

Abstract
Whether drugs that enhance cognition in healthy individuals will appear in the near future has become a topic of considerable interest. We address this possibility using a three variable system (psychological effect, neurobiological mechanism, efficiency vs. capabilities) for classifying candidates. Ritalin and modafinil, two currently available compounds, operate on primary psychological states that in turn affect cognitive operations (attention, memory), but there is little evidence that these effects translate into improvements in complex cognitive processing. A second category of potential enhancers includes agents that improve memory encoding, generally without large changes in primary psychological states. Unfortunately, there is little information on how these compounds affect cognitive performance in standard psychological tests. Recent experiments have identified a number of sites at which memory drugs could, in principle, manipulate the cell biological systems underlying the learning-related long-term potentiation (LTP) effect; this may explain the remarkable diversity of memory promoting compounds. Indeed, many of these agents are known to have positive effects on LTP. A possible third category of enhancement drugs directed specifically at integrated cognitive operations is nearly empty. From a neurobiological perspective, two plausible candidate classes have emerged that both target the fast excitatory transmission responsible for communication within cortical networks. One acts on nicotinic receptors (alpha7, alpha4) that regulate release of the neurotransmitter glutamate while the other (‘ampakines’) allosterically modulates the glutamate receptors mediating the post-synaptic response (EPSCs). Brain imaging in primates has shown that ampakines expand cortical networks engaged by a complex task; coupled with behavioral data, these findings provide evidence for the possibility of generating new cognitive capabilities. Finally, we suggest that continuing advances in behavioral sciences provide new opportunities for translational work, and that discussions of the social impact of cognitive enhancers have failed to consider the distinction between effects on efficiency vs. new capabilities.
Keywords: ampakine, learning, arousal, methylphenidate, modafinil, social issues
1. Introduction
Debates about the feasibility of cognitive enhancement rarely begin with what seems to be a pertinent question: How effective are cortical networks in performing the complex steps underlying serial thought, planning, memory retrieval, and other operations that go into cognition? If the substrates are not particularly efficient, then there should be numerous opportunities for improvement. Conversely, networks that are finely tuned with regard to cognition would presumably not be amenable to selective enhancement, at least with current technologies. Another natural question is whether improvements in one dimension of performance (e.g. speed, accuracy) will necessarily lead to improvements in others (e.g. creativity, or judgment). The reason that these points are not generally discussed is, of course, that, despite enormous advances in neuroscience over the past few years, we still know very little about the neurobiology and operating characteristics of cognition-related networks. But perhaps the ‘room for improvement’ issue can be recast in terms of brain evolution by asking whether comparative anatomical evidence points to strong adaptive pressures for designs that are logically related to improved cognitive performance.
Anatomists often resort to allometry when dealing with questions of selective pressures on brain regions. Applied to brain proportions, this involves collecting measurements for the region of interest – e.g., frontal cortex -- for a series of animals within a given taxonomic group and then relating it to the volume or weight of the brains of those animals. This can establish with a relatively small degree of error whether a brain component in a particular species is larger than would be predicted from that species’ brain size. While there is not a great deal of evidence, studies of this type point to the conclusion that cortical subdivisions in humans, including association regions, are about as large as expected for an anthropoid primate with a 1350cc brain. The volume of area 10 of human frontal cortex, for example, fits on the regression line (area 10 vs. whole brain) calculated from published data (Semendeferi et al., 2001) for a series composed of gibbons, apes and humans (Lynch and Granger, 2008). Given that this region is widely assumed to play a central role in executive functions and working memory, these observations do not encourage the idea that selective pressures for cognition have differentially shaped the proportions of human cortex. Importantly, this does not mean that those proportions are in any sense typical. The allometric equations involve different exponents for different regions, meaning that absolute proportions (e.g., primary sensory cortex vs. association cortex) change as brains grow larger. The balance of parts in the cortex of the enormous human brain is dramatically different than found in the much smaller monkey brain: area 10, for instance, occupies a much greater percentage of the cortex in man. But these effects seem to reflect expansion according to rules embedded in a conserved brain plan rather than selection for the specific pattern found in humans (Finlay et al., 2001).
In all, the explosive expansion of brain over the last 2 million years of hominid evolution resulted in a cortex with proportions that are greatly different than those found in laboratory animals. We can assume that this is responsible for the emergence of the unique capabilities incorporated into human mentation. But our argument here is that these expanded cortical areas are likely to use generic network designs shared by most primates; if so, then it appears unlikely that the designs are in any sense ‘optimized’ for cognition. We take this as a starting position for the assumption that the designs are far from being maximally effective for specialized human functions, and therefore that it is realistic to expect that cognition-related operations can be significantly enhanced.
But what is the likelihood that current lines of research will succeed in exploiting the assumed room for improvement over the next several years? The present review addresses this question beginning with a provisional scheme for classifying candidate cognitive enhancers, an exercise that we think will be useful in discussing what enhancement means. We will use the scheme to classify a restricted sample of compounds, and then employ the results as a starting point for asking if their effects constitute cognitive enhancement. The last two segments of the paper take up various problems surrounding translation, some of which relate directly to the above introductory material, and social issues that could arise if new generation drugs do in fact reach clinical application.
2. A classification scheme for cognitive enhancers
What constitutes a cognitive enhancer? Would this include agents that only secondarily affect cognition via actions on broader psychological variables? Should distinctions be made between drugs influencing psychological processes (e.g., short term memory) that feed into cognition vs. those acting on higher, integrative activities? Rather than trying to reach agreement on such questions, it may be more useful to classify potential enhancers according to multiple dimensions of action as illustrated in Figure 1.
Figure 1. A provisional classification scheme for candidate cognitive enhancers.
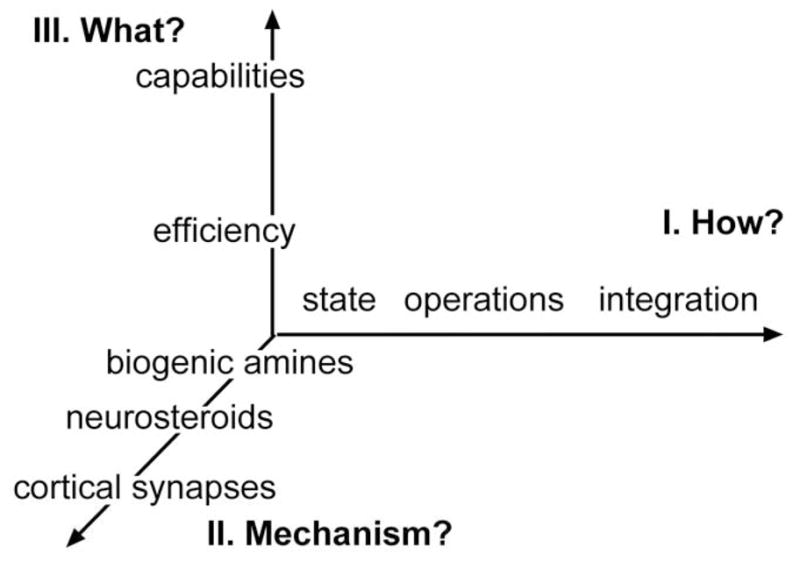
The ‘x’ axis (dimension “I”) lists possible psychological processes affected by the compound. The other two axes indicate neurobiological mechanisms (y; dimension II), and the question of whether the compound affects the efficiency of cognition or allows the subject to exceed normal boundaries (z; dimension III). The axes are collections of associated variables [e.g., brain mechanisms] that have no quantitative relationship with each other.
A critical first dimension involves the issue of ‘how’, in psychological terms, the treatment acts to change cognition (dimension I). There can be little question that fundamental states such as arousal and alertness (‘state’ in Fig. 1) affect complex cognitive operations; similarly, it seems only reasonable to assume that drugs with positive effects on psychological operations subsidiary to cognition, such as attention and the encoding of memory (‘operations’ in the figure), would have positive effects on cognitive performance. Finally, enhancement could, in principle at least, be achieved by actions on the integrated mental activities incorporating planning, cataloguing, memory retrieval, etc. that underlie seconds-long cognitive episodes (‘integration’).
Neurobiological ‘mechanisms’ provide a second dimension for defining enhancers (dimension II) and one that helps deal with dimension I problems that have long plagued preclinical attempts to develop such drugs. Specifically, how can one be confident that behaviors used to assess cognition in animals engage the same psychological processes employed by humans? Most learning tests involve pre-determined (by the experimenter) optimal behavior; similarities between species could result from forcing of very different brain processes to reach the same computational end points. We would argue, as others have (Sarter et al., 2009b) that an appropriate definition of an enhancer will include descriptions of its actions on behavior and on cognition-related networks shared by laboratory animals and humans.
Finally, there is the question of whether a proposed treatment affects the efficiency vs. the capabilities of cognition (dimension III). Consider, for example, a complex problem that an alert, healthy individual solves with a given accuracy and with expected improvements over successive trials. An effective enhancer in this instance could reduce errors during early testing and/or the amount of sampling needed to reach asymptotic performance -- in essence an increase in efficiency. A somewhat different case concerns the effects of the treatment on asymptotic scores that hold for a large population of over-trained subjects, values that might be thought of as a species limit. Such a limit could also be described as an empirically defined level of problem difficulty at which no member of a large population achieves more than a minimal level of performance. We suggest that distinguishing agents that allow subjects to more rapidly reach asymptotic or maximal complexity levels of performance from those that result in supra-normal behavior is a necessary step in classifying cognitive enhancers.
The flow of activity along the dimensions illustrated in figure 1 is likely to be bidirectional, creating a system in which different levels work together in loops. While fundamental psychological states in dimension I will influence memory encoding and attention (‘operations’), which in turn provide essential ingredients for integrated cognitive activities, we can also assume that those cognitive activities will feed back to operations and states. A prominent example involves the ‘forcing’ of attention towards specific cues by higher-level processes dealing with demands imposed by a complex problem (Sarter et al., 2009b). And, while the point is rarely discussed, cognition is accompanied by various, hard to define but familiar ‘feelings’; e.g., the discordant ‘sense’ that a particular argument is wrong, or the excitement and appreciation generated by a sudden insight. Conceivably, then, cognition periodically triggers the assembly of particular mixtures and intensities of fundamental psychological states -- an inner sense -- that alters the flow of thought. (The idea of a special sense associated with cognition was first advanced by Immanuel Kant (1987); see Palmer (2008)).
The idea of loops is also pertinent to the levels included in dimension II (neurobiological mechanisms). The associational cortices assumed to play a central role in integrating cognitive operations are notable in having bi-directional connections with subcortical telencephalic structures critical to memory encoding (hippocampus) and emotion (amygdala). They also project densely into the ventral striatum, a structure that di-synaptically, via the habenula, innervates brainstem biogenic amine nuclei that send fibers back to the hippocampus, amygdala, and associational regions.
The likely existence of psychological and neurobiological loops should be held in mind when considering the locus of action for a potential cognitive enhancer. Drugs discussed below that act on biogenic amines may not affect performance by producing basic psychological states (e.g., arousal) associated with their use, but rather by subtly changing the response of such states to descending signals from ongoing cognitive processing. Similarly, compounds that enhance communication within cortical regions might affect success in solving difficult problems by adjusting output to lower brain areas rather than by their primary action on complex networks. These points are meant as caveats to what follows.
3. Functional categories of candidate enhancers
This section uses specific instances to consider the problem of classifying putative enhancers, and thus to deal with the intertwined question of what enhancement means. It is organized along the dimension of psychological action (dimension I in Fig 1), but also includes information about neurobiological mechanism of action and efficiency vs. capability (dimensions II and III).
3.1 State variables
3.1.1 Methylphenidate
(Ritalin), a much-discussed compound with regard to the usage of enhancers by healthy individuals, is a good example of a drug whose actions can be classified with regard to psychological variables. Its effects on arousal are extensively documented beginning with a marked, amphetamine-like increase in rat exploratory behavior (Wanchoo et al., 2009). Differences between Ritalin’s influence on behavior relative to classical stimulants are subtle at best. That a drug whose primary effect is to increase arousal would produce positive effects on components of cognition, including attention and memory encoding, would not be surprising given the long history of work pointing to an inverted ‘U-curve’ relationship between arousal levels and performance on complex problems (e.g., the Yerkes-Dodson Law) (Yerkes and Dodson, 1908; Hebb, 1955; Malmo, 1959). But whether Ritalin does in fact produce such effects in the absence of disturbances to other aspects of cognition, the latter a possibility inherent in the inverted ‘U’ of the Yerkes-Dodson Law, is debatable. In large measure this reflects the fact that the great majority of animal and human studies on Ritalin are concerned with its effects on attention deficit hyperactivity disorder and other psychiatric problems; a much smaller number of experiments deal with control animals or healthy humans. Surveys of the older literature indicate that Ritalin and other stimulants increase attention or ‘vigilance’, although a recent study did not obtain this result in a test in which human subjects were required to discriminate significant stimuli from distractors (Agay et al., 2010). It seems likely that Ritalin’s positive effects on attention are most apparent in relatively simple tasks requiring sustained engagement and less evident for more difficult problems requiring selective attention (Advokat, 2010).
There is also evidence that Ritalin can improve spatial working memory in healthy humans under some but not all conditions. Specifically, performance was improved on novel spatial problems but impaired in repeat tests (Elliott et al., 1997). Other studies confirmed the improvements in spatial working memory but found them to be evident only in subjects with low baseline scores (Mehta et al., 2000); the latter point, as it happens, also arises in discussions of modafinil’s effects on cognitive operations (see below). Ritalin is also reported to improve digit span test score in healthy individuals, so its influence on working memory is not restricted to spatial problems (Agay et al., 2010). There is, however, little evidence for a positive effect on the encoding of long-term episodic or data memory. An early paper found that low to moderate doses have no effect on retention of various types of information on tests carried out 24 hours post-learning, while the highest concentration tested produced an impairment (Wetzel et al., 1981). Others provided evidence for improved scores shortly after learning but not at later time points (Camp-Bruno and Herting, 1994). In all, a limited set of results suggest that Ritalin’s pronounced effects on arousal can facilitate the attentional and working memory components of cognition but that these effects are situation- and perhaps subject-dependent.
Results for what might be thought of as integrated cognitive processes are generally negative (Advokat, 2010). An interesting study using a complex video game that requires the evolution of strategies for optimizing performance found that Ritalin disrupted normally occurring improvements in scores (Schroeder et al., 1987). Ritalin appears to have little effect on verbal fluency, set shifting (Elliott et al., 1997), decision making or selecting shapes to fit within a matrix (Raven’s test) (Agay et al., 2010). Perhaps a fair, though necessarily tentative, conclusion for this collection of findings is that enhancing arousal and some components of cognition do not produce positive outcomes on integrated cognitive operations in fully engaged subjects.
Ritalin also provides an illustrative case with regard to neurobiological mechanism of action (dimension III). It increases the concentration of dopamine and noradrenaline at synapses by blocking their reuptake (Scheel-Krüger, 1971; Seeman and Madras, 1998; Volkow et al., 2005), an action that fits well with its generalized effects on arousal. The drug thus belongs to a sizable group of therapeutically useful compounds acting on diffuse, ascending biogenic amine systems (ACh, 5-HT, NE, DA). As implied by the name, these projections originate from a relatively small number of cells in the lower brain that generate widely branching projections to broad areas of the forebrain. Such anatomical arrangements constitute a convergent/divergent system in which multiple inputs innervate relatively small targets (the biogenic amine nuclei) that disperse their outputs across much larger target regions.
Cortical models assume that computation depends on the pattern and relative strengths of activated synaptic inputs received by individual members of a very large population of neurons (McCulloch and Pitts, 1943; Hopfield, 1984). Candidate enhancers operating on the biogenic amine systems will not directly affect these variables, if for no other reason than that the diffuse projections are far too sparse to modulate the vast population of excitatory glutamatergic synapses on a contact-by-contact basis. Their effects instead are likely to be executed by changes in the firing thresholds of neurons (excitability), rhythmic patterns of activity within forebrain, or the magnitude of the excitatory drive from subcortex; e.g., one study shows that Ritalin modulates the net activity of frontal and parietal cortical regions in the human brain (Mehta et al., 2000). While it would seem that such effects must differ qualitatively from those produced by drugs that directly modulate the operations of excitatory cortical synapses (see below), describing what this difference might be will require work using biologically realistic simulations of cortical networks.
A further comment is needed about state drugs such as Ritalin that potently affect the nigro-striatal dopaminergic system. The pertinent projections from basal mesencephalon are largely, although not exclusively, directed to the striatum where they generate an extraordinarily (given the number of originating neurons) dense innervation. It follows from this that dopaminergic drugs will modulate the response of striatal cells to their cortical inputs and thus alter the fundamental ‘loop’ (frontal cortex > ventral striatum > thalamus > frontal cortex) that regulates timing in cortical regions subserving planning and organizing components of cognition. Thus, dopaminergic projections may exert one type of effect in cortical, particularly frontal, areas where they are comparatively sparse and another in the striatum where they are present in high numbers [see (Elliott et al., 1997) for a valuable discussion of these cortical vs. striatal effects].
3.1.2 Modafinil
(Provigil) is a second example of a drug operating on state variables that is widely discussed with regard to cognitive enhancement. The compound was first marketed nearly 20 years ago in Europe as an agent to offset excessive sleepiness associated with narcolepsy; it was approved by the Food and Drug Administration for this use and for the treatment of certain sleep disorders. While the effects of modafinil on sleep and its EEG correlates are well-documented (see Minzenberg and Carter (2008) for review), only a few studies have examined its effects on locomotor activity. This is surprising given that the drug is often referred to as a psychostimulant. In any event, modafinil causes a marked increase in activity in mice (Simon et al., 1995; Stone et al., 2002) but only modest effects in rats (Edgar and Seidel, 1997) or on daytime locomotion by monkeys (Andersen et al., 2010). The reasons for these discrepancies are an important topic for future work, but the results as they stand indicate that the psychological state variables affected by modafinil are quite different from those targeted by Ritalin. There is a large and often conflicting literature on the effects of modafinil on components of cognition. Some studies obtained a clear improvement in sustained attention in healthy human subjects (Randall et al., 2005) but others failed to find such effects (Turner et al., 2003). Similar discrepancies occur in the literature on animals (Waters et al., 2005). A recent, multi-factorial analysis provided convincing evidence that moderate doses of modafinil improve attention in healthy middle-aged rats without affecting motivation or locomotor activity (Morgan et al., 2007). Importantly, these effects became evident only as attentional demands were increased. In all, it seems reasonable at this point to conclude that modafinil’s effects on basic psychological state variables -- wakefulness -- can translate into selective improvements in attention.
There is also a sizable literature suggesting that the above conclusion can be extended to memory encoding. An intriguing aspect of these studies in rodents (Beracochea et al., 2002) and humans (Turner et al., 2003; Baranski et al., 2004; Muller et al., 2004; Randall et al., 2005) is that they generally point to a drug influence on working memory as opposed to the encoding of long-term memory for specific information (Minzenberg and Carter, 2008). (A similar argument was made earlier for Ritalin.) For example, the above noted work on middle-aged rats found no evidence for accelerated acquisition of a visual discrimination problem, with minimal demands on working memory, despite clear improvements in attention. There are, however, studies showing that modafinil accelerates the acquisition of simple rules (‘win-stay) (Beracochea et al., 2003), a spatial learning protocol (Shuman et al., 2009), and a non-match to position problem (Ward et al., 2004) in rodents. It is tempting to speculate that we are here seeing hierarchical effects of modafinil such that enhanced wakefulness produces greater attention that in turn improves both working memory and simple rule learning.
But does the above sequence in fact improve the integrative psychological processes that constitute cognition? By far the greater part of the human studies with modafinil involves subjects with impairments to performance (sleep deprivation) or psychiatric disorders. None of the animal studies used recently developed tests (see below) that are explicitly intended to assess variables such as recall vs. recognition or ‘top down’ forcing of attention. This leaves a small set of experiments involving performance by healthy human subjects on relatively simple learning/perceptual problems. A retrospective analysis of several studies led to the conclusion that modafinil does not produce a ‘global’ enhancement of cognition (Randall et al., 2005).
Uncertainties about the cellular effects of modafinil limit the utility of the neurobiological dimension in evaluating the drug’s likely effects on cognition. Recent work has reinforced earlier arguments (Minzenberg and Carter, 2008) that modafinil binds to forebrain dopamine transporters in rats (Zolkowska et al., 2009), monkeys (Andersen et al., 2010), and humans (Volkow et al., 2009), and that this is accompanied by the expected increases in extracellular dopamine concentrations. There is also evidence for binding to norepinephrine transporters in human thalamus (Madras et al., 2006). These effects would be expected to promote wakefulness via mechanisms engaged by classical stimulants. But then the question arises as to why modafinil’s behavioral profile (e.g., locomotor activity) differs so clearly from those for amphetamine, Ritalin, and cocaine. One possibility is that the dopamine binding is less potent than seen with the stimulants but is supplemented by other, more unusual effects. Studies using Fos expression as an index of neuronal activity have established that modafinil activates the perifornical region and the tuberomammilary nucleus of the hypothalamus, two areas that are of primary importance to sleep-wakefulness (Chemelli et al., 1999; Scammell et al., 2000). The first of these contains orexin synthesizing neurons that have been directly implicated in narcolepsy while the latter region is the exclusive location of wake-promoting histaminergic neurons. Modafinil still causes wakefulness as well as its other behavioral, EEG, and Fos expression effects in orexin null mice (Willie et al., 2005) and so is not likely to produce its major effects via the sleep suppressing peptide. It does however increase extracellular histamine levels in the posterior hypothalamus (Ishizuka et al., 2003) and there is considerable evidence that histamine plays an important role in maintaining the vigilant, wakeful state (Thakkar, 2010). It seems reasonable then to assume that modafinil, at moderate doses, exerts its effects via a combined activation of histaminergic and catecholaminergic projections into telencephalon.
With regard to cortical networks, modafinil is a compound that enhances inputs that are diffusely distributed and whose terminal populations are vastly outnumbered by the glutamatergic endings generated by the cortical neurons themselves. As argued with regard to Ritalin, these arrangements are suggestive of a modulatory role wherein the activated inputs change global parameters of networks as opposed to discrete changes in communication patterns within and between networks, or in the various synaptic plasticity effects used to store information. It is also highly likely that, as with Ritalin, the pertinent lower brain areas focus some component of their projections on subcortical areas that exert strong effects on particular cortical areas. For examine, the histaminergic cells in posterior hypothalamus give rise to projections to the relatively small medial septum; it can be imagined that this results in a strong histaminergic influence over cholinergic and GABAergic septal projection neurons that regulate hippocampal rhythms. Indeed, there are results (Luo and Leung, 2010) indicating that histaminergic-septal connections modify hippocampal activity during wakefulness in such a way as to facilitate the induction of long-term potentiation (LTP) in intrahippocampal pathways previously suggested to be involved in working memory (Kramar and Lynch, 2003).
3.2 Cognitive operations
In this section we will consider the possibility of developing enhancers that act directly on cognitive operations (e.g., memory, attention) as opposed to secondarily facilitating those operations via actions on fundamental psychological states. Encoding and retrieval of memory are critical elements of cognition and it is conceivable that facilitating either or both activities would cause a measurable degree of enhancement. Most work in this area is directed at encoding as it is the better understood and more easily studied process. These studies have produced a remarkable number of drug or hormonal treatments that improve retention scores, mostly in healthy rodents and monkeys but in some cases humans subjects as well. For example, neuropeptides (Lee et al., 2004), sex steroids (Markowski et al., 2001), and agents acting on nicotine (Bontempi et al., 2003), norepinephrine (Franowicz and Arnsten, 1999), glutamate (Staubli et al., 1994; Uslaner et al., 2009; Bernard et al., 2010), serotonin (Lamirault and Simon, 2001), or GABA receptor subtypes (Maubach, 2003; Escher and Mittleman, 2004), as well as compounds actin on specific signaling processes (Lynch, 2002) are all reported to enhance memory in multiple studies. While in some instances (e.g., nicotine) these effects are described as having sizable effects on psychological state variables, this is not likely to hold for most of the cases. Very few studies have tested the compounds in traditional tests of attention and it is possible that they operate via this route to enhance encoding. However, as discussed below, evaluation with regard to mechanism of action (dimension II) suggests that at least some of the widely discussed memory enhancers are more likely to act directly on synaptic substrates of encoding.
While the state drugs discussed earlier act by facilitating diffuse forebrain projections, the potential memory enhancers are either agonists that do not depend on the presence of particular neuroanatomical inputs, or drugs that modulate excitatory transmission within cortical networks. Note that the agonists and modulators are not limited in their range of action by the sparseness of non-glutamatergic cortical afferents; they can, in principle, act directly on each member of the enormous population of synapses that both interconnect cortical neurons and act as encoding elements. Relevant to this, a now very substantial literature links the LTP effect to commonplace types of memory (Bliss and Collingridge, 1993; Lynch, 2004b; Lynch et al., 2009). Almost as a byproduct, the great complexity of the cell biological systems involved in the induction, expression, and stabilization of LTP points to many sites at which pharmacological manipulations could, in principle at least, promote the synaptic modifications relating to memory (Lynch, 2002; Lynch et al., 2008). This may be why so many different types of treatments (see above) produce positive effects on memory in animals, and suggests a means for organizing them into a priori, functional subcategories. Specifically, the production of stable LTP requires a sequence beginning with learning-related patterns of afferent activity and ending with a reorganization of the spine actin cytoskeleton and synaptic morphology (Lynch et al., 2008, Lynch et al., 2009) (Fig 2). Many candidate memory enhancers have known relationships with these steps. For example, drugs acting on ascending biogenic amine or cholinergic systems influence the rhythmic activities of cortical networks and thus can potentially enhance the generation of afferent patterns needed to induce LTP. Excitatory synapses also possess multiple types of ‘modifier’ receptors that serve to regulate the formation and stabilization of the cytoskeletal changes that are essential for LTP consolidation (Kramar et al., 2009). These receptors are plausible targets for memory enhancing neuropeptides, hormones, and neurosteroids (see Fig 2). Experimental work has confirmed that nearly all of the agonist and modulatory compounds thought to have memory enhancing properties do in fact promote the induction of LTP. This observation adds support to the idea that these compounds operate on encoding, as opposed to state or attentional variables, to produce their observed improvements in memory scores.
Fig. 2. Summary of the events responsible for the consolidation of LTP and their relationship to sites of action for candidate enhancers.

Experimental work shows that the learning-related theta bursting pattern of afferent activity is particularly well suited for inducing stable LTP. Drugs acting on the ascending biogenic amine systems, including methylphenidrate, potently affect the generation of this rhythm [circled ‘1’]. Theta causes enhanced release of the neurotransmitter glutamate, a process that is increased by other types of possible cognitive enhancers [circled ‘2’]; a compound of this type is discussed in the text. The post-synaptic responses of AMPARs to theta can be pharmacologically amplified with ampakines [circled ‘3’], thereby promoting the post-synaptic (NMDA receptor triggered) signaling pathways that lead to stable LTP. Theta also causes the release of compounds that act on ‘modifier’ receptors that regulate the post-synaptic events that stabilize LTP; various candidate enhancers act at this level [circled ‘4’]. The glutamate transmitter receptors engage two co-localized classes of receptors: the above mentioned modifier receptors and a group of synaptic adhesion receptors; these two groups then combine to initiate and regulate two actin signaling pathways that respectively initiate the assembly and then the stabilization of a new subsynaptic cytoskeleton (Rex et al., 2009). This last event consolidates the synaptic changes that constitute LTP. Potential enhancers, directed at the actin signaling pathways [circled ‘4’], are beyond the scope of this review.
Given the above argument, should we assume that the positive effects of various drugs on memory formation lead to cognitive enhancement? Unfortunately, studies testing the point with conventional measures of cognitive performance, such as those used in work on Ritalin and modafinil, are generally lacking. And while it would seem that better acquisition will result in improvements in cognition, this necessarily depends on the degree to which encoding is a rate-limiting step in reasoning, planning, etc. Possibly, then, the compounds will have their best effect in complex problems involving heavy reliance on working memory, as in most laboratory tests of cognition, and will have more modest consequences in real world circumstances (Parikh and Sarter, 2008; Howe et al., 2010).
But assuming that enhanced encoding improves cognition, we can ask if such an effect will be limited to efficiency or, instead, will extend to capabilities. As noted, one way of investigating this is ask if the treatment causes subjects to exceed asymptotic levels performance in a series of complex problems. Deadwyler and colleagues have reported evidence for such an effect (Hampson et al., 1998b). Rats were trained on a delayed non-match to sample problem in which they were required to press one of two bars (sample phase) and then to move to a new location, wait for varying intervals for a signal, and then return to the test site and select the bar not previously pressed (test phase) (Fig 3A). After weeks of training the animals perform well in this test but show no further improvements with additional testing. Every-other-day injections with a positive modulator of AMPA-type glutamate receptors (an ‘ampakine’) (Lynch, 2006; Lynch and Gall, 2006) prior to the start of a day’s trial produced steadily increasing improvements in scores over a 2 week period (Fig 3B). Thus, the animals exceeded what appeared to be a species limit in dealing with a cognitively demanding problem. An important clue about how this was accomplished came with the surprising observations that the improvements were still present on days in which the rats did not receive the drug and indeed persisted for many days after the treatments were stopped. This indicates that the animals learned and retained something about the problem under the influence of the ampakine that was normally beyond their capabilities. Close examination of the data suggested that this involved a learned suppression of proactive interference. It thus appears that enhanced learning can lead to the acquisition of complex rules that would otherwise be beyond the rat’s abilities.
Fig. 3. An ampakine causes well trained rats to exceed asymptotic performance levels in a complex, delayed non-match to sample problem.

(Top panels) Rats were trained over several weeks to press one extended bar, then move to a nose poke location where they activate a light, and wait (for varying intervals) for the light to go out (left side). They then returned to the test location where two bars were extended. Pressing the bar not previously extended resulted in a water reward. (Bottom panel) Baseline performance over weeks of testing was about 77%. The graph summarizes changes in this performance level over 3 additional weeks for two groups that received daily vehicle injections (controls: open circles) or every other day injections of the ampakine CX516 (see fig. 5 below). The drug (“ampakine days”, filled circles) caused a gradual increase in performance that carried over into vehicle treated days and persisted after the treatments were terminated (gray circles) (Hampson et al., 1998a).
3.3 Integrated cognitive activities
There appears to be no substantial body of work seeking to pharmacologically enhance cognitive operations in the absence of, or in addition to, selective actions on neurobiological mechanisms of attention or learning. Continuing development of novel behavioral tests (see below) may set the stage for such attempts in rodents but has not as yet resulted in claims for discrete facilitation of analogues of human cognition. However, there are neurobiological reasons to suspect that a subclass of the drugs discussed in the previous section could have effects on cognition in addition to actions as memory enhancers. It is important here to make a distinction between agonist-type memory drugs vs. those that modulate transmission at glutamatergic junctions. While there is no reason to think that the agonists for steroid (etc.) receptors will affect the moment-to-moment operations of elaborate cortical networks, modulators of glutamatergic transmission certainly will. Although the modulators promote induction of LTP by increasing net depolarization during learning-related patterns of afferent activity (Lynch and Gall, 2006; Arai and Kessler, 2007), they exert their primary influence on the neuron-to-neuron communication within polysynaptic cortical networks (Sirvio et al., 1996; Lynch and Gall, 2006). Put another way, drugs that facilitate glutamatergic transmission within cortex may both improve memory and modify complex network-level computations.
Positive AMPA receptor modulators (Lynch 2002, 2004a, 2006; Wezenberg et al., 2007; Oertel et al, 2010) and agonists for α4β2 and α7 nicotinic receptors (Dunbar et al., 2007; Jiang and Role, 2008; Loughead et al., 2010) are the principle examples of drugs in clinical development that increase fast (glutamatergic) EPSCs in cortex. The compounds facilitate memory encoding but, as noted, have not been extensively tested with regard to complex cognitive operations. However, two recent studies have provided evidence for effects on higher cognitive functions. Selective α4β2 nicotinic receptor agonists produced substantial improvements in a rat sustained attention test that requires integration of information from signaled and non-signaled trials (Demeter et al., 2008). Detailed analyses suggest that the improvements were due to a top-down shift in processing mode rather than to a change in signal detection; this effect that might well be considered to be a selective enhancement of rat-level cognition. Other work showed that ampakines cause marked improvements in the performance of monkeys on a complex task in which the animals observe a ‘clip art’ image on a computer monitor and then after a varying delay are required to select it with a computer mouse from a set of 2–6 comparable figures (Porrino et al., 2005; Hampson et al., 2009). The positive effects of the drugs were most evident on trials involving long delays and the maximal number of possible choices (Fig 4A). While these results could reflect improvements in attention or short-term memory, it is tempting to speculate that they arise from improved use of cortical resources for evaluating a recent cue against a complex environment.
Fig. 4. Performance scores and correlated imaging results for rhesus monkeys in a complex match-to-sample problem in the absence and presence of an ampakine.
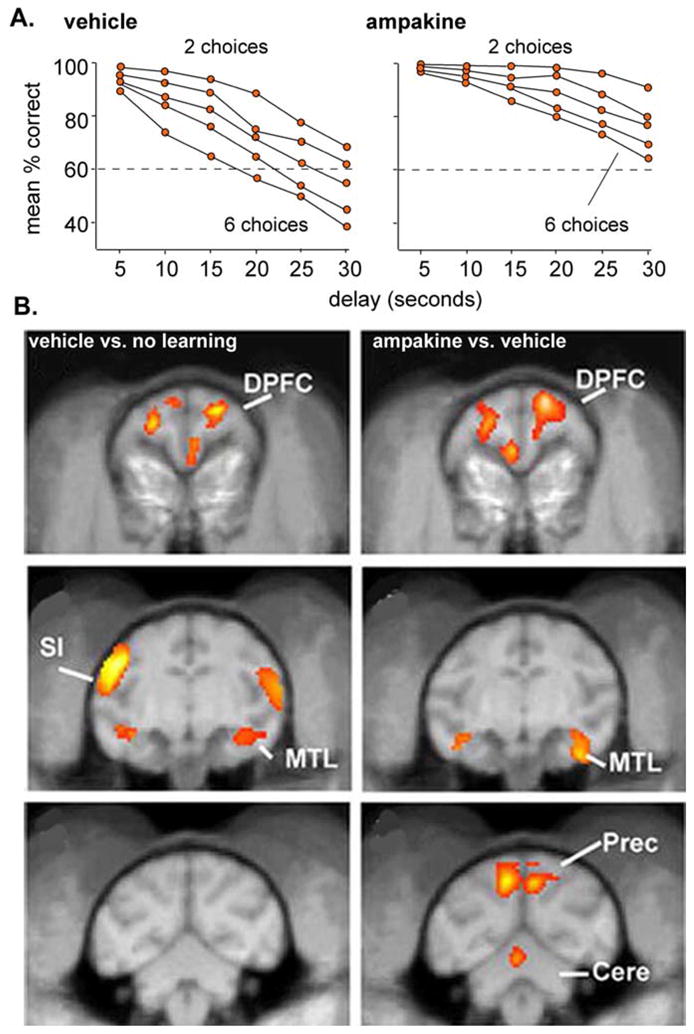
(A). Correct scores by the monkeys when they were required to select, after a variable delay, a previously seen object from a group of 2–6 similar objects. Note that the ampakine caused a marked improvement in scores that was most pronounced for the most difficult part of the problem (6 choices) and with the longest delays. (B) Coronal PET images of glucose metabolism during performance of the match to sample problem. Images at left show the difference between vehicle-treated monkeys before and during performance; note the behaviorally linked increases in activity in the dorsal prefrontal cortex (DPFC), medial temporal lobe (MTL), and primary sensorimotor cortex (SI). Images in the right column summarize the increase above the vehicle-learning effect produced by ampakine injection. The DPFC and MTL were enhanced above the vehicle level but the SI was not. The precuneus, which was not activated during performance in the vehicle condition, is engaged by the task following drug treatment (modified from Porrino et al., 2005).
Neurobiological approaches provide means for asking if drugs are likely to be acting at the level of complex cognitive processing, as opposed to facilitating operations that go into cognition. Pertinent to this, brain imaging of the monkeys performing the match to sample task showed that the ampakine increased activity in frontal and temporal cortex (Porrino et al., 2005), two areas that are differentially activated during normal performance of the task, without affecting regions driving the sensori-motor components of the task. These findings, first, confirm that the ampakine did not cause global changes and, second, strongly suggest that it expanded the local networks normally engaged in dealing with the complex problem. But the most dramatic result from imaging was one showing that the monkeys activated cortical areas that do not normally participate in the match to sample problem. Specifically, the ampakine treatment added the precuneus to the cortical zones that were intensely activated during performance (Fig 4B). This is particularly intriguing given work from humans indicating that the precuneus participates in the visualization of movements yet to happen (Cavanna and Trimble, 2006). In all, these results indicate that the ampakine allowed the cortex to incorporate additional association regions and thus to modify the network substrates of cognition. Note that this result also points to new capabilities, as opposed to simple increases in efficiency, because the monkeys were able to mobilize cortical resources that were not normally available to them in the absence of the drug.
Chronic unit recordings in rats carrying out a non-match to sample task (see fig 2A) also suggest that ampakines can result in an expansion of the cortical networks engaged by a difficult problem, although in a somewhat different manner than found in the monkeys. In particular, daily testing of rats that had just received an injection of an ampakine demonstrated a gradual increase in numbers of hippocampal neurons that fire in response to particular components of the test problem (Hampson et al., 1998b). These effects were still present in tests carried out after the ampakine treatments were stopped, suggesting that they were due to lasting changes in synaptic strength. Moreover, the expansion of the responsive cell population over trials was temporally correlated with the emergence of enhanced performance.
It is worth noting that the evidence for network expansion accords with well-established synaptic effects of the ampakines (Lynch, 2004a; Lynch, 2006; Lynch and Gall, 2006; Arai and Kessler, 2007). The drugs increase AMPA receptor (AMPAR)-mediated synaptic currents by slowing the two biophysical processes that terminate fast EPSCs (Arai et al., 1996); mechanically, this involves the occupation of a binding pocket that governs both the behavior of the receptor’s extracellular domains (deactivation) and the dimerization of its subunits (desensitization) (Fig 5A). Functionally, the enhanced depolarization resulting from an increase in AMPAR currents reduces the number of activated inputs needed to cause a target cell to cross the threshold for spiking (Fig 5B). It follows from this that ampakines will enhance subthreshold connections between cortical association regions to a point at which the global network expands beyond limits normally imposed by connectivity (Fig 5C). If this interpretation of the imaging and unit recording results is correct, then there is a real possibility that the drugs act on the substrates of integrated cognitive activities (complex cortical networks) to produce qualitatively new capabilities.
Fig 5. Effects of ampakines at three levels of analysis.

(A) Shown to the left are chemical structures for two functionally different types of ampakines (A-type: CX516; B-type: CX614). The middle schematic illustrates the binding pocket for the drugs as deduced from work using site directed mutagenesis (Partin, 2001) and X-Ray crystallography (Jin et al., 2005). The two structures represent two of the four subunits (GluR1,2, etc.) that comprise the tetrameric receptor. The glutamate binding site is located in the extracellular domains (circled ‘1’ and ‘2’) of each subunit. The subunits pair up to form two dimers; the ampakine pocket (orange circle) lies in the dimer interface. The traces to the right show representative effects of ampakines on the inward current flux produced by applying a one millesecond pulse of glutamate to a patch excised from a hippocampal pyramidal cell. Note that the drug slows deactivation of the AMPAR gated current (Arai et al., 1996). Synaptic responses from hippocampus are shown in the lower two records. A-Type ampakines increase the amplitude of the response while B-Types produce this effect along with a broadening of the EPSP (Arai et al., 2002). (B) Two neurons receiving different numbers of contacts from an activated (2 axon) input are illustrated (control). The density of the afferent connections is sufficient to spike the cell to the left but not that to the right. An ampakine increases AMPAR mediated EPSCs at each activated synapse, thereby increasing the spike output form the one cell and causing the other to cross spike threshold (+ampakine). The non-linear nature of spiking thus amplifies the functional effect of a drug causing relatively modest increases in transmission at individual synapses. (C) Four separate cortical regions that form a serial network are schematized in these panels. A strong input activates a set of neurons (solid dots) in the first network and brings several others (yellow dots) close to firing threshold. This causes a slightly weaker response (number of activated cells) in the next stage of the network, which then produces a still weaker response in the next stage. This gradual weakening of throughput results in a failure to engage the fourth potential component of the circuit. An ampakine, by increasing EPSCs at individual synapses, causes near threshold cells to fire (blue dots). This causes greater throughput, thus allowing the original input to activate an additional cortical region (Porrino et al., 2005).
4. Translation issues
It is striking that the many compounds found to improve retention scores in rodents or monkeys have yet to translate into a drug with clear-cut enhancing effects in healthy humans operating in real world environments. Of course, the absence of potent memory enhancers could simply reflect the fact that many of the most promising candidates are still winding their way through the multiple stages of clinical development. But the point remains that older agents multiply reported to produce positive effects in different animal species are not widely considered as enhancers. Understanding why this is so would be very useful in evaluating the likelihood that true cognitive enhancers will emerge in the not distant future. Below we consider some possible reasons why animal studies are not more predictive of human results, and discuss new behavioral strategies for reducing this problem.
4.1 Rate limiting steps in cognitive processing
The acquisition of information by humans in natural settings involves a host of activities such as shaping inputs, cataloguing, cross-referencing, etc. in addition to the synaptic modifications presumed to underlie actual storage. Conceivably, then, the synaptic changes underlying encoding of new memory could occur with excess capacity relative to the complex network-level processes. Facilitating synaptic encoding would not under these circumstances result in an overall improvement in learning performance. As an aside, this need not lessen the value of the compounds in the treatment of disorders involving memory impairments. Recent work has found that the complex cell biological systems responsible for LTP are defective in rodent models of several such disorders (Chapman et al., 1999; Gureviciene et al., 2003; Moretti et al., 2006; Lauterborn et al., 2007; Simmons et al., 2009; Kramar et al., 2010) and with aging (Rex et al., 2005); in these instances, drugs directed at synaptic encoding represent a logical therapeutic strategy. But, if encoding is not a rate-limiting step in normals, then the same compounds might have only slight positive effects on cognitive functions.
Moreover, encoding would in this argument occupy a much larger percentage of total processing in rodents simply because the complexity of their cognitive processing is greatly reduced relative to humans; encoding could therefore become a rate limiting step in the overall handling of new information. It is also possible that the need for well-controlled experiments and readily quantified outcome measures may have caused experimenters to employ paradigms in which complex processing is minimized, steps that would create a pre-eminent role for encoding. These considerations suggest that drug effects would be larger in laboratory settings than in much more complex real world environments, something that indeed appears to be the case for at least some memory enhancing drugs (Sarter et al., 2009b).
4.2 Regional differences in memory processing
The enormous encephalization of human brain inevitably results in differences between human and rodents, and indeed between human and monkey (see Introduction), in the relative contribution of specific regions to memory processing. It follows that the behavioral roles of structures such as amygdala and hippocampus, that exhibit flatter allometric relationships to brain size than does association neocortex, will not be the same in small and large brained mammals. To cite one example, hippocampal damage creates hyperactivity and reduced response inhibition in rats while such effects, if present, are minimal in humans. If this general theme extends to memory processing, then pharmaceutical effects arising from actions on a particular structure could easily differ between the two species.
Related to this, the cortical telencephalon is regionally differentiated with regard to the various receptors targeted by most candidate enhancers. Even the ubiquitous AMPA-type glutamate receptors that mediate transmission at the great majority of cortical synapses differ in their subunit composition between layers and regions (Gold et al., 1997). This increases the risk that a drug exerting strong effects in regions that are prominent in a small brain could have much less influence in the vastly expanded cortical areas of humans.
4.3 Models for translation: a conserved cortical system
Given the above issues, it is curious that little enhancement research has been directed to the cortical system with the smallest difference between rodents and humans. Olfactory cortex receives direct input from the phylogenetically conservative olfactory bulb which itself is innervated by sensory receptors of a type common to all mammals. This cortex does not possess specializations found in neocortical sensory areas (e.g., layer IV) and indeed has been argued to share basic design features with association regions (Lynch, 1986; Haberly and Bower, 1989; Johnson et al., 2000), a feature that, if true, would certainly recommend it for translational work. Olfactory cortex also has monosynaptic projections to hippocampus, resulting in a highly conserved, relatively simple network relating environmental cues to a region critical to encoding. Notably, discrete lesions that separate the olfactory cortex from hippocampus produce an anterograde amnesia in rats resembling that found in humans with temporal lobe damage (Staubli et al., 1986). Finally, olfaction remains the only case in which LTP induction has been observed as an animal learns a defined environmental cue (Roman et al., 1987).
Despite these seeming advantages, there appears to be only one set of studies in the literature comparing the effects of a candidate enhancer on memory for odors in rats and humans. The rat experiments used a large open field in which novel odors could be ejected from any of several positions with well-trained rats required to discriminate between rewarded and non-rewarded cues (Larson et al., 1995). The ampakine CX516 caused a substantial reduction in the number of trials required to form long-term memory. The human study required young adults to sample two odors and then 1) pick them out of a set of five 4-hours later and 2) identify the order in which they had been presented during the sample phase. CX516 again produced a sizable improvement in retention scores. Non-olfactory forms of memory were also improved in these tests (Ingvar et al., 1997).
The major drawback to using olfaction in comparative tests of candidate enhancers lies in the strong likelihood that the processing of odor memory is a much less complex (cognitive) process than is the case for other modalities. That human subjects found difficult an olfactory task that would be trivial for other sensory systems provides evidence of this. Nonetheless, comparative testing of potential enhancers on cues processed by the same cortical system would be good first step in translation.
4.4 Models for translation: ‘cognitive behaviors’
Another, more popular, strategy for dealing with translation problems is to develop tests for components of cognition that appear similar in animals and humans. We will here consider only two particularly interesting examples. The first involves the process of directing attention in such a way as to isolate task relevant cues for further cognitive processing, an activity that when extended to memory retrieval can be assumed to be important for many if not most cognitive activities. This is thought to be a top-down process regulated by fronto-parietal circuits with recent work suggesting a critical role for cholinergic inputs to the frontal cortex (Parikh and Sarter, 2008). It is also one that is disturbed in many schizophrenics (Sarter et al., 2009a). A gradually evolved rat test for directed attention uses a signal detection paradigm in which signal and non-signal presentations during an alerting event are responded to by pressing an appropriate lever (one for signal, another for non-signal). Factors such as variable signal duration, competing response tendencies, distracting cues, and different inter-trial intervals are used to increase the processing load. Work with a human version of the test found that healthy adults show effects similar to rats in response to several attention-related variables as well as to distractors. As noted earlier, agonists for a subclass of nicotinic receptors located in frontal cortex produce clear evidence of enhancement in this task in rat studies. It will be of great interest to see if this also holds for humans and, if so, if the improvements carry over into everyday cognitive behavior, as would be expected if ‘attention forcing’ is a rate limiting step on cognitive functioning.
The second example comes from work attempting to dissect the contributions of recognition vs. recall in a complex behavioral problem. These studies (Sauvage et al., 2008, Sauvage, 2010) used an olfactory task in which rats learned a set of odors during a sample phase and then some minutes later were presented with a series of odors that were either novel or repeats of those in the sample phase. Correct responses were to approach novel odors and avoid familiar ones. Tests over several days were carried out with different levels of response biasing (effort to obtain reward to the novel cue; magnitude of reward for avoiding old cues), a step that allowed the investigators to employ a signal detection technique known as receiver operating characteristics (ROC) (Sauvage, 2010). This involves a plot of the hit rate (percent correct responses to old items) vs. the false alarm rate (percent incorrect responses to new items) across the different biasing conditions. The ROC results led the authors to conclude that normal rats use recollection along with simple recognition to deal with the problem. These findings provide evidence that rats can employ what has to be considered as a fundamental cognitive operation (memory retrieval) and, in addition, link its measurement to a technology (ROC curves) employed in an extensive body of human studies -- in all, a set of conditions that seems to be particularly appropriate for comparative work. However, other investigators have challenged the recognition vs. recall interpretation of the ROC curves in these studies (Wixted and Squire, 2008). Whatever the ultimate resolution of this debate, the very idea that familiarity and recall can be studied with similar signal detection methods in rats, monkeys, and humans (Sauvage, 2010) has to be viewed as an important step in the evolution of integrated assays for cognitive enhancers.
A very different approach to integrated animal–human studies of cognitive enhancers begins with the assumption that the net output of telencephalic systems engaged by complex problems in rats and humans, although quantitatively quite different, will nonetheless have the same overall limiting conditions. Tests of this require placing rats and humans in situations that, while markedly different in their absolute complexity, place similar demands on the resources available to small vs. large brains. This approach also involves unsupervised learning (USL), a ubiquitous part of human life, and thus moves testing closer to real world conditions. We have, in collaboration with other groups, initiated studies of this kind using complex environments (Fedulov et al., 2007, Simmons et al., 2009). Well-handled rats are first exposed to an open area with opaque walls and no internal structure, and then on subsequent days are returned to the same environment, which now has compartments, interactive objects, a dark ‘refuge’, and a transparent wall opening onto a view of distant cues. Learning is assessed as changes in exploratory behavior within and between days as well as by the reaction to changes in the objects or shifts in their location (Fig 6A). An equivalent form of USL in humans is achieved using total immersion virtual reality (VR) in which subjects move through a complex environment (e.g, a village, a ship) with enormous numbers of local and distant cues; ultimately, interactive objects will be added (Fig 6B) (Makeig et al., 2009; also see: http://inc.ucsd.edu/~poizner/). Learning is again measured by within and between session changes in behavior, verbal report, and the subject’s success in detecting subtle changes to the environment.
Fig 6. Studying unsupervised learning (USL) in matched, complex environments by rats and humans.

Shown are the novel environments faced by a rat leaving a darkened ‘refuge’ (left) and a human subject entering a total immersion virtual reality ‘village’ (right; the simulation includes additional streets and distant buildings). In both cases, the subjects are allowed unrestricted exploration through the real (rats) and simulated environments. The experimental questions are the same for both cases: Does a candidate enhancer increase the amount of information encoded during a single exploration session in a novel, non-threatening commonplace world and to what extent is this accompanied by changes in baseline behaviors (selectivity)?
USL generates a large number of behavioral variables (speed of individual movements, duration of pauses, preferences for locales, exploratory patterns, etc.), making it possible to build high dimensional descriptions of behavior to supplement the multiple measures of learning. Comparisons can then be made between how a treatment affects the magnitude and speed of learning (e.g., switch between behaviors associated with novel vs. familiar environments) with its actions on general behavior – in essence, a selectivity index. Comparative (rat vs. human) tests of how candidate enhancers affect USL in complex, naturalistic environments are possible with these arrangements.
5. Social issues
The introduction of cognitive enhancers would have profound and unpredictable consequences for society, as usefully discussed in recent reviews (Greely et al., 2008; Farah et al., 2009; Sahakian and Morein-Zamir, 2010). Greely and colleagues (2008) take a generally positive stance towards the potential for such drugs to enhance human life, arguing they should be generally classed with “education, good health habits, and information technology” as means of cognitive enhancement, but warn that their risks must be identified and managed (neither simply left to the mercies of market forces nor addressed primarily through legislation). They discuss potential problems including questions of fairness (whether to those unenhanced in competitive situations, or cost and availability considerations), of safety (both for healthy and non-healthy individuals over time, and particularly for developing brains of children), and of personal freedom: whether the freedom to choose to use cognitive enhancing drugs on the one hand, or the potential for explicit or implicit coercion to improve performance (e.g. by employers, in the military, or in the classroom) on the other. They recommend a four-fold approach to managing the risks and promoting the benefits of cognitive enhancement, including increased research into their effects; collaboration among doctors, educators, and regulators to develop appropriate policies; public dissemination of their risks, benefits, and alternatives; and “careful and limited legislative action”. Below we will consider the topic in light of the classification arguments that run through the above sections.
Drugs that act on ascending biogenic amine projections and thereby change psychological state will be limited in their use because these state variables are, for reasons discussed, likely to be greater than their actions on cognitive efficiency. The state effects in other words may be the larger, and more familiar, issue for society. But compounds such as modafinil that couple moderate effects on catecholamines with actions on hypothalamic systems and thereby improve attention and working memory without pronounced arousal effects are good fits for the issues raised by in the above discussions of social issues. Our survey suggests that the advantages associated with such drugs are somewhat limited, mainly involving conditions in which sleepiness is factor or artificial testing circumstances that involve heavy loads on operations that feed into cognition. But there is little reason to think that such drugs will in any sense constitute ‘smart pills’ -- something that will give healthy, alert individuals any intellectual advantages in real world circumstances.
Agents that selectively enhance components of cognition, and in particular memory encoding, also raise regulatory questions that are described in the above noted reviews. But we would suggest that computational neuroscience points to additional, even more difficult to evaluate societal issues. Specifically, modeling work using biologically realistic networks and empirically derived synaptic learning rules based on LTP strongly suggest that accelerated learning will change the manner in which newly acquired information is organized. The simulated networks not only store memory but also place it into hierarchical categories (e.g., animal > bird > robin), without outside supervision (Ambros-Ingerson et al, 1990). Changing the synaptic rules, the functional effect of selective memory enhancing drugs, not only increases the speed of acquisition in the models but also alters the size of categories (does this item belong or not) and the number of exemplars needed to form them. These observations lead to the idea that the use of selective memory enhancing drugs could cause people to create cognitive structures of a type that do not occur within the range of normal human experience.
Given the argument that the cortex is composed of generic mammalian networks (see Introduction), there is no necessary reason to expect that these novel cognitive organizations would not be capable of dealing with the demands of a human-dominated environment that emerged in the last few thousand years. Regulatory considerations in this case would not be restricted to acute advantages (e.g., learning specific material for a test) gained by using memory enhancers but would also require weighing the consequences to society of individuals who gradually develop a world view that may differ in unique ways from that shared by the general population. There is no way, given our current very limited understanding of how networks generate cognition, of predicting whether these internal constructions would be more or less effective than the baseline human condition. If benign or positive, then we would have agents that both accelerate learning and have the potential for producing what would literally be a new way of seeing things. Why would society prevent the introduction of such drugs? Perhaps the most obvious reason goes to the predictability problem noted earlier: would the steady accumulation of novel cognitive architecture eventually affect inter-personal relations, a person’s integration into society, emotional life, and so forth? Since cognitive neuroscience can’t begin to answer such questions, it will not be possible to develop a risk/benefit analysis for drug effects on normal subjects. But the point can be accommodated empirically by running long-term tests on a population of carefully monitored individuals. Furthermore (as Greely and colleagues have noted), devices with cognition enhancing properties have been introduced into the public at least since paleolithic times (e.g., painting) and never more so than today. There can be no question that such inventions have altered the human experience in ways that were unforeseen at their introduction. In the end, only further research and long-term trials can provide the material needed for an evaluation of the opportunities and problems that will accompany the arrival of memory enhancers.
The possibility of drugs that add cognitive capabilities brings with it an additional set of social impact questions. We described behavioral results indicating that positive modulation of cortical EPSCs allows rats and monkeys to go beyond normal limits on performance in very complex learning paradigms. The available data from chronic recording in rats and PET imaging in monkeys suggest that the behavioral improvements are accompanied by an expansion of neural networks engaged by a given task, a result that reinforces the idea that the compounds permit the emergence of new and functionally effective types of cognitive processes. It remains the case, however, that there are no studies showing that the drugs allow animals to master tasks that are beyond their normal capabilities.
Despite this last caveat, the data as they stand, obtained as they were with drugs that do not cause notable side effects in humans, suggest that we should begin considering the possible societal impact of compounds that add new cognitive capabilities. It is hard to avoid the conclusion that allowing the cortex to assemble larger, more elaborate functional networks will lead to an increase in computational power. Physical expansion of associational cortex networks via the addition of more interconnected neurons, is the presumed basis of the great intelligence in humans relative to other primates. Further increases in functional networks could therefore result in still greater intelligence.
Others have considered the potential impact of a ‘smart pill’ with regard to fairness – who will get the pill? – and related issues (see above). But perhaps we should not assume that the effects of drug-induced network elaboration would be restricted to what is commonly meant by intelligence. Humans have mental abilities such as language that are barely detectable in closely related primates and, again, most researchers assume that this reflects the physical expansion of networks in large areas of cortex. Shouldn’t we then expect functional increases in network size to similarly result in new mental phenomena? The sudden appearance of new abilities would likely have profound and quite possibly irreversible effects on society. They could, for example, lead to concepts and arguments that would be difficult to translate back into the world of normal cognition. And once having experienced new abilities, wouldn’t a person be reluctant to give these up and return to what might be perceived as a more limited form of mental life? Questions of this kind could prove to be the most difficult issues in any debate about the regulation of cognitive enhancers.
The above, very speculative remarks bring us again to the need for experimental data; we simply cannot, at the present state of knowledge, develop a priori arguments about how dramatically drugs that affect network operations will affect mental capacities. Perhaps there will be no new abilities and only a modest improvement in cognitive performance. There are very few published results on the effects on healthy individuals of new generation drugs that modulate transmission within cortex, and those studies used early versions of the compounds as well as supervised learning in highly restricted testing paradigms. These are not conditions conducive to the expression of novel mental operations. As discussed, we feel that future studies will need to include unsupervised behavior in very complex environments, something that is becoming increasingly feasible with the continuing evolution of quantitative testing in virtual reality.
Acknowledgments
The authors thank Cheryl Cotman for preparation of Figure 6 and to Drs. Linda Porrino and Sam Deadwyler for permission to use material in Figure 4.
Funding: Research from the author’s laboratories was supported, in part, by grants from the National Institutes of Health (NS045260 to GL and CMG; NS051823 to GL), an Office of Naval Research Multidisciplinary University Research Initiative Award N00014-10-1-0072 to G.L., and reagents provided by Cortex Pharmaceuticals (Irvine, CA).
Abbreviations
- LTP
Long Term Potentiation
- ROC
receiver operating characteristics
- USL
unsupervised learning
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Advokat C. What are the cognitive effects of stimulant medications? Emphasis on adults with attention-deficit/hyperactivity disorder (ADHD) Neurosci Biobehav Rev. 2010;34:1256–1266. doi: 10.1016/j.neubiorev.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Agay N, Yechiam E, Carmel Z, Levkovitz Y. Non-specific effects of methylphenidate (Ritalin) on cognitive ability and decision-making of ADHD and healthy adults. Psychopharmacology (Berl) 2010;210:511–519. doi: 10.1007/s00213-010-1853-4. [DOI] [PubMed] [Google Scholar]
- Ambros-Ingerson J, Granger R, Lynch G. Simulation of paleocortex performs hierarchical clustering. Science. 1990;247:1344–1348. doi: 10.1126/science.2315702. [DOI] [PubMed] [Google Scholar]
- Andersen ML, Kessler E, Murnane KS, McClung JC, Tufik S, Howell LL. Dopamine transporter-related effects of modafinil in rhesus monkeys. Psychopharmacology (Berl) 2010;210:439–448. doi: 10.1007/s00213-010-1839-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai A, Kessler M, Ambros-Ingerson J, Quan A, Yigiter E, Rogers G, Lynch G. Effects of a centrally active benzoylpyrrolidine drug on AMPA receptor kinetics. Neuroscience. 1996;75:573–585. doi: 10.1016/0306-4522(96)00263-1. [DOI] [PubMed] [Google Scholar]
- Arai AC, Kessler M. Pharmacology of ampakine modulators: from AMPA receptors to synapses and behavior. Curr Drug Targets. 2007;8:583–602. doi: 10.2174/138945007780618490. [DOI] [PubMed] [Google Scholar]
- Arai AC, Xia YF, Rogers G, Lynch G, Kessler M. Benzamide-type AMPA receptor modulators form two subfamilies with distinct modes of action. J Pharmacol Exp Ther. 2002;303:1075–1085. doi: 10.1124/jpet.102.040360. [DOI] [PubMed] [Google Scholar]
- Baranski JV, Pigeau R, Dinich P, Jacobs I. Effects of modafinil on cognitive and meta-cognitive performance. Hum Psychopharmacol. 2004;19:323–332. doi: 10.1002/hup.596. [DOI] [PubMed] [Google Scholar]
- Beracochea D, Celerier A, Borde N, Valleau M, Peres M, Pierard C. Improvement of learning processes following chronic systemic administration of modafinil in mice. Pharmacol Biochem Behav. 2002;73:723–728. doi: 10.1016/s0091-3057(02)00877-8. [DOI] [PubMed] [Google Scholar]
- Beracochea D, Celerier A, Peres M, Pierard C. Enhancement of learning processes following an acute modafinil injection in mice. Pharmacol Biochem Behav. 2003;76:473–479. doi: 10.1016/j.pbb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Bernard K, Danober L, Thomas JY, Lebrun C, Munoz C, Cordi A, Desos P, Lestage P, Morain P. DRUG FOCUS: S 18986: A positive allosteric modulator of AMPA-Type glutamate receptors pharmacological profile of a novel cognitive enhancer. CNS Neurosci Ther. 2010;16:e193–212. doi: 10.1111/j.1755-5949.2009.00088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bontempi B, Whelan KT, Risbrough VB, Lloyd GK, Menzaghi F. Cognitive enhancing properties and tolerability of cholinergic agents in mice: a comparative study of nicotine, donepezil, and SIB-1553A, a subtype-selective ligand for nicotinic acetylcholine receptors. Neuropsychopharmacology. 2003;28:1235–1246. doi: 10.1038/sj.npp.1300150. [DOI] [PubMed] [Google Scholar]
- Camp-Bruno JA, Herting RL. Cognitive effects of milacemide and methylphenidate in healthy young adults. Psychopharmacology (Berl) 1994;115:46–52. doi: 10.1007/BF02244750. [DOI] [PubMed] [Google Scholar]
- Cavanna AE, Trimble MR. The precuneus: a review of its functional anatomy and behavioural correlates. Brain. 2006;129:564–583. doi: 10.1093/brain/awl004. [DOI] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Demeter E, Sarter M, Lustig C. Rats and humans paying attention: cross-species task development for translational research. Neuropsychology. 2008;22:787–799. doi: 10.1037/a0013712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar GC, Inglis F, Kuchibhatla R, Sharma T, Tomlinson M, Wamsley J. Effect of ispronicline, a neuronal nicotinic acetylcholine receptor partial agonist, in subjects with age associated memory impairment (AAMI) J Psychopharmacol. 2007;21:171–178. doi: 10.1177/0269881107066855. [DOI] [PubMed] [Google Scholar]
- Edgar DM, Seidel WF. Modafinil induces wakefulness without intensifying motor activity or subsequent rebound hypersomnolence in the rat. J Pharmacol Exp Ther. 1997;283:757–769. [PubMed] [Google Scholar]
- Elliott R, Sahakian BJ, Matthews K, Bannerjea A, Rimmer J, Robbins TW. Effects of methylphenidate on spatial working memory and planning in healthy young adults. Psychopharmacology (Berl) 1997;131:196–206. doi: 10.1007/s002130050284. [DOI] [PubMed] [Google Scholar]
- Escher T, Mittleman G. Effects of ethanol and GABAB drugs on working memory in C57BL/6J and DBA/2J mice. Psychopharmacology (Berl) 2004;176:166–174. doi: 10.1007/s00213-004-1875-x. [DOI] [PubMed] [Google Scholar]
- Farah MJ, Haimm C, Sankoorikal G, Smith ME, Chatterjee A. When we enhance cognition with Adderall, do we sacrifice creativity? A preliminary study. Psychopharmacology (Berl) 2009;202:541–547. doi: 10.1007/s00213-008-1369-3. [DOI] [PubMed] [Google Scholar]
- Fedulov V, Rex CS, Simmons DA, Palmer L, Gall CM, Lynch G. Evidence that long-term potentiation occurs within individual hippocampal synapses during learning. J Neurosci. 2007;27:8031–8039. doi: 10.1523/JNEUROSCI.2003-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay BL, Darlington RB, Nicastro N. Developmental structure in brain evolution. Behav Brain Sci. 2001;24:263–278. discussion 278–308. [PubMed] [Google Scholar]
- Franowicz JS, Arnsten AF. Treatment with the noradrenergic alpha-2 agonist clonidine, but not diazepam, improves spatial working memory in normal young rhesus monkeys. Neuropsychopharmacology. 1999;21:611–621. doi: 10.1016/S0893-133X(99)00060-3. [DOI] [PubMed] [Google Scholar]
- Gold SJ, Ambros-Ingerson J, Horowitz JR, Lynch G, Gall CM. Stoiciometries of AMPA receptor subunit mRNAs in rat brain fall into discrete categories. J Comp Neurol. 1997;385:491–502. doi: 10.1002/(sici)1096-9861(19970908)385:4<491::aid-cne1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Greely H, Sahakian B, Harris J, Kessler RC, Gazzaniga M, Campbell P, Farah MJ. Towards responsible use of cognitive-enhancing drugs by the healthy. Nature. 2008;456:702–705. doi: 10.1038/456702a. [DOI] [PubMed] [Google Scholar]
- Gureviciene I, Puolivali J, Pussinen R, Wang J, Tanila H, Ylinen A. Estrogen treatment alleviates NMDA-antagonist induced hippocampal LTP blockade and cognitive deficits in ovariectomized mice. Neurobiol Learn Mem. 2003;79:72–80. doi: 10.1016/s1074-7427(02)00012-6. [DOI] [PubMed] [Google Scholar]
- Haberly LB, Bower JM. Olfactory cortex: model circuit for study of associative memory? Trends Neurosci. 1989;12:258–264. doi: 10.1016/0166-2236(89)90025-8. [DOI] [PubMed] [Google Scholar]
- Hampson R, Rogers G, Lynch G, Deadwyler S. Facilitative effects of the ampakine CX516 on short-term memory in rats: enhancement of delayed-nonmatch-to-sample performance. J Neurosci. 1998a;18:2740–2747. doi: 10.1523/JNEUROSCI.18-07-02740.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson R, Rogers G, Lynch G, Deadwyler S. Facilitative effects of the ampakine CX516 on short-term memory in rats: correlations with hippocampal neuronal activity. J Neurosci. 1998b;18:2748–2763. doi: 10.1523/JNEUROSCI.18-07-02748.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson RE, Espana RA, Rogers GA, Porrino LJ, Deadwyler SA. Mechanisms underlying cognitive enhancement and reversal of cognitive deficits in nonhuman primates by the ampakine CX717. Psychopharmacology (Berl) 2009;202:355–369. doi: 10.1007/s00213-008-1360-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebb DO. Drives and the C.N.S. (central nervous system) Psych Rev. 1955;62:243–254. doi: 10.1037/h0041823. [DOI] [PubMed] [Google Scholar]
- Hopfield JJ. Neurons with graded response have collective computational properties like those of two-state neurons. Proc Natl Acad Sci U S A. 1984;81:3088–3092. doi: 10.1073/pnas.81.10.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe WM, Ji J, Parikh V, Williams S, Mocaer E, Trocme-Thibierge C, Sarter M. Enhancement of attentional performance by selective stimulation of alpha4beta2(*) nAChRs: underlying cholinergic mechanisms. Neuropsychopharmacology. 2010;35:1391–1401. doi: 10.1038/npp.2010.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingvar M, Ambros-Ingerson J, Davis M, Granger R, Kessler M, Rogers GA, Schehr RS, Lynch G. Enhancement by an ampakine of memory encoding in humans. Exp Neurol. 1997;146:553–559. doi: 10.1006/exnr.1997.6581. [DOI] [PubMed] [Google Scholar]
- Ishizuka T, Sakamoto Y, Sakurai T, Yamatodani A. Modafinil increases histamine release in the anterior hypothalamus of rats. Neurosci Lett. 2003;339:143–146. doi: 10.1016/s0304-3940(03)00006-5. [DOI] [PubMed] [Google Scholar]
- Jiang L, Role LW. Facilitation of cortico-amygdala synapses by nicotine: activity-dependent modulation of glutamatergic transmission. J Neurophysiol. 2008;99:1988–1999. doi: 10.1152/jn.00933.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R, Clark S, Weeks AM, Dudman JT, Gouaux E, Partin KM. Mechanism of positive allosteric modulators acting on AMPA receptors. J Neurosci. 2005;25:9027–9036. doi: 10.1523/JNEUROSCI.2567-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DM, Illig KR, Behan M, Haberly LB. New features of connectivity in piriform cortex visualized by intracellular injection of pyramidal cells suggest that “primary” olfactory cortex functions like “association” cortex in other sensory systems. J Neurosci. 2000;20:6974–6982. doi: 10.1523/JNEUROSCI.20-18-06974.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kant I. In: Critique of Judgement. Werner S, translator. Pluhar: Hackett Publishing Co; 1987. [Google Scholar]
- Kramar EA, Chen LY, Lauterborn JC, Simmons DA, Gall CM, Lynch G. BDNF upregulation rescues synaptic plasticity in middle-aged ovariectomized rats. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramar EA, Chen LY, Rex CS, Gall CM, Lynch G. Estrogen’s Place in the Family of Synaptic Modulators. Molecular and Cellular Pharmacology Mol and Cell Pharmacol. 2009;1:258–262. [PMC free article] [PubMed] [Google Scholar]
- Kramar EA, Lynch G. Developmental and regional differences in the consolidation of long-term potentiation. Neuroscience. 2003;118:387–398. doi: 10.1016/s0306-4522(02)00916-8. [DOI] [PubMed] [Google Scholar]
- Lamirault L, Simon H. Enhancement of place and object recognition memory in young adult and old rats by RS 67333, a partial agonist of 5-HT4 receptors. Neuropharmacology. 2001;41:844–853. doi: 10.1016/s0028-3908(01)00123-x. [DOI] [PubMed] [Google Scholar]
- Larson J, Lieu T, Petchpradub V, LeDuc B, Ngo H, Rogers GA, Lynch G. Facilitation of olfactory learning by a modulator of AMPA receptors. J Neurosci. 1995;15:8023–8030. doi: 10.1523/JNEUROSCI.15-12-08023.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterborn JC, Rex CS, Kramar E, Chen LY, Pandyarajan V, Lynch G, Gall CM. Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J Neurosci. 2007;27:10685–10694. doi: 10.1523/JNEUROSCI.2624-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Albiston AL, Allen AM, Mendelsohn FA, Ping SE, Barrett GL, Murphy M, Morris MJ, McDowall SG, Chai SY. Effect of I.C.V. injection of AT4 receptor ligands, NLE1-angiotensin IV and LVV-hemorphin 7, on spatial learning in rats. Neuroscience. 2004;124:341–349. doi: 10.1016/j.neuroscience.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Loughead J, Ray R, Wileyto EP, Ruparel K, Sanborn P, Siegel S, Gur RC, Lerman C. Effects of the alpha4beta2 partial agonist varenicline on brain activity and working memory in abstinent smokers. Biol Psychiatry. 2010;67:715–721. doi: 10.1016/j.biopsych.2010.01.016. [DOI] [PubMed] [Google Scholar]
- Luo T, Leung LS. Endogenous histamine facilitates long-term potentiation in the hippocampus during walking. J Neurosci. 2010;30:7845–7852. doi: 10.1523/JNEUROSCI.1127-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G. Synapses, Circuits and the Beginnings of Memory. Cambridge, MA: MIT Press; 1986. [Google Scholar]
- Lynch G. Memory enhancement: the search for mechanism-based drugs. Nature Neurosci. 2002;5:1035–1038. doi: 10.1038/nn935. [DOI] [PubMed] [Google Scholar]
- Lynch G. AMPA receptor modulators as cognitive enhancers. Curr Opin Pharmacol. 2004a;4:4–11. doi: 10.1016/j.coph.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Lynch G. Glutamate-based therapeutic approaches: ampakines. Curr Opin Pharmacol. 2006;6:82–88. doi: 10.1016/j.coph.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Lynch G. Memory enhancement: the search for mechanism-based drugs. Nature Neurosci. 2002;5:1035–1038. doi: 10.1038/nn935. [DOI] [PubMed] [Google Scholar]
- Lynch G, Gall CM. Ampakines and the threefold path to cognitive enhancement. Trends Neurosci. 2006;29:554–562. doi: 10.1016/j.tins.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Lynch G, Granger R. Big Brain: The Origins and Future of Human Intelligence. New York: Palgrave Macmillan Press; 2008. [Google Scholar]
- Lynch G, Rex CS, Chen LY, Gall CM. The substrates of memory: defects, treatments, and enhancement. Eur J Pharmacol. 2008;585:2–13. doi: 10.1016/j.ejphar.2007.11.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Chen LY, Gall CM. Synaptic Mechanisms for Encoding Memory Encyclopedia of Behavioral Neuroscience. In: Koob G, Thompson RF, LeMoal M, editors. T Shors. 2. Academic Press, an imprint of Elsevier Ltd; London: 2009. [Google Scholar]
- Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004b;84:87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- Madras BK, Xie Z, Lin Z, Jassen A, Panas H, Lynch L, Johnson R, Livni E, Spencer TJ, Bonab AA, Miller GM, Fischman AJ. Modafinil occupies dopamine and norepinephrine transporters in vivo and modulates the transporters and trace amine activity in vitro. J Pharmacol Exp Ther. 2006;319:561–569. doi: 10.1124/jpet.106.106583. [DOI] [PubMed] [Google Scholar]
- Makeig S, Gramann K, Jung TP, Sejnowski TJ, Poizner H. Linking brain, mind and behavior. Int J Psychophysiol. 2009;73:95–100. doi: 10.1016/j.ijpsycho.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmo RB. Activation: a neuropsychological dimension. Rass Giuliana Med. 1959;11:86–114. [PubMed] [Google Scholar]
- Markowski M, Ungeheuer M, Bitran D, Locurto C. Memory-enhancing effects of DHEAS in aged mice on a win-shift water escape task. Physiol Behav. 2001;72:521–525. doi: 10.1016/s0031-9384(00)00446-7. [DOI] [PubMed] [Google Scholar]
- Maubach K. GABA(A) receptor subtype selective cognition enhancers. Curr Drug Targets CNS Neurol Disord. 2003;2:233–239. doi: 10.2174/1568007033482779. [DOI] [PubMed] [Google Scholar]
- McCulloch WS, Pitts WH. A logical calculus of the ideas immanent in nervous activity. Bulletin of Mathematical Biophysics. 1943;5:115–133. [PubMed] [Google Scholar]
- Mehta MA, Owen AM, Sahakian BJ, Mavaddat N, Pickard JD, Robbins TW. Methylphenidate enhances working memory by modulating discrete frontal and parietal lobe regions in the human brain. J Neurosci. 2000;20:RC65. doi: 10.1523/JNEUROSCI.20-06-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minzenberg MJ, Carter CS. Modafinil: a review of neurochemical actions and effects on cognition. Neuropsychopharmacology. 2008;33:1477–1502. doi: 10.1038/sj.npp.1301534. [DOI] [PubMed] [Google Scholar]
- Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26:319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RE, Crowley JM, Smith RH, LaRoche RB, Dopheide MM. Modafinil improves attention, inhibitory control, and reaction time in healthy, middle-aged rats. Pharmacol Biochem Behav. 2007;86:531–541. doi: 10.1016/j.pbb.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Muller U, Steffenhagen N, Regenthal R, Bublak P. Effects of modafinil on working memory processes in humans. Psychopharmacology (Berl) 2004;177:161–169. doi: 10.1007/s00213-004-1926-3. [DOI] [PubMed] [Google Scholar]
- Oertel BG, Felden L, Tran PV, Bradshaw MH, Angst MS, Schmidt H, Johnson S, Greer JJ, Geisslinger G, Varney MA, Lotsch J. Selective antagonism of opioid-induced ventilatory depression by an ampakine molecule in humans without loss of opioid analgesia. Clin Pharmacol Ther. 2010;87:204–21. doi: 10.1038/clpt.2009.194. [DOI] [PubMed] [Google Scholar]
- Palmer L. Kant and the Brain: A New Empirical Hypothesis. General Review of Psychology. 2008;12:105–117. [Google Scholar]
- Parikh V, Sarter M. Cholinergic mediation of attention: contributions of phasic and tonic increases in prefrontal cholinergic activity. Ann N Y Acad Sci. 2008;1129:225–235. doi: 10.1196/annals.1417.021. [DOI] [PubMed] [Google Scholar]
- Partin KM. Domain interactions regulating ampa receptor desensitization. J Neurosci. 2001;21:1939–1948. doi: 10.1523/JNEUROSCI.21-06-01939.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrino L, Daunais J, Rogers G, Hampson R, Deadwyler S. Facilitation of task performance and removal of the effects of sleep deprivation by an ampakine (CX717) in nonhuman primates. PLoS Biol. 2005;3:e299. doi: 10.1371/journal.pbio.0030299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall DC, Viswanath A, Bharania P, Elsabagh SM, Hartley DE, Shneerson JM, File SE. Does modafinil enhance cognitive performance in young volunteers who are not sleep-deprived? J Clin Psychopharmacol. 2005;25:175–179. doi: 10.1097/01.jcp.0000155816.21467.25. [DOI] [PubMed] [Google Scholar]
- Rex CS, Chen LY, Sharma A, Liu J, Babayan AH, Gall CM, Lynch G. Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J Cell Biol. 2009;186:85–97. doi: 10.1083/jcb.200901084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Kramar EA, Colgin LL, Lin B, Gall CM, Lynch G. Long-term potentiation is impaired in middle-aged rats: regional specificity and reversal by adenosine receptor antagonists. J Neurosci. 2005;25:5956–5966. doi: 10.1523/JNEUROSCI.0880-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman F, Staubli U, Lynch G. Evidence for synaptic potentiation in a cortical network during learning. Brain Res. 1987;418:221–226. doi: 10.1016/0006-8993(87)90089-8. [DOI] [PubMed] [Google Scholar]
- Sahakian BJ, Morein-Zamir S. Neuroethical issues in cognitive enhancement. J Psychopharmacol. 2010 doi: 10.1177/0269881109106926. [DOI] [PubMed] [Google Scholar]
- Sarter M, Martinez V, Kozak R. A neurocognitive animal model dissociating between acute illness and remission periods of schizophrenia. Psychopharmacology (Berl) 2009a;202:237–258. doi: 10.1007/s00213-008-1216-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Parikh V, Howe WM. nAChR agonist-induced cognition enhancement: integration of cognitive and neuronal mechanisms. Biochem Pharmacol. 2009b;78:658–667. doi: 10.1016/j.bcp.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvage MM. ROC in animals: uncovering the neural substrates of recollection and familiarity in episodic recognition memory. Conscious Cogn. 2010;19:816–828. doi: 10.1016/j.concog.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvage MM, Fortin NJ, Owens CB, Yonelinas AP, Eichenbaum H. Recognition memory: opposite effects of hippocampal damage on recollection and familiarity. Nat Neurosci. 2008;11:16–18. doi: 10.1038/nn2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scammell TE, Estabrooke IV, McCarthy MT, Chemelli RM, Yanagisawa M, Miller MS, Saper CB. Hypothalamic arousal regions are activated during modafinil-induced wakefulness. J Neurosci. 2000;20:8620–8628. doi: 10.1523/JNEUROSCI.20-22-08620.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel-Krüger J. Comparative studies of various amphetamine analogues demonstrating different interactions with the metabolism of the catecholamines in the brain. Eur J Psychopharmacology. 1971;14:47–59. doi: 10.1016/0014-2999(71)90121-x. [DOI] [PubMed] [Google Scholar]
- Schroeder SR, Mann-Koepke K, Gualtieri CT, Eckerman DA, Breese GR. Methylphenidate affects strategic choice behavior in normal adult humans. Pharmacol Biochem Behav. 1987;28:213–217. doi: 10.1016/0091-3057(87)90217-6. [DOI] [PubMed] [Google Scholar]
- Seeman P, Madras BK. Anti-hyperactivity medication: methylphenidate and amphetamine. Mol Psychiatry. 1998;3:386–396. doi: 10.1038/sj.mp.4000421. [DOI] [PubMed] [Google Scholar]
- Semendeferi K, Armstrong E, Schleicher A, Zilles K, Van Hoesen GW. Prefrontal cortex in humans and apes: a comparative study of area 10. Am J Phys Anthropol. 2001;114:224–241. doi: 10.1002/1096-8644(200103)114:3<224::AID-AJPA1022>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Shuman T, Wood SC, GAS Modafinil and Memory: Effects of Modafinil on Morris Water Maze Learning and Pavlovian Fear Conditioning. Behavioral Neuroscience. 2009;123:257–266. doi: 10.1037/a0014366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DA, Rex CS, Palmer L, Pandyarajan V, Fedulov V, Gall CM, Lynch G. Up-regulating BDNF with an ampakine rescues synaptic plasticity and memory in Huntington’s disease knockin mice. Proc Natl Acad Sci U S A. 2009;106:4906–4911. doi: 10.1073/pnas.0811228106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon P, Hemet C, Ramassamy C, Costentin J. Non-amphetaminic mechanism of stimulant locomotor effect of modafinil in mice. Eur Neuropsychopharmacol. 1995;5:509–514. doi: 10.1016/0924-977x(95)00041-m. [DOI] [PubMed] [Google Scholar]
- Sirvio J, Larson J, Quach CN, Rogers GA, Lynch G. Effects of pharmacologically facilitating glutamatergic transmission in the trisynaptic intrahippocampal circuit. Neuroscience. 1996;74:1025–1035. doi: 10.1016/0306-4522(96)00170-4. [DOI] [PubMed] [Google Scholar]
- Staubli U, Fraser D, Kessler M, Lynch G. Studies on retrograde and anterograde amnesia of olfactory memory after denervation of the hippocampus by entorhinal cortex lesions. Beh and Neural Biol. 1986;46:432–444. doi: 10.1016/s0163-1047(86)90464-4. [DOI] [PubMed] [Google Scholar]
- Staubli U, Perez Y, Xu F, Rogers G, Ingvar M, Stone-Elander S, Lynch G. Centrally active modulators of glutamate (AMPA) receptors facilitate the induction of LTP in vivo. Proc Natl Acad Sci USA. 1994;91:11158–11162. doi: 10.1073/pnas.91.23.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone EA, Cotecchia S, Lin Y, Quartermain D. Role of brain alpha 1B-adrenoceptors in modafinil-induced behavioral activity. Synapse. 2002;46:269–270. doi: 10.1002/syn.10127. [DOI] [PubMed] [Google Scholar]
- Thakkar MM. Histamine in the regulation of wakefulness. Sleep Med Rev. 2010 doi: 10.1016/j.smrv.2010.06.004. ePub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DC, Robbins TW, Clark L, Aron AR, Dowson J, Sahakian BJ. Cognitive enhancing effects of modafinil in healthy volunteers. Psychopharmacology (Berl) 2003;165:260–269. doi: 10.1007/s00213-002-1250-8. [DOI] [PubMed] [Google Scholar]
- Uslaner JM, Parmentier-Batteur S, Flick RB, Surles NO, Lam JS, McNaughton CH, Jacobson MA, Hutson PH. Dose-dependent effect of CDPPB, the mGluR5 positive allosteric modulator, on recognition memory is associated with GluR1 and CREB phosphorylation in the prefrontal cortex and hippocampus. Neuropharm. 2009;57:531–538. doi: 10.1016/j.neuropharm.2009.07.022. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Logan J, Alexoff D, Zhu W, Telang F, Wang GJ, Jayne M, Hooker JM, Wong C, Hubbard B, Carter P, Warner D, King P, Shea C, Xu Y, Muench L, Apelskog-Torres K. Effects of modafinil on dopamine and dopamine transporters in the male human brain: clinical implications. JAMA. 2009;301:1148–1154. doi: 10.1001/jama.2009.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Ding YS. Imaging the effects of methylphenidate on brain dopamine: new model on its therapeutic actions for attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57:1410–1415. doi: 10.1016/j.biopsych.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Wanchoo SJ, Swann AC, Dafny N. Descending glutamatergic pathways of PFC are involved in acute and chronic action of methylphenidate. Brain Res. 2009;1301:68–79. doi: 10.1016/j.brainres.2009.08.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CP, Harsh JR, York KM, Stewart KL, McCoy JG. Modafinil facilitates performance on a delayed nonmatching to position swim task in rats. Pharmacol Biochem Behav. 2004;78:735–741. doi: 10.1016/j.pbb.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Waters KA, Burnham KE, O’Connor D, Dawson GR, Dias R. Assessment of modafinil on attentional processes in a five-choice serial reaction time test in the rat. J Psychopharmacol. 2005;19:149–158. doi: 10.1177/0269881105048995. [DOI] [PubMed] [Google Scholar]
- Wetzel CD, Squire LR, Janowsky DS. Methylphenidate impairs learning and memory in normal adults. Behav Neural Biol. 1981;31:413–424. doi: 10.1016/s0163-1047(81)91481-3. [DOI] [PubMed] [Google Scholar]
- Wezenberg E, Verkes RJ, Ruigt GS, Hulstijn W, Sabbe BG. Acute effects of the ampakine farampator on memory and information processing in healthy elderly volunteers. Neuropsychopharmacology. 2007;32:1272–1283. doi: 10.1038/sj.npp.1301257. [DOI] [PubMed] [Google Scholar]
- Willie JT, Renthal W, Chemelli RM, Miller MS, Scammell TE, Yanagisawa M, Sinton CM. Modafinil more effectively induces wakefulness in orexin-null mice than in wild-type littermates. Neuroscience. 2005;130:983–995. doi: 10.1016/j.neuroscience.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Wixted JT, Squire LR. Constructing receiver operating characteristics (ROCs) with experimental animals: cautionary notes. Learn Mem. 2008;15:687–690. doi: 10.1101/lm.1077708. [DOI] [PubMed] [Google Scholar]
- Yerkes RM, Dodson JD. The relation of strength of stimulus to rapidity of habit formation. J Comp Physiol Psych. 1908;18:459–482. [Google Scholar]
- Zolkowska D, Jain R, Rothman RB, Partilla JS, Roth BL, Setola V, Prisinzano TE, Baumann MH. Evidence for the involvement of dopamine transporters in behavioral stimulant effects of modafinil. J Pharmacol Exp Ther. 2009;329:738–746. doi: 10.1124/jpet.108.146142. [DOI] [PMC free article] [PubMed] [Google Scholar]