65 deficient at either early or late time points. Cultures made p65 deficient by OHT addition at either DIV 5–8 (early) or DIV 13–16 (late) time points showed no difference in spine density, compared with p65 wild-type controls, when evaluated at DIV 19–21 (Fig. 2G); this finding further supports differential regulation of basal spine density by p65 during neuronal maturation.
Dendritic spine regulation is cell autonomous
Lentiviral infection was highly efficient and achieved Cre recombinase expression in ≥95% of cultured hippocampal cells. To determine whether the effects of p65 deficiency on dendritic spine density were cell autonomous or, alternatively, attributable to potential effects on either the network activity of cultures or loss of p65 in glial cells, isolated neurons in cultures from RelAF/F mice were made p65 deficient by transient transfection. Cultures were cotransfected with GFP and either mCherry or constitutively active Cre recombinase coexpressing the dsRed fluorophore (Cre-IRES-dsRed) and were analyzed for spine density 48 h later. Transient transfection achieved expression in 1–2% of cells and readily permitted analysis of single isolated neurons. This duration of Cre expression was sufficient to cause a loss of NF-κB-dependent gene expression as monitored by luciferase assay (data not shown). The loss of dendritic spines generated by p65 deficiency in either a population (lentiviral) or individual cell (transfection) was not significantly different (Fig. 2H) (30.7 ± 7.5; 34.0 ± 4.5% loss of spines, respectively). This indicates that cell-autonomous actions in the postsynaptic neuron can mediate p65-dependent regulation of dendritic spine density.
NF-κB deficiency decreases dendritic spine head size
The size of dendritic spines at glutamatergic synapses is highly variable with a robust positive correlation between spine size and both receptor content and excitatory synapse strength (Bonhoeffer and Yuste, 2002; Holtmaat and Svoboda, 2009). A comparison of spine morphology in control or p65-deficient (OHT) DIV 16 pyramidal neurons revealed that NF-κB modestly shortens dendritic protrusion length (spines and filopodia) (Fig. 2I) while enhancing the diameter and volume of dendritic spine heads (Fig. 2J,K). Loss of p65 decreased average spine head volume by 33.9% (Fig. 2K) [control (−OHT): 0.081 ± 0.0028 μm3, n = 1133 spines; p65-deficient (OHT): 0.0537 ± 0.0034, n = 516; p = 4.7 × 10−10]. There was a left shift in the cumulative distribution of spine head diameter (Fig. 2J), indicating that all classes of dendritic spines in p65-deficient pyramidal neurons have smaller head diameters. Collectively, these data show that loss of NF-κB function leads to a decrease in dendritic spine density, whereas the remaining spines are longer in length and have diminished spine head size. These effects are consistent with weakened excitatory synaptic connectivity and fewer mature dendritic spines in the absence of p65.
NF-κB regulates dendritic spine density and mEPSC amplitude in vivo
To examine the role of NF-κB in vivo, we generated a conditional transgenic line by crossing RelAF/F mice with the SLICK-V line that expresses YFP and CreERT2 from bicistronic Thy1 promoters in a subset (∼5% by positional variegation) of hippocampal pyramidal neurons (Young et al., 2008). Recombination could then be induced in vivo by treating animals with OHT. This allowed morphology of individual neurons to be distinguished and for individual neurons to be manipulated in a wild-type background.
Endogenous NF-κB regulates spine density in the hippocampus during periods of rapid synapse formation. The time course of synaptogenesis in our cultures resembled the rate and duration of synaptogenesis in vivo as has been previously described (Steward and Falk, 1991). RelAF/F × SLICK-V pups were treated with OHT or vehicle several days before spine analysis at P12 and successful recombination verified by PCR assay (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Consistent with our in vitro studies, loss of p65 in hippocampal pyramidal neurons significantly reduced spine density by 32.8% in vivo (control: 11.6 ± 0.39 spines/10 μm, n = 35 dendrites; OHT: 7.8 ± 0.33, n = 36) (Fig. 3A,B). This experiment indicates that NF-κB regulates spine density during a period of rapid synaptogenesis in vivo as well as in a culture system.
Figure 3.

Endogenous NF-κB regulates spine formation and mEPSC amplitude during synaptogenesis in vivo. A, Representative confocal projections of dendrites from YFP/CreERT2-expressing pyramidal neurons from hippocampal sections of RelAF/F × SLICK-V mice that were treated with OHT or vehicle. Scale bar, 10 μm. B, Loss of p65 (OHT) significantly decreases spine density at P12 in vivo (*p = 1.8 × 10−10; control, n = 35 dendrites; OHT, n = 36 dendrites; 3 brains per condition). C, Whole-cell voltage-clamp recordings in acute brain slices from RelAF/F × SLICK-V mice treated with OHT; shown are representative traces from YFP/CreERT2 (−) and YFP/CreERT2 (+) hippocampal pyramidal neurons; mEPSCs identified by asterisks are shown at expanded timescale below. D, Top, Overlay of average mEPSCs from five representative YFP/CreERT2 (−) and five representative YFP/CreER T2 (+) neurons. Bottom, Average mEPSC for all cells in each condition. E, Cumulative percentage plot of the mEPSC amplitude from all recorded neurons [n = 12 YFP/CreERT2 (−) and 15 YFP/CreER T2 (+) neurons; K-S test, p = 3.7 × 10−17].
To directly test whether the structural changes in spine density and morphology correlate with reduced synaptic function, we performed whole-cell voltage-clamp recordings from hippocampal pyramidal neurons in acute brain slices from OHT-treated RelAF/F × SLICK-V mice during a developmental period of active hippocampal synaptogenesis. mEPSC recordings in the presence of TTX, gabazine (GABA receptor antagonist), and RS-CPP (NMDA receptor antagonist) were made from SLICK-V YFP/CreERT2-positive neurons (p65-deficient; n = 15) and YFP/CreERT2 negative neurons (p65-wild-type; n = 12) within the same brains. The amplitude of mEPSCs recorded from YFP/CreERT2-positive neurons was significantly reduced compared with the amplitude of mEPSCs from YFP/CreERT2-negative neurons (p = 3.7 × 10−17) (Fig. 3C–E), indicating that endogenous NF-κB positively regulates AMPA receptor responses. The observed enhancement of synaptic function is consistent with our data that NF-κB positively regulates spine head volume (Fig. 2K).
Given that a decrease in spine density was observed in OHT-treated YFP/CreERT2-positive neurons, a reduction in the frequency of mEPSCs might also be anticipated. Recordings from YFP/CreERT2-positive neurons demonstrated a trend toward reduced mEPSC frequency, but these results did not reach significance. One potential explanation for this finding lies in the pronounced cell-to-cell variability of mEPSC frequency in acute brain slices at this developmental age. Indeed, the coefficient of variation for mEPSC frequency within control (YFP/CreERT2-negative) neurons (CV = 0.90) far exceeds that for mEPSC amplitude (CV = 0.31), indicating that experimental sensitivity for changes in amplitude is much greater.
Basal NF-κB activity is highest before and during rapid synapse development
Under our culture conditions, the rate of new spine formation was greatest before DIV 17 and increases in spine density began to plateau after DIV 18. To determine whether and how endogenous NF-κB activity changed throughout spine and synapse development, we assayed the activity of an NF-κB-responsive reporter, Igκ2-IFN-LUC, during culture maturation at DIV 3, 5, 7, 10, 16, and 20 (Fig. 4A). Specificity for NF-κB was demonstrated with a reporter containing mutant κB sites (mtκB), which did not undergo significant change in activity during culture maturation. Basal NF-κB-dependent reporter activity was lowest at DIV 20, and reporter values for all other days were normalized to the DIV 20 value (set as 1) for comparison. During the period of rapid new spine and synapse formation, DIV 7, 10, and 16, NF-κB activity was 8.2, 4.6, and 4.9 times higher, respectively, than at DIV 20 (n = 3 independent culture time courses each with three wells per condition; p = 0.018, 0.006, and 0.047, respectively). Interestingly, relative NF-κB activity was highest at DIV 5, before the detection of visible dendritic spines in the period leading up to rapid synaptic development. Immunoblot of whole-cell lysates demonstrated no significant change in either p65 or IκBα protein content during culture development (Fig. 4B), indicating that the developmental changes in NF-κB-dependent gene expression likely result from increased posttranslational activation of the transcription factor. As IκBα is a gene target of NF-κB, activated NF-κB regenerates steady-state levels of the IκB inhibitor over time.
Figure 4.

Response to increased synaptic demand in mature neurons requires NF-κB. A, NF-κB activation was monitored by luciferase assay at time points in culture maturation preceding (DIV 3, 5), during (DIV 7, 10, 16), and at the conclusion of rapid spine and synapse formation (DIV 20). Comparison of activity from a reporter containing a wild-type NF-κB consensus binding sequence (wtκB) or a mutant binding sequence (mtκB) demonstrates specificity for NF-κB. Relative activity was plotted by normalizing fold induction to DIV 20 levels, which were the lowest observed. B, Equivalent total protein amounts from culture lysates at the indicated DIV were immunoblotted for p65, IκBα, and eIF4E (loading control); quantitated p65 levels relative to DIV 5 from two independent experiments (average ± SEM) indicate minimal changes in p65 expression. C, The effects of blockade of NMDA and AMPA receptors with APV (100 μm) and CNQX (40 μm) on NF-κB transcriptional activity at DIV 5 before rapid synapse formation and at DIV 16 (*p = 0.001) during rapid synapse formation. D, Loss of p65 after OHT addition does not decrease spine density in CreERT2-expressing DIV 20 cultured hippocampal pyramidal neurons from RelAF/F mice. Dendritic spine density in DIV 20 pyramidal neurons was increased in response to E2 (0.3 μm; 48–72 h) or bicuculline (25 μm; 24 h) by 34.1 ± 7.7 or 25.5 ± 3.6%, respectively, from untreated. Loss of p65 prevented neurons from increasing spine density in response to either E2 or bicuculline. Differences in dendritic spine density in neurons exposed to E2 or bicuculline in the presence or absence of OHT were highly significant (E2, *p = 0.003; Bic, *p = 5.5 × 10−7). E, NF-κB-dependent gene expression as determined by luciferase assay with wild-type (wtκB) or mutant (mtκB) NF-κB reporters in neurons stimulated with E2 or bicuculline for the indicated dose and duration; fold induction is relative to unstimulated. F, Loss of p65 (OHT) eliminated the induction of NF-κB activity by E2 in CreERT2-expressing hippocampal cultures from RelAF/F mice. Data are means ± SEM.
Excitatory neurotransmission is a major NF-κB-activating stimulus in healthy brain; however, the elevated NF-κB levels at DIV 3 and DIV 5 occur before robust excitatory synapse formation. Inhibiting excitatory glutamatergic transmission decreased NF-κB activation at DIV 16 but not at DIV 5 (Fig. 4C). These results show that NF-κB activity is elevated during synaptogenesis (at DIV 16) by excitatory neurotransmission.
Increased synaptic demand reinstates the requirement for NF-κB
Loss of NF-κB function during the period of rapid dendritic spine and synapse development significantly attenuated basal dendritic spine density (DIV 12–16) (Fig. 2E,F), whereas no effect was observed in mature cultures when dendritic spine density had mostly plateaued (Figs. 2F, 4D) (−OHT, n = 102 dendrites; OHT, n = 65). Under conditions of plasticity, such as during learning, mature neurons can significantly increase the number of dendritic spines and excitatory synapses from basal levels. To test whether increasing the demand for new synapses might reinstate a requirement for NF-κB in mature neurons, DIV 20 cultures were exposed to low-dose bicuculline (25 μm; 24 h; −OHT, n = 163 dendrites; OHT, n = 129) or 17β-estradiol (E2; 0.3 μm; 48–72 h; −OHT, n = 33; OHT, n = 32). Short-term bicuculline exposure (Papa and Segal, 1996) and E2 (Murphy and Segal, 1996; Liu et al., 2008) have both been previously characterized to enhance dendritic spine and synapse numbers in hippocampal neurons. Increased demand for new synapses by exposure to either E2 or bicuculline resulted in robust increases in dendritic spine density in DIV 20 pyramidal neurons (Fig. 4D; supplemental Fig. S3, available at www.jneurosci.org as supplemental material), similar in magnitude to spine density increases observed during experience-dependent plasticity in adult mammals (Holtmaat and Svoboda, 2009). Increases in spine density caused by E2 and bicuculline were both entirely absent in p65-deficient neurons (Fig. 4D; supplemental Fig. S3, available at www.jneurosci.org as supplemental material). These results indicate that the observed structural plasticity of dendritic spines in response to both E2 and bicuculline requires NF-κB.
Under unstimulated conditions, the lack of effect of p65 loss on dendritic spine density at DIV 19–21 correlated with the reduced basal NF-κB-dependent transcriptional activity in mature cultures relative to cultures during the active period of spinogenesis (Figs. 2E,F, 4A). To investigate the possibility that the NF-κB requirement for stimulated increases in dendritic spine density in mature cultures might reflect E2 and bicuculline-induced NF-κB activation, time and dose-dependent NF-κB reporter assays were conducted. Long-term E2 exposure (36 h) at both high and low doses (0.3, 1.7 μm) robustly activated NF-κB-dependent gene expression, whereas short-term E2 exposure at either dose was relatively ineffective (Fig. 4E). Interestingly, E2-mediated increases in spine density are also apparent only after several days of E2 treatment (Murphy and Segal, 1996) (48–72 h) (Fig. 4D; supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Failure of E2 to activate the mtκB-reporter after either long- or short-term treatment demonstrates specificity for NF-κB (Fig. 4E); given the required E2 treatment duration, the activation pathway is likely indirect. In agreement with previously published findings, bicuculline robustly activated NF-κB after short-term exposure (Meffert et al., 2003) (Figs. 2B, 4E). Long-term treatment with low-dose bicuculline (25 μm) generated weaker, but significant, activation of NF-κB-dependent transcription (Fig. 4E); long-term treatment with high-dose bicuculline (50 μm) was toxic in preliminary experiments. Importantly, induction of NF-κB activity by either E2 or bicuculline depended on the presence of the p65 NF-κB subunit (Figs. 2B, 4F).
NF-κB effects on spine density require transcription
Loss of p65 in mature neurons when NF-κB activation was low (DIV 19–21) did not alter basal dendritic spine and synapse number, in contrast to the effects of p65 deficiency at earlier time points when NF-κB transcriptional activity was higher. This suggests that NF-κB-dependent gene expression could regulate dendritic spines and synapses; however, in addition to controlling transcription in the nucleus, NF-κB transcription factors are also present at synapses and could potentially function by local protein–protein interactions (Kaltschmidt et al., 1993; Suzuki et al., 1997; Meffert et al., 2003). A nontranscriptional mechanism of action has been suggested for Drosophila NF-κB at the neuromuscular junction (Heckscher et al., 2007). To test whether NF-κB might regulate dendritic spine density alternatively through either transcription or through local protein–protein interactions, two transcriptionally inactive mutants of p65 were designed. C-terminal truncation deleted the two transactivation domains of p65 to make GFPp65ΔTAD; loss of the transactivation domains has been previously characterized to eliminate transcriptional activity (Schmitz and Baeuerle, 1991; Schmitz et al., 1995). The crystal structures of the p65:p50 and p65:p65 dimers bound to DNA (F. E. Chen et al., 1998; Y. Q. Chen et al., 1998) were used to design a DNA-binding domain mutant in which two DNA-contacting N-terminal residues of p65 were changed to alanines (GFPp65R33,35A). As anticipated, expression of GFPp65, but not GFPp65ΔTAD or GFPp65R33,35A, in HEK 293T cells resulted in a dose-dependent NF-κB activation (Fig. 5A). At the highest tested doses, none of the p65 constructs activated a reporter with mutant consensus NF-κB binding sites (mκB-lucif) (Fig. 5A).
Live confocal imaging and quantitation revealed that tagged p65 (GFPp65, n = 206 spines; GFP, n = 158; p = 8.8 × 10−52) (mCherryp65, n = 101 spines; mCherry, n = 137; p = 1.1 × 10−22) is not only present but is significantly enriched in the dendritic spines of hippocampal pyramidal neurons (Fig. 5B,C) (see Materials and Methods). GFPp65 was enriched in spines relative to dendritic shafts by 42 ± 2.2% compared with nontagged GFP. p65 tagged with an alternative fluorophore, mCherry (mCherryp65), was similarly, but slightly less, enriched in dendritic spines relative to nontagged mCherry (Fig. 5B,C). To test that mutations with loss of transcriptional activity did not inadvertently affect spine localization, the percentage enrichment in dendritic spines of the p65 mutants was quantitated and found not significantly different from GFPp65 (Fig. 5B,C) (GFPp65R33,35A: 43.9 ± 7.5%, n = 88 spines, p = 0.811; and GFPp65ΔTAD: 40.8 ± 4.9%, n = 147, p = 0.813). Transfections were titrated to express equivalent amounts of each p65 variant. These findings indicated that the two structurally distinct transcriptionally inactive mutants of p65 could be used to test whether NF-κB might control dendritic spines through local actions or by transcriptional regulation.
Cultured hippocampal pyramidal neurons from RelAF/F mice were made p65 deficient by transient transfection of Cre-IRES-dsRed in combination with low levels of GFP, GFPp65, GFPp65R33,35A, or GFPp65ΔTAD, and spine density at DIV 16 was compared with pyramidal neurons coexpressing mCherry and GFP. Loss of endogenous p65 produced a deficiency in dendritic spine density that was rescued only by coexpression of transcriptionally active p65 (Fig. 5D) (mCherry alone: 4.5 ± 0.28 spines/10 μm, n = 27 dendrites; Cre: 3.0 ± 0.20, n = 22; GFPp65 plus Cre: 4.6 ± 0.24, n = 26; GFPp65ΔTAD 2.9 ± 0.14, n = 14; GFPp65R33, 35A 3.2 ± 0.25, n = 23). Spine densities in cells expressing Cre with either GFPp65R33,35A or GFPp65ΔTAD were not significantly different from the densities in cells expressing Cre alone. Low levels of expression and the use of high-density cultures prevented GFPp65 from substantially increasing spine density above controls. These results indicate that transcriptional activity is required for the function of p65 in regulating spine density.
PSD-95 is a critical transcriptional target of NF-κB
To begin to identify critical NF-κB targets in excitatory synaptogenesis, we considered the regulation of genes previously known to be important in this process. The gene for PSD-95 was initially identified as a potential NF-κB target in unpublished results of a microarray-based screen using nfκb1−/−rela−/− doubly deficient murine embryonic fibroblast cell lines (Hoffmann et al., 2003). Analysis of the 5′ upstream region of mouse Dlg4 (encoding PSD-95) (see Materials and Methods) reveals multiple potential κB sites, including two conserved tandem consensus κB-sites at 213 and 350 bp (just outside the predicted transcriptional start site) upstream of the ATG, respectively. The effect of p65 deficiency on PSD-95 protein levels was evaluated in lysates of cultured RelAF/F mouse hippocampal neurons during (DIV 16) and after (DIV 21) the most active period of synaptogenesis (Fig. 6A). Loss of p65 significantly decreased PSD-95 protein levels at DIV 16 (n = 3 independent cultures with two wells per condition; p = 0.005) when basal NF-κB is active, and insignificantly decreased PSD-95 protein in mature neurons (DIV 21) when basal NF-κB activity is low. The effects of p65 deficiency on levels of PSD-95 mRNA was assessed using ribonuclease protection assays. Consistent with results for PSD-95 protein, mRNA levels for PSD-95 were significantly decreased by p65 deficiency during periods of elevated basal NF-κB activity (DIV 14) (Fig. 6B), but not in mature neurons when basal NF-κB activity is relatively low (DIV 21) (Fig. 6C). Enhancing excitatory synaptic activity (bicuculline, 50 μm) in mature neurons activates NF-κB and p65-dependent transcription of PSD-95 mRNA (DIV 21) (Fig. 6C).
Figure 6.

NF-κB-dependent increase in spine density involves the novel gene target, PSD-95. A, p65-deficient neurons have significantly decreased levels of PSD-95 at DIV 16 (p = 0.005), but not at DIV 21 (p = 0.09). Representative immunoblot (left) and quantitated data normalized to GAPDH (right). B, Basal expression of PSD-95 mRNA is decreased in DIV 14 p65 deficient neuronal cultures (p65−/−) compared with p65 wild-type cultures (p65+/+) (p = 0.001). C, At DIV 21, p65−/− cultures no longer have significantly lower basal PSD-95 mRNA levels, but fail to increase PSD-95 expression in response to bicuculline (Bic)-induced activation of NF-κB (p = 0.001). D, PSD-95 is required for the rescue of decreased spine density in p65-deficient neurons by p65 expression. DIV 16 pyramidal neurons from RelAF/F mice expressing Cre recombinase show a significant loss of spine density that is rescued by p65 expression in the presence of control mutant siRNA, but not in the presence of siRNA for PSD-95 (Cre/p65/mut.siPSD95, n = 8, p = 0.037; Cre/p65/siPSD95, n = 13). Data are means ± SEM. See also supplemental Figure S4 (available at www.jneurosci.org as supplemental material).
PSD-95 promotes dendritic spine and excitatory postsynaptic formation and can modulate and recruit presynaptic components (El-Husseini et al., 2000; Futai et al., 2007). The coordination of both presynaptic and postsynaptic development by NF-κB of PSD-95 (Fig. 1), as well as the pattern of NF-κB-dependent expression (Fig. 6A–C), are consistent with PSD-95 as an endogenous gene target for the regulation of spine and excitatory synapse formation by p65. To test a requirement for PSD-95 in mediating the effects of NF-κB, experiments to rescue loss of p65 were performed in the presence or absence of PSD-95 knockdown. Cre recombinase was cotransfected in DIV 14 neurons with siRNA oligos for PSD-95 (Elias et al., 2006) or control mutant RNA oligos in the presence or absence of cotransfected p65 and neurons were analyzed at DIV 16. siRNA against PSD-95 successfully knocked down cotransfected GFP-PSD-95 (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Loss of endogenous p65 by Cre transfection reduced spine density (Fig. 6D) comparably with levels observed in the rescue experiments of Figure 5D. In the presence of the mutant RNA oligos, expression of p65 produced a robust rescue of dendritic spine density. In contrast, expression of p65 was unable to rescue dendritic spine density when elevations of endogenous PSD-95 were prevented by siRNA targeting PSD-95 (Fig. 6D). PSD-95 knockdown did not further reduce dendritic spine number below that seen with loss of p65 alone (Fig. 6D) (Cre plus mut.siPSD95 compared with Cre plus siPSD-95). These results indicate that PSD-95 is regulated by NF-κB in a developmental and activity-dependent manner and that PSD-95 is required for the transcriptionally mediated enhancement of dendritic spine density by NF-κB.
Discussion
Despite the characterization of many molecules involved in the development, function, and plasticity of excitatory synapses, a striking deficit remains in knowledge of the endogenous mechanisms controlling the coordinated expression of these molecules. Few transcription factors have been implicated in dendritic spine and excitatory synapse formation and the relationship between pathways regulating initial synapse development and adult synaptic plasticity is also unclear. This study demonstrates that a subunit of the NF-κB transcription factor, p65, regulates dendritic spine morphology and spine and excitatory synapse density both during developmental synaptogenesis and during increased demand for new synapses in mature neurons.
NF-κB activity is highly regulated during development with activity peaking before and during rapid synaptogenesis and dropping to low levels in mature neurons. Developmental changes in NF-κB activity do not appear to result from changes in the expression of NF-κB components, and are instead attributed to the posttranslational activation pathway characteristic of this transcription factor. During developmental synaptogenesis, NF-κB activation can be blocked by the acute inhibition of glutamate receptors, indicating a dependence on excitatory neurotransmission.
In mature neurons, basal NF-κB activity is low and loss of p65 no longer attenuates basal spine density. Instead, stimuli such as estrogen and short-term bicuculline that induce demand for new synapses, both activate NF-κB and require NF-κB to upregulate spine density in mature neurons. Collectively, these data reveal that NF-κB may fulfill a unique function in the regulation of excitatory synapse number and function. Other transcription factors, such as Cux1 and Cux2, have been reported to regulate the basal density of spine and excitatory synapses in the mature nervous system (Cubelos et al., 2010). In contrast, NF-κB does not appear to affect the maintenance of basal spine density in mature neurons and is instead specifically required for stimulus-induced upregulation of dendritic spine density during this period. This role is consistent with the requirement for NF-κB in behavioral learning paradigms and in vitro assays of plasticity, as well as the absence of reported brain structural defects in mice lacking subunits of the NF-κB transcription factor.
Structural plasticity of dendritic spines and synapses is increasingly appreciated as a candidate for an enduring memory trace or engram (Hübener and Bonhoeffer, 2010). The magnitude of changes in spine density induced by p65 deficiency, near 30% loss both in vitro and in vivo, closely approximates structural plasticity associated with both learning and regulation by estrogen. Neurons from control animals have been reported to have between 10 and 40% fewer spines than neurons from animals undergoing learning paradigms or estrogen exposure (Gould et al., 1990; Moser et al., 1994; Restivo et al., 2009). Structural plasticity also encompasses changes in dendritic spine morphology, in which spine head size is highly responsive to activity. In p65-deficient neurons, we observe a reduction in spine head volume as well as reduced mEPSC amplitudes carried by AMPA receptors. These findings are consistent with previous reports that larger spine heads and larger synapses correlate with increased numbers of AMPA receptors and increased AMPA receptor-mediated currents (Nusser et al., 1998; Matsuzaki et al., 2001). NF-κB specifically regulates the density of excitatory, but not inhibitory, synapses and influences spine head volume and, to a lesser extent, spine length (Figs. 1, 2).
Remarkably, the p65 subunit of NF-κB is enriched in the dendritic spines whose structural plasticity it regulates. Our data clearly demonstrate, however, that p65-mediated regulation of spine density absolutely depends on the ability of p65 to bind DNA and activate transcription of target genes rather than being mediated by local protein–protein interactions. Point mutation of the DNA-binding domain of p65 as well as deletion of the transactivation domain both eliminate NF-κB transcriptional activity and regulation of dendritic spines, while leaving spine enrichment unaffected. The importance of spine enrichment for initial NF-κB activation or other cellular functions is not precluded by these data. In fact, other studies indicate that NF-κB can be activated locally at the synapse and that retrograde motor transport can mediate NF-κB-dependent gene expression in response to synaptic stimulation (Wellmann et al., 2001; Meffert et al., 2003; Mikenberg et al., 2007; Shrum et al., 2009). The p65 subunit thus appears to be both localized in spines and able to provide feedback to control spine density and morphology and the recruitment of presynaptic elements in a cell-autonomous manner.
Other subunits of mammalian NF-κB, including p50 and c-Rel, have also been implicated in synaptic plasticity and may have additional uncharacterized mechanisms in the regulation of synapse structure and function. A role for the Drosophila homologs of NF-κB and IκB, Dorsal and Cactus, in regulating postsynaptic glutamate receptor clustering at the neuromuscular junction has been reported (Heckscher et al., 2007). Another component of the NF-κB pathway, the IκB-kinase (IKK), was recently reported to alter spine number in medium spiny neurons of the nucleus accumbens in a cocaine reward model (Russo et al., 2009). IKK can activate NF-κB as well as perform NF-κB-independent functions, including chromatin remodeling (Perkins, 2007). Although a requirement for IKK-induced chromatin remodeling during plasticity has been demonstrated (Lubin and Sweatt, 2007), downstream NF-κB activation by this kinase could also regulate nucleus accumbens plasticity in response to cocaine. Additional studies will be required to evaluate a generalized role of NF-κB in promoting dendritic spine and excitatory synapse formation in brain regions outside of the hippocampus.
We identify PSD-95 as a transcriptional target that is critical for NF-κB enhancement of dendritic spine density and is regulated in a p65-dependent manner both during neuronal development and in response to synaptic activity in mature neurons (Fig. 6). PSD-95 is a key component of the postsynaptic density with an ability to organize both postsynaptic and presynaptic machinery (El-Husseini et al., 2000; Futai et al., 2007; de Wit et al., 2009) that might permit postsynaptic NF-κB activation to recruit excitatory presynaptic components as observed in our data (Fig. 1). Through its many binding partners, PSD-95 also influences glutamate receptor trafficking and synaptic strength (Fitzjohn et al., 2006), making it a highly relevant target. PSD-95 is reported to regulate spine and synapse density during periods of rapid synaptogenesis (El-Husseini et al., 2000; Ehrlich et al., 2007), but not after synaptogenesis has plateaued (Elias et al., 2006). These studies phenocopy NF-κB manipulations and further support a functional link between NF-κB and PSD-95. Nonetheless, it is likely that complex functions, such as regulating synaptic networks, will be achieved by the coordinated control of multiple gene targets. NF-κB-dependent genes remain incompletely characterized within the mammalian CNS, but several reported target genes that could act in concert with PSD-95 are GluR1, BDNF, NGF, nNOS (neuronal nitric oxide synthase), and several adhesion molecules [e.g., NCAM (neural cell adhesion molecule), P-selectin, and ephrin-A1] (Deregowski et al., 2002; Shrum and Meffert, 2008). The regulation of diffusible targets such as nitric oxide and neurotrophins could be speculated to contribute to the capacity of postsynaptic NF-κB manipulations to influence presynaptic elements.
Given the fundamental role of dendritic spines and associated synapses in information acquisition and retention, understanding the mechanisms responsible for their transcriptional regulation could provide insights to control points during development and learning and potential sources of synaptic pathology in disorders such as Alzheimer's and Parkinson's disease in which NF-κB activation is dysregulated (Mattson and Meffert, 2006). The data presented here demonstrate a conserved pathway requiring the p65 subunit of NF-κB to coordinately regulate excitatory synapse formation both during initial synaptogenesis and in mature neurons in response to increased demand for new synapses. This underscores the capacity of neurons to use the same downstream pathway in the context of both developmental and mature stimulus-dependent upregulation of excitatory synapses. These findings also present a novel pathway regulating PSD-95 expression to mediate structural plasticity of dendritic spines and highlight transcription factor-specific roles in excitatory synapse regulation. NF-κB-dependent regulation of the density, size, and function of dendritic spines and excitatory synapses provides a potential cellular mechanism to underlie the importance of this transcription factor in learning and memory.
Footnotes
This work was supported by the Braude Foundation, an American Cancer Society Research Scholar Grant, and National Institute of Mental Health Grants R01 MH080740 (M.K.M.) and MH084020 (D.E.B. and L.M.D.). We thank R. Schmid for creating and providing the RelAF/F mouse line that was critical to our experiments. We thank A. Cho for assistance with experiments, J. Pomerantz and members of the Meffert Laboratory for discussion and critical reading of this manuscript, and M. Caterina and R. Huganir for scientific suggestions.
References
- Ahn HJ, Hernandez CM, Levenson JM, Lubin FD, Liou HC, Sweatt JD. c-Rel, an NF-kappaB family transcription factor, is required for hippocampal long-term synaptic plasticity and memory formation. Learn Mem. 2008;15:539–549. doi: 10.1101/lm.866408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai HW, Shaner NC, Cheng Z, Tsien RY, Campbell RE. Exploration of new chromophore structures leads to the identification of improved blue fluorescent proteins. Biochemistry. 2007;46:5904–5910. doi: 10.1021/bi700199g. [DOI] [PubMed] [Google Scholar]
- Bhatt DH, Zhang S, Gan WB. Dendritic spine dynamics. Annu Rev Physiol. 2009;71:261–282. doi: 10.1146/annurev.physiol.010908.163140. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer T, Yuste R. Spine motility. Phenomenology, mechanisms, and function. Neuron. 2002;35:1019–1027. doi: 10.1016/s0896-6273(02)00906-6. [DOI] [PubMed] [Google Scholar]
- Chen FE, Huang DB, Chen YQ, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature. 1998;391:410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- Chen YQ, Ghosh S, Ghosh G. A novel DNA recognition mode by the NF-kappaB p65 homodimer. Nat Struct Biol. 1998;5:67–73. doi: 10.1038/nsb0198-67. [DOI] [PubMed] [Google Scholar]
- Cubelos B, Sebastián-Serrano A, Beccari L, Calcagnotto ME, Cisneros E, Kim S, Dopazo A, Alvarez-Dolado M, Redondo JM, Bovolenta P, Walsh CA, Nieto M. Cux1 and Cux2 regulate dendritic branching, spine morphology, and synapses of the upper layer neurons of the cortex. Neuron. 2010;66:523–535. doi: 10.1016/j.neuron.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Biase LM, Nishiyama A, Bergles DE. Excitability and synaptic communication within the oligodendrocyte lineage. J Neurosci. 2010;30:3600–3611. doi: 10.1523/JNEUROSCI.6000-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deregowski V, Delhalle S, Benoit V, Bours V, Merville MP. Identification of cytokine-induced nuclear factor-kappaB target genes in ovarian and breast cancer cells. Biochem Pharmacol. 2002;64:873–881. doi: 10.1016/s0006-2952(02)01151-6. [DOI] [PubMed] [Google Scholar]
- de Wit J, Sylwestrak E, O'Sullivan ML, Otto S, Tiglio K, Savas JN, Yates JR, 3rd, Comoletti D, Taylor P, Ghosh A. LRRTM2 interacts with Neurexin1 and regulates excitatory synapse formation. Neuron. 2009;64:799–806. doi: 10.1016/j.neuron.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Klein M, Rumpel S, Malinow R. PSD-95 is required for activity-driven synapse stabilization. Proc Natl Acad Sci U S A. 2007;104:4176–4181. doi: 10.1073/pnas.0609307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52:307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Feil R, Wagner J, Metzger D, Chambon P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun. 1997;237:752–757. doi: 10.1006/bbrc.1997.7124. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Doherty AJ, Collingridge GL. Promiscuous interactions between AMPA-Rs and MAGUKs. Neuron. 2006;52:222–224. doi: 10.1016/j.neuron.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Freudenthal R, Romano A. Participation of Rel/NF-kappaB transcription factors in long-term memory in the crab Chasmagnathus. Brain Res. 2000;855:274–281. doi: 10.1016/s0006-8993(99)02358-6. [DOI] [PubMed] [Google Scholar]
- Futai K, Kim MJ, Hashikawa T, Scheiffele P, Sheng M, Hayashi Y. Retrograde modulation of presynaptic release probability through signaling mediated by PSD-95-neuroligin. Nat Neurosci. 2007;10:186–195. doi: 10.1038/nn1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler F, Algül H, Paxian S, Schmid RM. Genetic inactivation of RelA/p65 sensitizes adult mouse hepatocytes to TNF-induced apoptosis in vivo and in vitro. Gastroenterology. 2007;132:2489–2503. doi: 10.1053/j.gastro.2007.03.033. [DOI] [PubMed] [Google Scholar]
- Gould E, Woolley CS, Frankfurt M, McEwen BS. Gonadal steroids regulate dendritic spine density in hippocampal pyramidal cells in adulthood. J Neurosci. 1990;10:1286–1291. doi: 10.1523/JNEUROSCI.10-04-01286.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckscher ES, Fetter RD, Marek KW, Albin SD, Davis GW. NF-kappaB, IkappaB, and IRAK control glutamate receptor density at the Drosophila NMJ. Neuron. 2007;55:859–873. doi: 10.1016/j.neuron.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer SB, Mrsic-Flogel TD, Bonhoeffer T, Hübener M. Experience leaves a lasting structural trace in cortical circuits. Nature. 2009;457:313–317. doi: 10.1038/nature07487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Leung TH, Baltimore D. Genetic analysis of NF-kappaB/Rel transcription factors defines functional specificities. EMBO J. 2003;22:5530–5539. doi: 10.1093/emboj/cdg534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Hübener M, Bonhoeffer T. Searching for engrams. Neuron. 2010;67:363–371. doi: 10.1016/j.neuron.2010.06.033. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Ndiaye D, Korte M, Pothion S, Arbibe L, Prüllage M, Pfeiffer J, Lindecke A, Staiger V, Israël A, Kaltschmidt C, Mémet S. NF-kappaB regulates spatial memory formation and synaptic plasticity through protein kinase A/CREB signaling. Mol Cell Biol. 2006;26:2936–2946. doi: 10.1128/MCB.26.8.2936-2946.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt C, Kaltschmidt B, Baeuerle PA. Brain synapses contain inducible forms of the transcription factor NF-kappaB. Mech Dev. 1993;43:135–147. doi: 10.1016/0925-4773(93)90031-r. [DOI] [PubMed] [Google Scholar]
- Kassed CA, Willing AE, Garbuzova-Davis S, Sanberg PR, Pennypacker KR. Lack of NF-kappaB p50 exacerbates degeneration of hippocampal neurons after chemical exposure and impairs learning. Exp Neurol. 2002;176:277–288. doi: 10.1006/exnr.2002.7967. [DOI] [PubMed] [Google Scholar]
- Kopec CD, Real E, Kessels HW, Malinow R. GluR1 links structural and functional plasticity at excitatory synapses. J Neurosci. 2007;27:13706–13718. doi: 10.1523/JNEUROSCI.3503-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, Choi S, Lee SY, Cao YA, Ahn HJ, Worley KC, Pizzi M, Liou HC, Sweatt JD. A bioinformatics analysis of memory consolidation reveals involvement of the transcription factor c-rel. J Neurosci. 2004;24:3933–3943. doi: 10.1523/JNEUROSCI.5646-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Day M, Muñiz LC, Bitran D, Arias R, Revilla-Sanchez R, Grauer S, Zhang G, Kelley C, Pulito V, Sung A, Mervis RF, Navarra R, Hirst WD, Reinhart PH, Marquis KL, Moss SJ, Pangalos MN, Brandon NJ. Activation of estrogen receptor-beta regulates hippocampal synaptic plasticity and improves memory. Nat Neurosci. 2008;11:334–343. doi: 10.1038/nn2057. [DOI] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Lubin FD, Sweatt JD. The IkappaB kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron. 2007;55:942–957. doi: 10.1016/j.neuron.2007.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006;13:852–860. doi: 10.1038/sj.cdd.4401837. [DOI] [PubMed] [Google Scholar]
- Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-kappaB functions in synaptic signaling and behavior. Nat Neurosci. 2003;6:1072–1078. doi: 10.1038/nn1110. [DOI] [PubMed] [Google Scholar]
- Mikenberg I, Widera D, Kaus A, Kaltschmidt B, Kaltschmidt C. Transcription factor NF-kappaB is transported to the nucleus via cytoplasmic dynein/dynactin motor complex in hippocampal neurons. PLoS ONE. 2007;2:e589. doi: 10.1371/journal.pone.0000589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser MB, Trommald M, Andersen P. An increase in dendritic spine density on hippocampal CA1 pyramidal cells following spatial learning in adult rats suggests the formation of new synapses. Proc Natl Acad Sci U S A. 1994;91:12673–12675. doi: 10.1073/pnas.91.26.12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DD, Segal M. Regulation of dendritic spine density in cultured rat hippocampal neurons by steroid hormones. J Neurosci. 1996;16:4059–4068. doi: 10.1523/JNEUROSCI.16-13-04059.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K. Structure and function of dendritic spines. Annu Rev Physiol. 2002;64:313–353. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Lujan R, Laube G, Roberts JD, Molnar E, Somogyi P. Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron. 1998;21:545–559. doi: 10.1016/s0896-6273(00)80565-6. [DOI] [PubMed] [Google Scholar]
- O'Riordan KJ, Huang IC, Pizzi M, Spano P, Boroni F, Egli R, Desai P, Fitch O, Malone L, Ahn HJ, Liou HC, Sweatt JD, Levenson JM. Regulation of nuclear factor-κB in the hippocampus by group I metabotropic glutamate receptors. J Neurosci. 2006;26:4870–4879. doi: 10.1523/JNEUROSCI.4527-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan NC, McGettigan PA, Sheridan GK, Pickering M, Conboy L, O'Connor JJ, Moynagh PN, Higgins DG, Regan CM, Murphy KJ. Temporal change in gene expression in the rat dentate gyrus following passive avoidance learning. J Neurochem. 2007;101:1085–1098. doi: 10.1111/j.1471-4159.2006.04418.x. [DOI] [PubMed] [Google Scholar]
- Papa M, Segal M. Morphological plasticity in dendritic spines of cultured hippocampal neurons. Neuroscience. 1996;71:1005–1011. doi: 10.1016/0306-4522(95)00490-4. [DOI] [PubMed] [Google Scholar]
- Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- Pomerantz JL, Denny EM, Baltimore D. CARD11 mediates factor-specific activation of NF-kappaB by the T cell receptor complex. EMBO J. 2002;21:5184–5194. doi: 10.1093/emboj/cdf505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restivo L, Vetere G, Bontempi B, Ammassari-Teule M. The formation of recent and remote memory is associated with time-dependent formation of dendritic spines in the hippocampus and anterior cingulate cortex. J Neurosci. 2009;29:8206–8214. doi: 10.1523/JNEUROSCI.0966-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, Wilkinson MB, Mazei-Robison MS, Dietz DM, Maze I, Krishnan V, Renthal W, Graham A, Birnbaum SG, Green TA, Robison B, Lesselyong A, Perrotti LI, Bolaños CA, Kumar A, Clark MS, Neumaier JF, Neve RL, Bhakar AL, Barker PA, et al. Nuclear factor-κB signaling regulates neuronal morphology and cocaine reward. J Neurosci. 2009;29:3529–3537. doi: 10.1523/JNEUROSCI.6173-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz ML, Baeuerle PA. The p65 subunit is responsible for the strong transcription activating potential of NF-kappaB. EMBO J. 1991;10:3805–3817. doi: 10.1002/j.1460-2075.1991.tb04950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz ML, Stelzer G, Altmann H, Meisterernst M, Baeuerle PA. Interaction of the COOH-terminal transactivation domain of p65 NF-kappaB with TATA-binding protein, transcription factor IIB, and coactivators. J Biol Chem. 1995;270:7219–7226. doi: 10.1074/jbc.270.13.7219. [DOI] [PubMed] [Google Scholar]
- Shrum CK, Meffert MK. The NF-κB family in learning and memory. In: Byrne J, editor. Learning and memory: a comprehensive reference. Oxford: Elsevier; 2008. pp. 567–586. [Google Scholar]
- Shrum CK, Defrancisco D, Meffert MK. Stimulated nuclear translocation of NF-kappaB and shuttling differentially depend on dynein and the dynactin complex. Proc Natl Acad Sci U S A. 2009;106:2647–2652. doi: 10.1073/pnas.0806677106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Falk PM. Selective localization of polyribosomes beneath developing synapses: a quantitative analysis of the relationships between polyribosomes and developing synapses in the hippocampus and dentate gyrus. J Comp Neurol. 1991;314:545–557. doi: 10.1002/cne.903140311. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Mitake S, Okumura-Noji K, Yang JP, Fujii T, Okamoto T. Presence of NF-kappaB-like and IkappaB-like immunoreactivities in postsynaptic densities. Neuroreport. 1997;8:2931–2935. doi: 10.1097/00001756-199709080-00025. [DOI] [PubMed] [Google Scholar]
- Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. Correction of multi-gene deficiency in vivo using a single “self-cleaving” 2A peptide-based retroviral vector. Nat Biotechnol. 2004;22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- Wellmann H, Kaltschmidt B, Kaltschmidt C. Retrograde transport of transcription factor NF-kappaB in living neurons. J Biol Chem. 2001;276:11821–11829. doi: 10.1074/jbc.M009253200. [DOI] [PubMed] [Google Scholar]
- Young P, Qiu L, Wang D, Zhao S, Gross J, Feng G. Single-neuron labeling with inducible Cre-mediated knockout in transgenic mice. Nat Neurosci. 2008;11:721–728. doi: 10.1038/nn.2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]