

Abstract
Exercise induces growth of heart muscle cells and heart size. A new report in Cell (Boström et al., 2010) suggests that mice also respond to exercise with increased cardiac myocyte proliferation, and the molecular regulators of this pathway are linked to maladaptive and pathologic responses to cardiac stresses such as pressure overload.
Shortly after birth, cardiomyocyte proliferation largely ceases, and continued growth of the heart is achieved by further increases in the size of pre-existing cardiomyocytes (hypertrophy). Conditions that place a physiologic gradual increase in demand on the heart, such as endurance training or pregnancy, can enhance cardiac growth rates and pump function (physiologic hypertrophy). On the other hand, pathologic stress such as sustained pressure overload resulting from high blood pressure or valvular abnormalities, can lead to a maladaptive form of cardiac growth (pathologic hypertrophy), which is associated with loss of cardiomyocytes, fibrosis, progressive cardiac dysfunction, and heart failure. At the molecular level, the similarities and distinctions between physiologic and pathologic hypertrophy have remained largely mysterious. Recently, Boström and colleagues reported a novel and provocative molecular relationship between these cellular pathways involving CCAAT/enhancer-binding protein (C/EBP) β, suggesting that one mechanism by which endurance exercise enhances cardiac function is by inducing cardiomyocyte proliferation (Boström et al., 2010).
To compare transcriptional programs regulating pathologic and physiologic hypertrophy in mice, Boström et al. utilized a quantitative PCR-based screening method, termed “Quanttrx” (Gupta et al., 2010), and cardiac samples from young adult mice (12 weeks of age) after thoracic aortic constriction (TAC) surgery, a model of pathologic hypertrophy, or swimming exercise, a model of physiologic hypertrophy. They observed little overlap between the transcriptional programs activated by these different forms of stimulation. After further validation, the authors chose to focus on C/EBPβ, which was dramatically reduced in exercised hearts (by about 60%), but not in those exposed to pressure overload.
C/EBPβ is a basic-helix-loop-helix transcription factor expressed in a variety of tissues including fat, liver, and heart. Since C/EBPβ levels were reduced with exercise, Boström et al. examined heterozygous mice with partial loss of C/EBPβ function. These animals exhibited signs of endurance training even without exercise. Partial loss of C/EBPβ was associated with an increase in Cbp/p300-interacting transactivator with ED-rich carboxy-terminal domain (CITED) 4 expression, and this increase was necessary for the manifestation of reduced C/EPβ activity. This suggests that reduced C/EBPβ function and resulting increases in CITED4 are at least partially responsible for the molecular consequences of endurance training on the heart.
Heterozygous C/EBPβ mice not only mimic aspects of exercise-trained animals, but are also resistant to the maladaptive responses normally seen after exposure to pressure overload. Heterozygous animals show a less steep decline in cardiac function and fewer signs of heart failure than wild-type animals after TAC. Although not examined in this study, it will be interesting to determine if endurance training provides the same protection to pathologic stress and if the degree of protection is enhanced by C/EBPβ haploinsufficiency. Furthermore, it will be important to confirm that the effects of C/EBPβ haploinsufficiency are due to loss of function within cardiomyocytes by analyzing inducible tissue-specific deletion in adult heart cells.
How is C/EBPβ-regulated in response to exercise, and how does its loss result in protection from pathologic stress? The insulin-like growth factor (IGF) pathway has been implicated in physiologic hypertrophy and control of cell size via activation of thymoma viral proto-oncogene (AKT) and downstream signaling components (DeBosch et al., 2006; Dorn and Force, 2005). Boström et al. showed that AKT activation results in downregulation of C/EBPβ (Figure 1). Furthermore, C/EBPβ can inhibit binding of serum response factor (SRF) to DNA, providing a potential mechanism for regulation of downstream signaling during hypertrophy. SRF activity in the heart is potently regulated by the myocardin family of transcriptional coactivators and by the homeodomain-containing protein Hopx. These transcriptional complexes are implicated in pathologic hypertrophy downstream of neurohormonal and calcium-mediated signals (Kook et al., 2003; Wang et al., 2001). SRF functionally interacts with other cardiac-specific transcription factors including Nkx2-5 and Gata4. Loss of C/EBPβ as a result of endurance training might result in enhanced SRF transcriptional activity and activation of pathways associated with pathologic hypertrophy; yet, pathologic hypertrophy is reduced in C/EBPβ heterozygotes. Clearly, a more detailed understanding of the relationship of C/EBPβ to other hypertrophic signaling cascades is required. In this regard, it is worth noting that C/EBPβ can interact with nuclear factor of activated T cells (NFAT) and modulate its activity (Oh et al., 2010), and NFAT can modulate pathologic hypertrophy downstream of calcium signaling.
Figure 1. Signaling Pathways Regulating Physiologic and Pathologic Cardiac Hypertrophy.
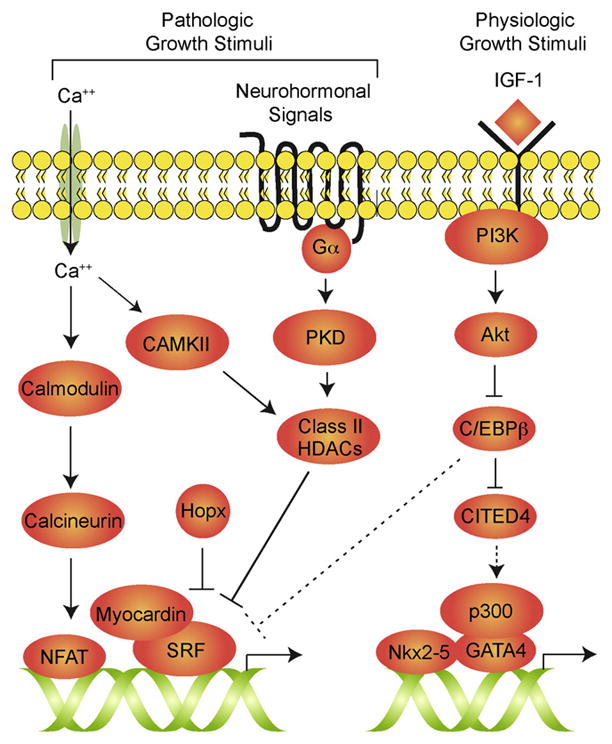
Neurohormonal and calcium (Ca2+) -mediated pathways can activate signaling cascades in cardiac myocytes that lead to activation of maladaptive hypertrophy. Exercise, pregnancy, and other physiologic growth stimuli can act through insulin-like growth factor (IGF) 1 to stimulate phosphoinositide-3 kinase (PI3K) and Akt, which inhibits C/EBPβ. C/EBPβ may function through inhibition of CITED4 to regulate physiologic hypertrophy and myocyte proliferation, and it may also affect the activity of serum response factor (SRF) to modulate pathologic responses (dotted lines).
Boström et al. also noted a marked increase in DNA synthesis within cardiomyocytes of endurance-trained animals. They suggest that cardiac myocyte proliferation is enhanced by exercise, and that myocyte hyperplasia (more cells) may account for some of the beneficial effects of exercise, in addition to effects mediated by myocyte hypertrophy.
The ability of adult cardiac myoctyes to proliferate remains controversial. Classic teaching suggests myocyte proliferation ceases shortly after birth, but recent reports indicate that this may not be the case (Bersell et al., 2009), and some turnover of adult myocytes may occur in both mice and humans (Bergmann et al., 2009; Hsieh et al., 2007). Studies of cardiomyocyte proliferation are complicated by the fact that these cells can be polyploid and undergo DNA synthesis and nuclear division to produce multinucleated cells without undergoing cellular division. DNA synthesis and polyploidy are increased under conditions of cardiac stress and hypertrophy, a response requiring cyclinG1 (Liu et al., 2010). Thus, the demonstration of DNA synthesis does not necessarily mean that more myocytes have been produced. Boström et al. addressed this issue by examining expression of aurora B kinase, which is a necessary component of the contractile ring mediating cytoplasmic separation and cell division. The paper identifies an increase in aurora B kinase expressing myocytes after exercise, although the numbers appear small (~0.2% of myocytes after exercise compared to >6% of myocytes that had incorporated BrdU). Hence, it remains unclear how many myocytes actually undergo cell division in response to exercise, how many new myocytes are produced, and to what degree this accounts for beneficial effects. It will be of interest to apply alternative quantitative approaches, such as those used to estimate myocyte turnover after myocardial infarction or pressure overload (Hsieh et al., 2007), to validate these findings. It is also important to determine if similar changes in myocyte proliferation occur in older animals, since the mice used in this study were relatively young (12 weeks old) and proliferation potential likely decreases with age.
If exercise truly enhances the heart’s ability to proliferate and regenerate, perhaps recovery after myocardial injury would be enhanced after endurance training or in C/EBPβ heterozygous mice. If so, C/EBPβ may be the tip of a targetable pathway for cardiac protection and regeneration in humans, and future studies may provide us with yet more incentive to exercise.
References
- Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, et al. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersell K, Arab S, Haring B, Kuhn B. Cell. 2009;138:257–270. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- Boström P, Mann N, Wu J, Quintero PA, Plovie ER, Panáková D, Gupta RK, Xiao C, MacRae CA, Rosenzweig A, Spiegelman BM. Cell. 2010;143:1072–1083. doi: 10.1016/j.cell.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, Muslin AJ. Circulation. 2006;113:2097–2104. doi: 10.1161/CIRCULATIONAHA.105.595231. [DOI] [PubMed] [Google Scholar]
- Dorn GW, 2nd, Force T. J Clin Invest. 2005;115:527–537. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Arany Z, Seale P, Mepani RJ, Ye L, Conroe HM, Roby YA, Kulaga H, Reed RR, Spiegelman BM. Nature. 2010;464:619–623. doi: 10.1038/nature08816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT. Nat Med. 2007;13:970–974. doi: 10.1038/nm1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kook H, Lepore JJ, Gitler AD, Lu MM, Wing-Man Yung W, Mackay J, Zhou R, Ferrari V, Gruber P, Epstein JA. J Clin Invest. 2003;112:863–871. doi: 10.1172/JCI19137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Yue S, Chen X, Kubin T, Braun T. Circ Res. 2010;106:1498–1506. doi: 10.1161/CIRCRESAHA.109.211888. [DOI] [PubMed] [Google Scholar]
- Oh M, Dey A, Gerard RD, Hill JA, Rothermel BA. J Biol Chem. 2010;285:16623–16631. doi: 10.1074/jbc.M109.098236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN. Cell. 2001;105:851–862. doi: 10.1016/s0092-8674(01)00404-4. [DOI] [PubMed] [Google Scholar]