

Abstract
In recent years, it has become evident that heart failure is not solely due to reduced contractile performance of the heart muscle as impaired relaxation is evident in almost all heart failure patients. In more than half of all heart failure patients, diastolic dysfunction is the major cardiac deficit. These heart failure patients have normal (or preserved) left ventricular ejection fraction, but impaired diastolic function evident from increased left ventricular end-diastolic pressure. Perturbations at the cellular level which cause impaired relaxation of the heart muscle involve changes in Ca2+-handling proteins, extracellular matrix components, and myofilament properties. The present review discusses the deficits in myofilament function observed in human heart failure and the most likely underlying causal protein changes. Moreover, the consequences of impaired myofilament function for in vivo diastolic dysfunction are discussed taking into account the reported changes in Ca2+ handling.
Keywords: Diastole, Myocardial contractility, Muscle stiffness, Myofilament, Phosphorylation, Heart
Systolic and diastolic function of the heart: role of the myofilaments
Every heart beat, the ventricles eject blood into the small and large circulation to provide organs with sufficient oxygen. Cardiac output depends on the amount of blood ejected per heart beat (i.e., stroke volume) and heart rate. Although myocardial muscle contraction is indispensible for proper cardiac output during the systolic (activation) phase of the cardiac cycle, filling of the ventricles during the diastolic (relaxation) phase heavily depends on proper cardiac muscle relaxation. The latter is even more important during increased cardiac stress as occurs during exercise. To match cardiac output to increased demands of the body, heart rate is increased by enhanced sympathetic drive. The magnitude of contraction is increased by increased Ca2+-induced Ca2+-release from the sarcoplasmic reticulum (SR) within the heart muscle cells. To match the increase in heart rate, a faster relaxation of the heart muscle is required which is achieved by increased re-uptake of Ca2+ into the SR and desensitization of the myofilaments to Ca2+ [6, 56].
Upon depolarization of the heart muscle cells, L-type Ca2+-channels are opened, which causes Ca2+ entry into the cytosol and triggers Ca2+ release from the SR via the ryanodine receptors (RyR2; so-called Ca2+-induced Ca2+-release). Subsequently, Ca2+ binds to troponin C and initiates myofilament contraction via interactions between the thick filament myosin heads and the thin filament component actin. Relaxation of the heart muscle cells occurs upon detachment of Ca2+ from the troponin complex and subsequent re-uptake of Ca2+ into the SR via the SR Ca2+-ATPase (SERCA2), which activity depends on the phosphorylation status of phospholamban (i.e., unphosphorylated phospholamban blocks SERCA2 activity). SERCA2 is responsible for re-uptake of ~70% of the Ca2+ involved in the Ca2+ transient and approximately 30% of the cytosolic Ca2+ is removed out of the cell via the Na+–Ca2+ exchanger (NCX) [6, 7]. Changes in cellular Ca2+ cycling and myofilament properties are under the tight control of kinases and phosphatases within the heart muscle cells, which respectively phosphorylate and dephosphorylate cellular target proteins that regulate contraction and relaxation [29, 51]. Upon increased sympathetic activation, β1-adrenergic receptors are activated which initiates protein kinase A (PKA)-mediated phosphorylation of proteins involved in Ca2+ handling (RyR2, phospholamban) and of the myofilament target proteins troponin I (cTnI), myosin binding protein C (cMyBP-C), and titin [6, 28, 40, 56, 75]. The predominant role of the myofilaments during increased β1-adrenergic receptor stimulation is enhancement of relaxation, which is caused by desensitization of the myofilaments to Ca2+ (Fig. 1a) and faster kinetics of cross-bridge cycling [17, 77]. The PKA-mediated phosphorylation of cTnI is thought to be the major contributor to myofilament Ca2+-desensitization [56]. More recent studies have indicated a modulating role for cMyBP-C in the PKA-mediated reduction of myofilament Ca2+-sensitivity [16, 17, 37], although the major effect exerted by phosphorylated cMyBP-C seems to be enhancement of the rate of contraction and relaxation [39, 44, 58, 59]. The third protein phosphorylated upon β1-adrenergic receptor activation is the giant protein titin [75], which, upon PKA-mediated phosphorylation, reduces passive stiffness of cardiac muscle cells [11, 40, 75]. Overall, the PKA-mediated enhancement of myofilament relaxation is critical to maintain proper cardiac performance at increased heart rates associated with β-adrenergic stimulation.
Fig. 1.
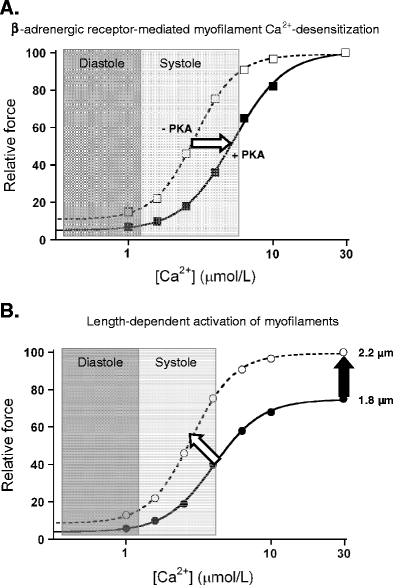
Myofilament responses to increased sympathetic activation and increased left ventricular filling during diastole (Frank–Starling mechanism). a Activation of protein kinase A (PKA) upon β-adrenergic receptor stimulation increases phosphorylation of myofilament proteins (troponin I, myosin binding protein C and titin) and thereby reduces myofilament Ca2+-sensitivity (indicated by the white arrow), enhances cross-bridge kinetics and lowers passive stiffness. The PKA-mediated changes in myofilament properties contribute to enhanced muscle relaxation, which is required for proper filling of the heart during diastole. b An increase in left ventricular filling (increased end-diastolic left ventricular volume) increases the maximal force-generating capacity (black arrow) and myofilament Ca2+-sensitivity (white arrow) and underlies increased cardiac output during the subsequent systolic phase
Apart from phosphorylation-induced changes in myofilament function, a change in sarcomere length upon increased filling of the ventricles during diastole increases the maximal force-generating capacity and the Ca2+-sensitivity of the myofilaments (Fig. 1b). This length-dependent activation is called the Frank–Starling mechanism of the heart and underlies increased cardiac output at increased left ventricular (LV) end-diastolic volumes. The exact mechanisms underlying the increased force-generating capacity of the myofilaments at higher sarcomere lengths are still controversial and have been extensively discussed in previous reviews [33, 36].
Lastly, changes in heart rate adjust myofilament properties to cardiac pump performance [2, 42, 43, 69]. Under physiological conditions, an increase in cardiac stimulation frequency results in enhanced systolic function (so-called positive force–frequency relation), which has been attributed to an increased Ca2+ influx into the cardiomyocytes. The increase in Ca2+ influx increases SR Ca2+ content and promotes the Ca2+-induced Ca2+-release. Varian and Janssen [69] observed a decrease in myofilament Ca2+-sensitivity with increased frequency in the healthy myocardium and suggested that, similar to β-adrenergic PKA-mediated Ca2+-desensitization, the frequency-induced myofilament Ca2+-desensitization accelerates relaxation of the heart muscle. The frequency-mediated alteration in myofilament Ca2+-sensitivity most likely involves changes in protein phosphorylation caused by Ca2+-activated kinases [42, 61, 70].
Systolic and diastolic heart failure
The amount of blood ejected as a fraction of total blood in the ventricles at the end of the diastolic phase is called the ejection fraction. In clinical practice, LV ejection fraction (LVEF) is used as a measure to define systolic cardiac performance. A patient with a LV ejection fraction <45% has heart failure with reduced ejection fraction (HFREF) or systolic heart failure. Over the last two decades, it became evident that more than 50% of all heart failure patients suffer of heart failure with normal (or preserved) ejection fraction (HFNEF) [53]. Compared to HFREF patients, these so-called HFNEF patients have a higher mortality and morbidity [1, 8]. The main perturbation in HFNEF patients is diastolic dysfunction. Moreover, frequently, patients with systolic dysfunction also show impaired diastolic function. The epidemiologic evidence that diastolic rather that systolic dysfunction is a major cause of cardiac failure in Western society has triggered scientists to investigate the underlying mechanisms of diastolic dysfunction in humans.
Perturbations at the cellular level which are thought to underlie diastolic dysfunction in human heart failure are: impaired Ca2+-handling [7], extracellular matrix modifications [11, 63], and myofilament dysfunction [29, 30, 67, 73]. In heart failure, the decline in the cytosolic Ca2+-transient is slowed, which is most likely caused by reduced re-uptake of Ca2+ into the SR due to reduced SERCA2 expression and reduced PKA-mediated phosphorylation of phospholamban. The reduced Ca2+-transient decline contributes to diastolic dysfunction and hampers ventricular filling during the relaxation phase of the heart. The reduced Ca2+ re-uptake also lowers SR Ca2+ content and thereby reduces the amount of Ca2+ available for the subsequent contraction. Thus, perturbations in Ca2+ handling contribute to both systolic and diastolic dysfunction of the heart. Cardiac dysfunction has been ascribed to alterations in the extracellular matrix. Stiffness of the extracellular matrix is largely determined by collagen through regulation of its total amount, the relative abundance of collagen type I and the degree of collagen cross-linking. Excessive collagen type I deposition results from an imbalance between an exaggerated synthesis and a depressed degradation. Comparison of myocardial structure of LV endomyocardial catheter biopsies from HFNEF and HFREF patients showed similar increases in collagen volume fraction in both groups (respectively, 12.2 ± 1.4% and 14.4 ± 1.5%) compared to normal values (5.4 ± 2.2%)[63]. In the HFREF group, collagen deposition was significantly enhanced by diabetes mellitus and associated with increased deposition of AGEs (advanced glycation end products) [64]. AGEs result from long-standing hyperglycemia and augment passive stiffness via cross-linking and enhanced collagen formation. Recently, Westermann et al. [72] provided evidence that enhanced deposition and remodelling of the extracellular matrix in HFNEF patients may involve myocardial inflammation. In addition to collagen deposition, intrinsic cardiomyocyte stiffness contributes to LV diastolic dysfunction evident from the high myofilament passive force observed in patients with HFNEF and HFREF [63].
Diastolic heart failure: role of the myofilaments
The first studies on myofilament function in membrane-permeabilized single cell preparations were already performed more than 30 years ago [22, 26]. Myofilament function is commonly measured in Triton-permeabilized cardiac muscle preparations, which allows investigation of myofilament properties without interference of extracellular matrix components and under well-controlled conditions (e.g., fixed sarcomere length and calcium concentration) [65]. Nowadays, single cells can be isolated from small needle biopsies, which are obtained during cardiac surgery or cardiac catheterization [11, 18]. The major limitation of the method may be the small size of the human cardiac tissue samples available for research, as throughout the heart, regional and transmural differences may exist in myofilament properties. Heterogeneity in myofilament function and protein phosphorylation may be larger in cardiac disease as the disease trigger may be localized to a certain area of the heart. Transmural differences in myofilament properties have been reported in rodent studies [15, 21]. To assess regional differences in myofilament properties in the human heart, LV subepi- and subendocardial biopsies were obtained during valve replacement surgery from patients with mitral valve or aortic valve stenosis or insufficiency [66]. In the latter study, we did not find evidence for regional differences in myofilament function and protein composition within the human ventricle. In addition, recent analysis of variability of the phosphorylation of the PKA target proteins cTnI and cMyBP-C showed that the intra-patient variability in protein phosphorylation was comparable between donor and cardiomyopathy samples [62]. Thus, our data indicate that within the precision of the measurements small, biopsy-sized cardiac human tissue samples are representative for the region of the free LV wall from which they are obtained.
The initial studies in humans were done in samples obtained during heart transplantation surgery from end-stage failing patients with idiopathic (IDCM) or ischemic (ISHD) cardiac disease [67, 73]. A comparison was made with cells isolated from non-failing donor hearts. Myofilament Ca2+-sensitivity was increased in end-stage failing compared to donor hearts as illustrated in Fig. 2a. The higher sensitivity for Ca2+ in end-stage failing hearts may exert a beneficial effect on systolic cardiac performance, but it may limit relaxation during diastole. Diastolic function may be further impaired by the combined changes in myofilament properties and perturbations in Ca2+ handling as discussed above. The blunted decline in Ca2+-transient decay in failing myocardium will increase diastolic Ca2+ levels and exaggerate diastolic dysfunction as shown in Fig. 2b.
Fig. 2.
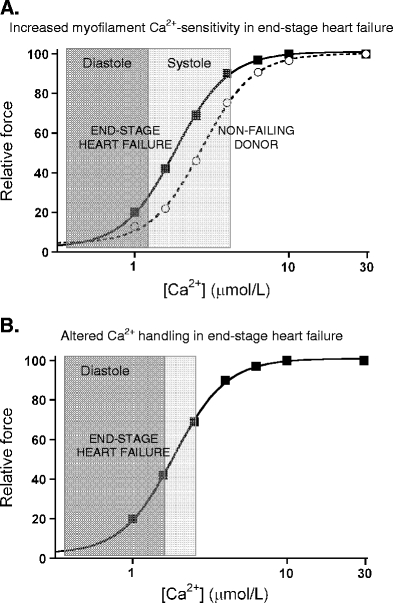
a Force measurements in single Triton-permeabilized cardiomyocytes from end-stage failing patients with ischemic and idiopathic cardiomyopathy showed increased myofilament Ca2+-sensitivity compared to non-failing donor hearts [67]. Translation of the altered responsiveness to Ca2+ to cardiac pump function suggests that the force-generating capacity of myofilaments will be higher at systolic [Ca2+] and improves cardiac output, while the enhanced myofilament Ca2+-sensitivity may impair relaxation of the heart muscle during diastole. b Taking into account the reported changes in the Ca2+ transient in human heart failure (i.e. reduced systolic peak Ca2+ and slowed diastolic Ca2+ decline [7]) would further worsen diastolic function. The combination of enhanced myofilament Ca2+-sensitivity and increased diastolic Ca2+ levels is illustrated in b
The enhanced myofilament Ca2+-sensitivity has been ascribed to defects in the β-adrenergic receptor pathway as reduced phosphorylation of the PKA target proteins, cTnI and cMyBP-C, has been reported in end-stage failing compared to non-failing donor myocardium [9, 20, 25, 48, 67]. In further support for defective β-adrenergic signaling was the observation that myofilament Ca2+-sensitivity was normalized to donor values after treatment of cells with exogenous PKA [67]. Enhanced myofilament Ca2+-sensitivity and correction to control values with PKA treatment have been observed in different animal models as well (e.g., post myocardial infarction or pressure overload) [23, 30, 68]. However, in all animal models, it has been difficult to find proof for reduced PKA-mediated phosphorylation of cTnI and cMyBP-C [30]. Only recently, we have observed a blunted cTnI phosphorylation at the PKA sites (Serines 23/24) upon dobutamine infusion in post-infarction compared to sham pigs [10]. In contrast, the dobutamine-induced phosphorylation of cMyBP-C at Ser282 (one of the PKA sites) was preserved in post-infarction hearts, and coincided with increased autophosphorylation of the cytosolic Ca2+-dependent calmodulin kinase II (CaMKII-δC) [10]. The exact cause of the enhanced myofilament Ca2+-sensitivity in cardiac disease models needs to be further investigated and requires analysis of site-specific protein phosphorylation using mass spectrometry as in addition to reduced PKA activity changes in other kinase (protein kinase C, CaMKII) and in phosphatases have been documented in cardiac disease development [4, 5, 13, 51]. In a recent study [32], we have observed that alterations in the β-adrenergic receptor pathway are more pronounced in human IDCM than in ISHD and may reflect sequential changes in cellular protein composition and function and indicates the need to evaluate changes in myofilament properties in the acute phase after the initial disease trigger (e.g., infarction, valve rupture) and at later stages during remodelling of the heart muscle.
The increased myofilament Ca2+-sensitivity reported in end-stage human heart failure is not a consistent observation in different experimental models of heart failure. Similar to humans, an increased myofilament Ca2+-sensitivity has been observed in pig and mice with a myocardial infarction [10, 23], while a reduction in Ca2+-sensitivity was found in rat models with congestive heart failure due to pressure overload or myocardial infarction [4, 5]. The direction of the Ca2+-sensitivity shift may involve the stage of cardiac disease (i.e., period after the initial cardiac insult). Possible explanations for the opposite changes in myofilament Ca2+-sensitivity in cardiac disease models have been discussed in recent papers [30, 47, 57].
Apart from reduced PKA-mediated protein phosphorylation other myofilament protein modifications have been reported which may underlie impaired diastolic function of the heart. Varian et al. [70] reported a lack of frequency-dependent Ca2+-desensitization in a rabbit model of pressure overload, which was attributed to lack of frequency-dependent cTnI phosphorylation. A reduction in myosin light chain 2 (MLC-2) phosphorylation has been reported in human end-stage heart failure [67], while loss of a transmural MLC-2 phosphorylation gradient has been described in rodent models [15, 21]. Phosphorylation of MLC-2 has been shown to enhance cross-bridge kinetics and force production per unit Ca2+ [55], while ablation of MLC-2 phosphorylation in mice resulted in a blunted response to β-adrenergic receptor stimulation [54]. Specific and selective proteolysis of cTnI at its C-terminus has been proposed to play a key role in human myocardial ischemic disease, including stunning [27, 52]. The C-terminally truncated cTnI protein has been reported to reduce the force-generating capacity upon ischemia-reperfusion in rodent studies. Incorporation of C-terminal truncated cTnI in rat cardiac muscle depressed maximal force and increased cross-bridge kinetics [60]. However, exchange of C-terminal truncated cTnI in human cardiomyocytes had no effect on maximal force development and increased Ca2+-sensitivity of the myofilaments [49]. This indicates that cTnI truncation at the C-terminus in human cardiomyocytes impairs diastolic rather than systolic function. In contrast, truncation of cTnI at the N-terminus has been shown to enhance ventricular diastolic function [3, 76].
Apart from the increased myofilament responsiveness to Ca2+ in end-stage failing human hearts, studies in cardiomyocytes isolated from cardiac catheter biopsies from HFNEF patients revealed significantly elevated passive stiffness compared to control cells (Fig. 3a) [11, 63]. The unique advantage of the catheter biopsies is that in vivo hemodynamic data are collected at the time of biopsy procurement and samples are directly frozen in liquid nitrogen, which will fix phosphorylation status of the myofilament proteome. This allowed us to show that the high intrinsic passive stiffness of the myofilaments correlated well with LV end-diastolic pressure (Fig. 3b) [11], which indicates that increased passive myofilament stiffness is an important contributor to diastolic dysfunction in HFNEF patients. Compared to control cells from individuals with normal LVEF and normal LV end-diastolic pressure, force development by the myofilaments was higher at low “diastolic” Ca2+ concentrations and slightly lower at higher “systolic” Ca2+ concentrations in HFNEF as illustrated in the force-calcium relations in Fig. 3c. Treatment with exogenous PKA significantly reduced passive stiffness and abolished the difference in myofilament passive force between HFNEF and control cells (Fig. 3a, d), while force development at systolic Ca2+ concentrations remained somewhat lower in HFNEF compared to the control group.
Fig. 3.
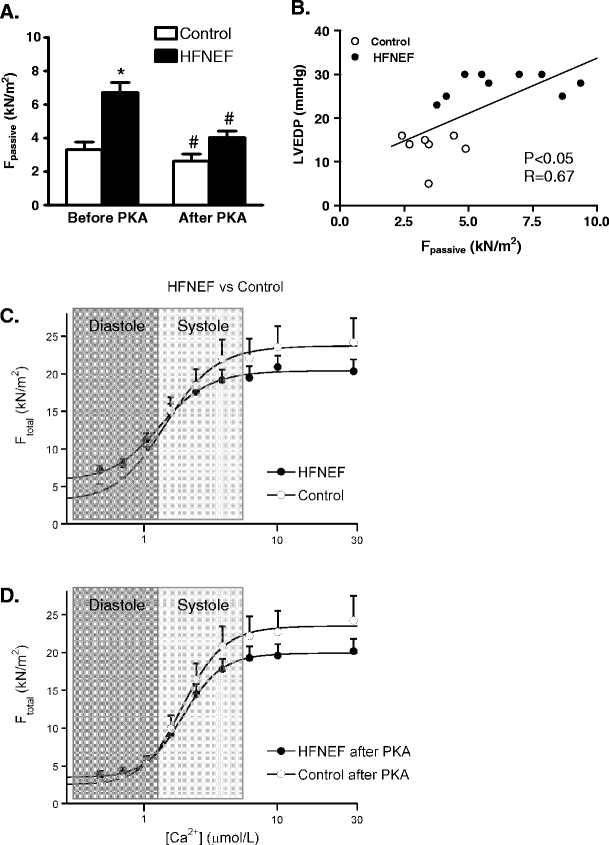
a Isolation of single Triton-permeabilized cardiomyocytes from biopsies taken during cardiac catheterization from patients with a diastolic dysfunction evident from increased left ventricular (LV) end-diastolic pressures (>16 mmHg: heart failure patients with normal (or preserved) LV ejection fraction, HFNEF) revealed increased passive stiffness compared to control cells from patients with normal LV ejection fraction and normal LV end-diastolic pressure [11]. The high passive stiffness was corrected to control values upon treatment with protein kinase A (PKA), suggesting that the high passive force is largely caused by hypophosphorylation of a sarcomeric protein (most likely titin [7]). b A significant relation was found between in vivo LV end-diastolic pressure and cardiomyocyte stiffness, indicating that the in vivo diastolic dysfunction in HFNEF patients is at least partly due to an intrinsic defect of the myofilaments. c Illustrates that the increased myofilament passive stiffness would impair diastolic function of the heart of HFNEF patients compared to controls, while the force-generating capacity of the myofilaments during systole may be somewhat lower compared to controls. d PKA treatment corrected myofilament diastolic dysfunction to values observed in controls
Correction of passive stiffness in HFNEF with PKA indicated that the myofilament dysfunction is caused by protein hypophosphorylation. As mentioned above, the main protein involved in myofilament stiffness it the giant sarcomeric protein titin. Previous studies have shown that titin is a target of PKA, PKG, and PKC [34, 41, 75]. Phosphorylation by PKA and PKG have been shown to reduce passive stiffness [41, 75], while PKCα treatment increased passive stiffness in mouse and pig myocardium [34]. In addition to reduced cTnI and cMyBP-C phosphorylation, end-stage failing human hearts showed a deficit in titin phosphorylation compared to non-failing donor hearts [40]. A study in catheter biopsies from HFNEF and HFREF patients indicated relative hypophosphorylation of the stiff N2B isoform compared to control samples [12]. These data support the hypothesis that hypophosphorylated titin causes increased passive stiffness in cardiac disease. Until present, no evidence for a detrimental effect of PKC-mediated titin phosphorylation has been found in human cardiac samples. Opposite to the expected increase in passive stiffness, PKC treatment of end-stage failing cardiomyocytes slightly reduced passive force [38]. However, the exact modulating role of PKC-mediated titin phosphorylation on passive stiffness in human myocardium should be more carefully assessed in cardiac tissue which is obtained after different stimuli, e.g., after alpha-adrenergic receptor stimulation, which is known to activate downstream PKC.
Phosphorylation deficits of titin may be counterbalanced by adaptations in titin isoform composition possibly aimed to lower passive myofibrillar stiffness [14, 35]. Titin isoform switching has been demonstrated in end-stage failing myocardium: a shift from the stiff N2B isoform to the compliant N2BA isoform coincided with lower passive stiffness [46, 50] and may rescue diastolic dysfunction. Alternatively, a maladaptive shift towards the stiff N2B isoform has been reported in human samples from patients with aortic stenosis and LV hypertrophy [71], which would exert a detrimental effect on diastolic function.
Figure 4 depicts passive stiffness measured in single cells isolated from catheter biopsies and from hearts which were obtained during heart transplantation surgery. Passive stiffness was highest in HFNEF and HFREF patients which were classified as relatively moderate forms of heart failure (NYHA II to III). Passive stiffness was lowest in end-stage failing human hearts classified as NYHA IV. Noteworthy, passive force development in control cardiac catheter biopsies and donor hearts are similar. The changes in passive stiffness in patient groups compared to the control groups may reflect altered passive stiffness during the progression of cardiac disease, characterized by an enhanced passive stiffness at relatively early stages of the disease where titin phosphorylation deficits are predominant, and lower passive stiffness in end-stage failing hearts in which the shift to more compliant titin balances the perturbations in titin phosphorylation.
Fig. 4.
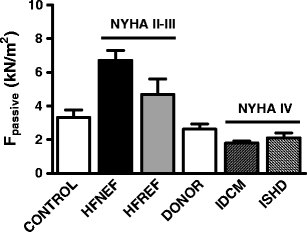
Comparison of passive force measurements in cells isolated from cardiac biopsies taken during cardiac catheterization (control, HFNEF, and HFREF) and from heart tissue obtained during heart transplantation surgery (donor, IDCM, and ISHD) illustrates high passive stiffness in HFNEF and HFREF patients, similar values in control and donor hearts, and low passive force in idiopathic and ischemic end-stage failing cardiomyopathy
Clinical perspectives
Large clinical trials have convincingly shown that β-blocker therapy reduces mortality and improves LV function in HFREF patients. In HFNEF patients, favourable effects of β-blocker therapy on mortality and LV function have not been convincingly demonstrated. Comparison of patients who received β-blocker therapy and patients untreated with β-blockers showed higher maximal force-generating capacity of myofilament in patients with β-blocker therapy, which may underlie improved systolic performance [31]. However, β-blocker therapy increased passive force in HFNEF patients (Fig. 5a), which may even worsen diastolic dysfunction. A positive effect of the β-blockers was observed on the extracellular matrix as the collagen volume fraction was significantly lower in HFNEF with β-blockers (Fig. 5b). The opposite effects of β-blocker therapy on two important determinants of cardiac diastolic function may partly explain the inconsistent results of β-blocker trials in HFNEF patients. Moreover, although collagen deposition may underlie diastolic dysfunction, within the physiologic sarcomere lengths, stiffness is largely determined by titin. In a recent study, Chung and Granzier [19] have shown that titin is the dominant contributor to LV passive pressure within physiological volumes, while the extracellular matrix exerts a dominant effect on LV pressure at larger volumes. As diastolic dysfunction is present in both HFNEF and HFREF patients and is also a major problem in the growing population of patients with diabetic cardiomyopathy [64], it is of great relevance to design a targeted treatment to titin-mediated passive stiffness. Benefits of therapy targeted at myofilament function may depend on the stage of cardiac disease. In pigs, β-blocker therapy directly initiated after a myocardial infarction reversed the increased myofilament Ca2+-sensitivity to values observed in sham animals and significantly reduced passive stiffness in infarct animals [24]. The latter observations are in contrast with the data from our human studies, which showed an increase in passive force in β-blocker treated HFNEF patients (Fig. 5a) and a minor increase in myofilament Ca2+-sensitivity (not shown) [31]. Although there may be species differences, these conflicting data may also be explained by timing at which therapy is initiated. Longitudinal studies in large animal models [45, 74] are warranted to disentangle adaptive from maladaptive myofilament protein changes in the initiation and progression of diastolic dysfunction in heart failure.
Fig 5.
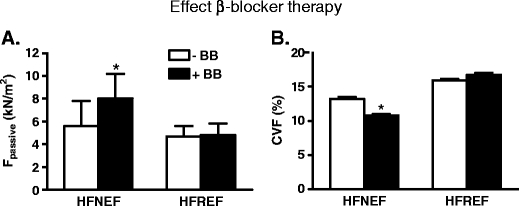
Comparison of heart failure patients with normal (HFNEF) and reduced (HFREF) left ventricular ejection fraction which received chronic β-blocker therapy +BB compared to patients untreated with β-blockers −BB showed higher passive stiffness in HFNEF patients treated with β-blockers a, while the percentage of collagen volume fraction (CVF) was significantly lower in the HFNEF patients with β-blockers b. Passive force was higher in HFNEF compared to HFREF patients, while CVF was higher in HFREF than in HFNEF
Acknowledgment
I would like to thank Diederik Kuster for his excellent advice on the design of the figures.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Ahmed A, Zile MR, Rich MW, et al. (2007) Hospitalizations due to unstable angina pectoris in diastolic and systolic heart failure. Am J Cardiol. 2007;99:460–464. doi: 10.1016/j.amjcard.2006.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antoons G, Mubagwa K, Nevelsteen I, et al. Mechanisms underlying the frequency dependence of contraction and [Ca2+]i transients in mouse ventricular myocytes. J Physiol. 2002;543:889–898. doi: 10.1113/jphysiol.2002.025619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbato JC, Huang QQ, Hossain MM, et al. Proteolytic N-terminal truncation of cardiac troponin I enhances ventricular diastolic function. J Biol Chem. 2005;280:6602–6609. doi: 10.1074/jbc.M408525200. [DOI] [PubMed] [Google Scholar]
- 4.Belin RJ, Sumandea MP, Kobayashi T, et al. Left ventricular myofilament dysfunction in rat experimental hypertrophy and congestive heart failure. Am J Physiol Heart Circ Physiol. 2006;291:H2344–H2353. doi: 10.1152/ajpheart.00541.2006. [DOI] [PubMed] [Google Scholar]
- 5.Belin RJ, Sumandea MP, Allen EJ, et al. Augmented protein kinase C-α-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ Res. 2007;101:195–204. doi: 10.1161/CIRCRESAHA.107.148288. [DOI] [PubMed] [Google Scholar]
- 6.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 7.Bers DM. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 2006;21(Bers DM):380–387. doi: 10.1152/physiol.00019.2006. [DOI] [PubMed] [Google Scholar]
- 8.Bhatia RS, Tu JV, Lee DS, et al. (2006) Outcome of heart failure with preserved ejection fraction in a population-based study. N Engl J Med. 2006;355:260–269. doi: 10.1056/NEJMoa051530. [DOI] [PubMed] [Google Scholar]
- 9.Bodor GS, Oakeley AE, Allen PD, et al. Troponin I phosphorylation in the normal and failing adult human heart. Circulation. 1997;96:1495–1500. doi: 10.1161/01.cir.96.5.1495. [DOI] [PubMed] [Google Scholar]
- 10.Boontje NM, Merkus D, Zaremba R, et al. Enhanced myofilament responsiveness upon β-adrenergic stimulation in post-infarct remodeled myocardium. J Mol Cell Cardiol. 2011;50:487–499. doi: 10.1016/j.yjmcc.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 11.Borbély A, van der Velden J, Bronzwaer JGF, et al. Cardiomyocyte stiffness in diastolic heart failure. Circulation. 2005;111:774–781. doi: 10.1161/01.CIR.0000155257.33485.6D. [DOI] [PubMed] [Google Scholar]
- 12.Borbely A, Falcao-Pires I, van Heerebeek L, et al. Hypophosphorylation of the stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res. 2009;104:780–786. doi: 10.1161/CIRCRESAHA.108.193326. [DOI] [PubMed] [Google Scholar]
- 13.Bowling N, Walsh RA, Song G, et al. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–391. doi: 10.1161/01.cir.99.3.384. [DOI] [PubMed] [Google Scholar]
- 14.Cazorla O, Freiburg A, Helmes M, et al. Differential expression of cardiac titin isoforms and modulation of cellular stiffness. Circ Res. 2000;86:59–67. doi: 10.1161/01.res.86.1.59. [DOI] [PubMed] [Google Scholar]
- 15.Cazorla O, Szilagyi S, Le Guennec JY, et al. Transmural stretch-dependent regulation of contractile properties in rat hearts and its alteration after myocardial infarction. FASEB J. 2005;19:88–90. doi: 10.1096/fj.04-2066fje. [DOI] [PubMed] [Google Scholar]
- 16.Cazorla O, Szilagyi S, Vignier N, et al. Length and protein kinase A modulations of myocytes in cardiac myosin binding protein C-deficient mice. Cardiovasc Res. 2006;69:370–380. doi: 10.1016/j.cardiores.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 17.Chen PP, Patel JR, Rybakova IN, et al. Protein kinase A-induced myofilament desensitization to Ca2+ as a result of phosphorylation of cardiac myosin-binding protein C. J Gen Physiol. 2010;136:615–627. doi: 10.1085/jgp.201010448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chimenti C, Hamdani N, Boontje NM, et al. Myofilament dysfunction in human cardiomyocytes with Fabry disease. Am J Pathol. 2008;172:1482–1490. doi: 10.2353/ajpath.2008.070576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung CS, Granzier HL. Contribution of titin and extracellular matrix to passive pressure and measurement of sarcomere length in the mouse left ventricle. J Mol Cell Cardiol. 2011;50:731–739. doi: 10.1016/j.yjmcc.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Copeland O, Sadayappan S, Messer AE, et al. Analysis of cardiac myosin binding protein-C phosphorylation in human heart muscle. J Mol Cell Cardiol. 2010;49:1003–1011. doi: 10.1016/j.yjmcc.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 21.Davis JS, Hassanzadeh S, Winitsky S, et al. The overall pattern of cardiac contraction depends on a spatial gradient of myosin regulatory light chain phosphorylation. Cell. 2001;107:631–641. doi: 10.1016/S0092-8674(01)00586-4. [DOI] [PubMed] [Google Scholar]
- 22.De Clerck NM, Claes VA, Brutsaert DL. Effect of temperature on the mechanical behaviour of single skinned cardiac cells. J Muscle Res Cell Motil. 1981;2:183–191. doi: 10.1007/BF00711868. [DOI] [PubMed] [Google Scholar]
- 23.De Waard MC, van der Velden J, Bito V, et al. Early exercise training normalizes myofilament function and attenuates left ventricular pump dysfunction in mice with a large myocardial infarction. Circ Res. 2007;100:1079–1088. doi: 10.1161/01.RES.0000262655.16373.37. [DOI] [PubMed] [Google Scholar]
- 24.Duncker DJ, Boontje NM, Merkus D, et al. Prevention of myofilament dysfunction by beta–blocker therapy in post-infarct remodeling. Circ Heart Failure. 2009;2:233–242. doi: 10.1161/CIRCHEARTFAILURE.108.806125. [DOI] [PubMed] [Google Scholar]
- 25.El-Armouche A, Pohlmann L, Schlossarek S, et al. Increased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. J Mol Cell Cardiol. 2007;43:223–229. doi: 10.1016/j.yjmcc.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 26.Fabiato A, Fabiato F. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J Physiol. 1975;3:469–495. doi: 10.1113/jphysiol.1975.sp011026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao WD, Atar D, Liu Y, et al. Role of troponin I proteolysis in the pathogenesis of stunned myocardium. Circ Res. 1997;80:393–399. [PubMed] [Google Scholar]
- 28.Gautel M, Zuffardi O, Freiburg A, et al. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J. 1995;14:1952–1960. doi: 10.1002/j.1460-2075.1995.tb07187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamdani N, Kooij V, Merkus D, et al. Sarcomeric dysfunction in heart failure. Cardiovasc Res. 2008;77:649–658. doi: 10.1093/cvr/cvm079. [DOI] [PubMed] [Google Scholar]
- 30.Hamdani N, de Waard MC, Messer AE, Boontje NM, Kooij V, van Dijk SJ, Versteilen A, Lamberts R, Merkus D, dos Remedios C, et al. Myofilament dysfunction in cardiac disease from mice to men. J Muscle Res Cell Motil. 2008;29:189–201. doi: 10.1007/s10974-008-9160-y. [DOI] [PubMed] [Google Scholar]
- 31.Hamdani N, Paulus WJ, van Heerebeek L, et al. Distinct myocardial effects of beta–blocker therapy in heart failure with normal and reduced left ventricular ejection fraction. Eur Heart J. 2009;30:1863–1872. doi: 10.1093/eurheartj/ehp189. [DOI] [PubMed] [Google Scholar]
- 32.Hamdani N, Borbely A, Veenstra SPGR, et al. Diverse alterations in sarcomeric protein composition and function in ischemic and idiopathic dilated cardiomyopathy. J Muscle Res Cell Motil. 2010;31:289–301. doi: 10.1007/s10974-010-9231-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanft LM, Korte FS, McDonald KS. Cardiac function and modulation of sarcomeric function by length. Cardiovasc Res. 2008;77:627–636. doi: 10.1093/cvr/cvm099. [DOI] [PubMed] [Google Scholar]
- 34.Hidalgo C, Hudson B, Bogomolovas J, et al. PKC phosphorylation of titin’s PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res. 2009;105:631–638. doi: 10.1161/CIRCRESAHA.109.198465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaber WA, Maniu C, Krysiak J, et al. Titin isoforms, extracellular matrix, and global chamber remodeling in experimental dilated cardiomyopathy: functional implications and mechanistic insight. Circ Heart Fail. 2008;3:192–199. doi: 10.1161/CIRCHEARTFAILURE.108.768465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Konhilas JP, Irving TC, de Tombe PP. Frank–Starling law of the heart and the cellular mechanisms of length-dependent activation. Pflugers Arch. 2002;445:305–310. doi: 10.1007/s00424-002-0902-1. [DOI] [PubMed] [Google Scholar]
- 37.Kooij V, Saes M, Jaquet K, et al. Effect of troponin I Ser23/24 phosphorylation on Ca2+-sensitivity in human myocardium depends on the phosphorylation background. J Mol Cell Cardiol. 2010;48:954–963. doi: 10.1016/j.yjmcc.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kooij V, Boontje NM, Zaremba R, et al. Protein kinase C α and ε phosphorylation of troponin and myosin binding protein C reduce Ca2+-sensitivity in human myocardium. Bas Res Cardiol. 2010;105:289–300. doi: 10.1007/s00395-009-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Korte FS, McDonald KS, Harris SP, et al. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ. Res. 2003;93:752–758. doi: 10.1161/01.RES.0000096363.85588.9A. [DOI] [PubMed] [Google Scholar]
- 40.Krüger M, Linke WA. Protein kinase-A phosphorylates titin in human heart muscle and reduces myofibrillar passive tension. J Muscle Res Cell Motil. 2006;27:435–444. doi: 10.1007/s10974-006-9090-5. [DOI] [PubMed] [Google Scholar]
- 41.Krüger M, Kötter S, Grützner A, et al. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res. 2009;104:87–94. doi: 10.1161/CIRCRESAHA.108.184408. [DOI] [PubMed] [Google Scholar]
- 42.Lamberts RR, Soekhoe TW, Hamdani NM, et al. Frequency-dependent Ca2+-desensitization in failing rat hearts. J Physiol London. 2007;582:695–709. doi: 10.1113/jphysiol.2007.134486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Layland J, Kentish JC. Positive force– and [Ca2+]i frequency relationships in rat ventricular trabeculae at physiological frequencies. Am J Physiol Heart Circ Physiol. 1999;276:H9–H18. doi: 10.1152/ajpheart.1999.276.1.H9. [DOI] [PubMed] [Google Scholar]
- 44.Lecarpentier Y, Vignier N, Oliviero P, et al. Cardiac Myosin-binding protein C modulates the tuning of the molecular motor in the heart. Biophys J. 2008;95:720–728. doi: 10.1529/biophysj.107.127787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lewinter MM, Popper J, McNabb M, et al. Extensible behavior of titin in the miniswine left ventricle. Circulation. 2010;121:768–774. doi: 10.1161/CIRCULATIONAHA.109.918151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Makarenko I, Opitz CA, Leake MC, et al. Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ Res. 2004;95:708–716. doi: 10.1161/01.RES.0000143901.37063.2f. [DOI] [PubMed] [Google Scholar]
- 47.Marston SB, de Tombe PP. Troponin phosphorylation and myofilament Ca2+–sensitivity in heart failure: increased or decreased? J Mol Cell Cardiol. 2008;45:603–607. doi: 10.1016/j.yjmcc.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Messer AE, Jacques AM, Marston SB. Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J Mol Cell Cardiol. 2007;42:247–259. doi: 10.1016/j.yjmcc.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 49.Narolska NA, Piroddi N, Belus A, et al. Impaired diastolic function after exchange of endogenous troponin I with C-terminal truncated troponin I in human cardiac muscle. Circ Res. 2006;99:1012–1020. doi: 10.1161/01.RES.0000248753.30340.af. [DOI] [PubMed] [Google Scholar]
- 50.Neagoe C, Kulke M, del Monte F, et al. Titin isoform switch in ischemic human heart disease. Circulation. 2002;106:1333–1341. doi: 10.1161/01.CIR.0000029803.93022.93. [DOI] [PubMed] [Google Scholar]
- 51.Neumann J, Eschenhagen T, Jones LR, et al. Increased expression of cardiac phosphatases in patients with end-stage heart failure. J Mol Cell Cardiol. 1997;29:265–272. doi: 10.1006/jmcc.1996.0271. [DOI] [PubMed] [Google Scholar]
- 52.Murphy AM, Kogler H, et al. Transgenic mouse model of stunned myocardium. Science. 2000;287:488–491. doi: 10.1126/science.287.5452.488. [DOI] [PubMed] [Google Scholar]
- 53.Owan TE, Hodge DO, Herges RM, et al. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355:251–259. doi: 10.1056/NEJMoa052256. [DOI] [PubMed] [Google Scholar]
- 54.Scruggs SB, Hinken AC, Thawornkaiwong A, Robbins J, Walker LA, de Tombe PP, et al. Ablation of ventricular myosin regulatory light chain phosphorylation in mice causes cardiac dysfunction in situ and affects neighboring myofilament protein phosphorylation. J Biol Chem. 2009;284:5097–5106. doi: 10.1074/jbc.M807414200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scruggs SB, Solaro RJ (2011) The significance of regulatory light chain phosphorylation in cardiac physiology. Arch Biochem Biophys. doi:10.1016/j.abb.2011.02.013 [DOI] [PMC free article] [PubMed]
- 56.Solaro RJ, Moir AJ, Perry SV. Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature. 1976;262:615–617. doi: 10.1038/262615a0. [DOI] [PubMed] [Google Scholar]
- 57.Solaro RJ, van der Velden J. Why does troponin have so many phosphorylation sites? Fact and fancy. Point—counterpoint. J Mol Cell Cardiol. 2010;48:810–816. doi: 10.1016/j.yjmcc.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stelzer JE, Patel JR, Moss RL. Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP-C. Circ Res. 2006;99:884–890. doi: 10.1161/01.RES.0000245191.34690.66. [DOI] [PubMed] [Google Scholar]
- 59.Stelzer JE, Dunning SB, Moss RL. Ablation of cardiac myosin-binding protein-C accelerates stretch activation in murine skinned myocardium. Circ Res. 2006;98:1212–1218. doi: 10.1161/01.RES.0000219863.94390.ce. [DOI] [PubMed] [Google Scholar]
- 60.Tachampa K, Kobayashi T, Wang H, et al. Increased cross-bridge cycling kinetics after exchange of C-terminal truncated troponin I in skinned rat cardiac muscle. J Biol Chem. 2008;283:15114–15121. doi: 10.1074/jbc.M801636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tong CW, Gaffin RD, Zawieja DC, et al. Roles of phosphorylation of myosin binding protein-C and troponin I in mouse cardiac muscle twitch dynamics. J Physiol. 2004;558:927–941. doi: 10.1113/jphysiol.2004.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Van Dijk SJ, Holewijn RA, Tebeest A, et al. A piece of the human heart. Variance of protein phosphorylation in left ventricular samples from end-stage primary cardiomyopathy patients. J Muscle Res Cell Motil. 2009;30:299–302. doi: 10.1007/s10974-010-9205-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van Heerebeek L, Borbély A, Niessen HW, et al. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation. 2006;113:1966–1973. doi: 10.1161/CIRCULATIONAHA.105.587519. [DOI] [PubMed] [Google Scholar]
- 64.Van Heerebeek L, Hamdani N, Handoko ML, et al. Diastolic stiffness of the failing diabetic heart. Importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation. 2008;117:43–51. doi: 10.1161/CIRCULATIONAHA.107.728550. [DOI] [PubMed] [Google Scholar]
- 65.Van der Velden J, Klein LJ, van der Bijl M, et al. Force production in mechanically isolated cardiac myocytes from human ventricular muscle tissue. Cardiovasc Res. 1998;38:414–423. doi: 10.1016/S0008-6363(98)00019-4. [DOI] [PubMed] [Google Scholar]
- 66.Van der Velden J, Klein LJ, van der Bijl M, et al. Isometric tension development and its calcium sensitivity in skinned myocyte-sized preparations from different regions of the human heart. Cardiovasc Res. 1999;42:706–719. doi: 10.1016/S0008-6363(98)00337-X. [DOI] [PubMed] [Google Scholar]
- 67.Van der Velden J, Papp Z, Zaremba R, et al. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res. 2003;57:37–47. doi: 10.1016/S0008-6363(02)00606-5. [DOI] [PubMed] [Google Scholar]
- 68.Van der Velden J, Merkus D, Klarenbeek BR, et al. Alterations in myofilament function contribute to left ventricular dysfunction in pigs early after myocardial infarction. Circ Res. 2004;95:e85–e95. doi: 10.1161/01.RES.0000149531.02904.09. [DOI] [PubMed] [Google Scholar]
- 69.Varian KD, Janssen PM. Frequency dependent acceleration of relaxation involves decreased myofilament calcium sensitivity. Am J Physiol Heart Circ Physiol. 2007;292:H2212–2219. doi: 10.1152/ajpheart.00778.2006. [DOI] [PubMed] [Google Scholar]
- 70.Varian KD, Kijtawornrat A, Gupta SC, et al. Impairment of diastolic function by lack of frequency-dependent myofilament desensitization rabbit right ventricular hypertrophy. Circ Heart Fail. 2009;2:472–81. doi: 10.1161/CIRCHEARTFAILURE.109.853200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Williams L, Howell N, Pagano D, et al. Titin isoform expression in aortic stenosis. Clin Sci (Lond) 2009;117:237–242. doi: 10.1042/CS20080248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Westermann D, Lindner D, Kasner M, et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. 2011;1(4):44–52. doi: 10.1161/CIRCHEARTFAILURE.109.931451. [DOI] [PubMed] [Google Scholar]
- 73.Wolff MR, Buck SH, Stoker SW, et al. Myofibrillar calcium sensitivity of isometric tension is increased in human dilated cardiomyopathies. J Clin Invest. 1996;98:167–176. doi: 10.1172/JCI118762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu Y, Bell SP, Trombitas K, Witt CC, et al. Changes in titin isoform expression in pacing-induced cardiac failure give rise to increased passive muscle stiffness. Circulation. 2002;106:1384–1389. doi: 10.1161/01.CIR.0000029804.61510.02. [DOI] [PubMed] [Google Scholar]
- 75.Yamasaki R, Wu Y, McNabb M, et al. Protein kinase A phosphorylates titin's cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ Res. 2002;90:1181–1188. doi: 10.1161/01.RES.0000021115.24712.99. [DOI] [PubMed] [Google Scholar]
- 76.Yu ZB, Zhang LF, Jin JP. A proteolytic NH2-terminal truncation of cardiac troponin I that is up–regulated in simulated microgravity. J Biol Chem. 2001;276:15753–15760. doi: 10.1074/jbc.M011048200. [DOI] [PubMed] [Google Scholar]
- 77.Zhang R, Zhao J, Mandveno A, et al. Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ Res. 1995;76:1028–1035. doi: 10.1161/01.res.76.6.1028. [DOI] [PubMed] [Google Scholar]