

Abstract
Endovascular infections with Staphylococcus aureus (S. aureus) are associated with high mortality. gC1qR/p33 (gC1qR), a receptor for the complement component C1q expressed on endothelial cells, interacts with protein A of S. aureus and gC1qR blockade reduces S. aureus colonization during infective endocarditis. The aim of this study was to analyze in vivo whether this observation is due to a decreased interaction of S. aureus with the microvascular endothelium. A dorsal skinfold chamber was prepared in Syrian golden hamsters, which were treated with the monoclonal antibody (MAb) 74.5.2 directed against gC1qR or vehicle. The interaction of fluorescein isothiocyanate (FITC)-labeled staphylococci and leukocytes with the endothelium was analyzed under physiological conditions as well as after TNF-α-induced inflammation using intravital fluorescence microscopy. Administration of MAb 74.5.2 significantly reduced adherence of S. aureus to the endothelium in untreated and TNF-α-exposed tissue. In addition, we could demonstrate in vitro that S. aureus adherence to human endothelial cells was inhibited by MAb 74.5.2. Blockade of gC1qR did not affect leukocyte-endothelial cell interaction. In conclusion, our findings indicate that immunological inhibition of gC1qR may be therapeutically used to decrease the interaction of S. aureus with the microvascular endothelium.
Keywords: Staphylococcus aureus, endothelium, gC1qR/p33, monoclonal antibody, adherence, infective endocarditis, dorsal skinfold chamber, intravital fluorescence microscopy
Introduction
Staphylococcus aureus (S. aureus) is a highly virulent pathogen causing a wide array of community- and hospital-acquired infections (Lowy, 1998). Today, antimicrobial resistance is a major issue in the treatment of invasive S. aureus disease, as methicillin-resistant S. aureus (MRSA) is pandemic worldwide (Westh et al., 2006; Wisplinghoff et al., 2004) and as the advent of community-acquired MRSA in many parts of the world now represents an additional serious public health problem (Boyle-Vavra and Daum, 2007; Fridkin et al., 2005; Stam-Bolink et al., 2007).
S. aureus-induced endovascular infections are of particular importance as they are associated with a high mortality rate (Madani, 2002; Yeaman and Bayer, 2000). Pathogenetically, endovascular S. aureus disease is a multistep process involving primary bacterial adherence to the endothelium, establishment and persistence of the infection, bacterial proliferation usually associated with tissue damage and finally dissemination of septic emboli and organic spread (Foster and McDevitt, 1994; Hartleib et al., 2000; Herrmann et al., 1988; Moreillon et al., 2002).
During the last few years, it has been reported that S. aureus expresses a number of bacterial cell wall-anchored proteins, which mediate bacterial adherence to host cells as well as extracellular matrix (ECM) components. These include FnBPA and FnBPB, which recognize fibronectin and fibrinogen (Foster and Hook, 1998; Wann et al., 2000), or ClfA and ClfB binding to fibrinogen (McDevitt et al., 1994; Ni et al., 1998). Another important adherence protein of S. aureus is protein A (Hartleib et al., 2000), which has been shown to bind to von Willebrand factor (vWF) and to gC1qR/p33 (gC1qR) (Hartleib et al., 2000; Nguyen et al., 2000).
The cellular protein gC1qR is expressed on activated platelets (Peerschke et al., 2003) and on endothelial cells (Guo et al., 1999). Furthermore, gC1qR is present in the extracellular matrix (Hasan et al., 1998) and circulates in a soluble form in the blood plasma (Peterson et al., 1997; van den Berg et al., 1997). Interestingly, recent studies have demonstrated that gC1qR enhances S. aureus-fibrinogen interactions and that administration of monoclonal antibodies (MAbs) directed against gC1qR inhibit S. aureus tissue colonization during infective endocarditis (Peerschke et al., 2006). Accordingly, gC1qR may play a crucial role in the pathogenesis of S. aureus endovascular infections and, thus, may represent a novel target for the establishment of therapies preventing colonization and subsequent infection.
To test this hypothesis, we used the dorsal skinfold chamber model in Syrian golden hamsters, which allows to analyze in vivo the interaction of fluorescein isothiocyanate (FITC)-labeled staphylococci with the microvascular endothelium by means of intravital fluorescence microscopy (Kerdudou et al., 2006; Laschke et al., 2005a; Roller et al., 2008). Using this animal model, we were able to study the effects of gC1qR blockade on S. aureus-endothelial cell interaction, which represents the initial step in the pathogenesis of S. aureus-associated endovascular diseases.
Materials and Methods
Animals
The experiments were conducted in accordance with the German legislation on protection of animals and the NIH Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Research Council, Washington, USA), and were approved by the local governmental animal care committee.
Eight- to ten-week-old male Syrian golden hamsters with a body weight of 60–80g were used for the study. The animals were housed one per cage and had free access to tap water and standard pellet food (Altromin, Lage, Germany) throughout the experiment.
Preparation of the dorsal skinfold chamber
Dorsal skinfold chambers were prepared in Syrian golden hamsters as described previously in detail (Menger et al., 2002). In brief, animals were anesthetized using sodium pentobarbital anesthesia (50 mg/kg body weight i.p.) and two symmetrical titanium frames were implanted on the extended dorsal skinfold of the hamsters, so that they sandwiched the double layer of skin. One layer of skin was then removed in a circular area of ~15mm in diameter, and the remaining layers (consisting of striated skin muscle, subcutaneous tissue and skin) were covered with a removable cover slip incorporated into one of the titanium frames (Figs. 1A and B). After the procedure, the animals were allowed to recover from anesthesia and surgery for at least 48h before the microcirculatory analyses.
Figure 1.
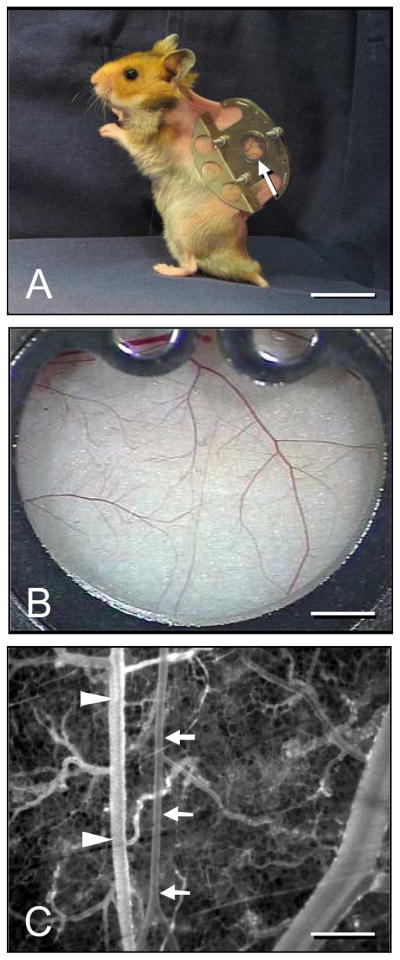
A: Syrian golden hamster equipped with a dorsal skinfold chamber (chamber weight ~ 4g). Within the observation window (arrow) all segments of the microcirculation including arterioles, capillaries and venules of the striated skin muscle and subcutaneous tissue can be analyzed using trans- and epi-illumination microscopy. B: Overview of the observation window. C: Intravital fluorescence microscopic image of the micro-angioarchitecture of the dorsal skinfold chamber displaying a representative venule (arrowheads) and arteriole (arrows). Note that the arteriole exhibits a straight-lined wall structure and much less bifurcations and branches than the venule. Blue light epi-illumination with intravascular plasma contrast enhancement by 5% FITC-labeled dextran 150,000 i.v.. Scale bars: A = 3cm, B = 1.7mm, C = 400μm.
Preparation of bacteria
For this study, we used the wild-type strain S. aureus Cowan 1 (ATCC 12598). Bacteria were prepared as described previously (Laschke et al., 2005a). To obtain 6h (late exponential phase) bacteria, S. aureus were cultured in 10 mL of Muller Hinton broth under constant rotation and then washed. This suspension yielded approximately 5 × 108 – 1 × 109 cfu/mL. The exact determination of each experiment’s bacterial number was performed by viable cell counting after vigorous vortexing of the washed, resuspended microorganisms. Subsequently, the bacteria were stained with FITC (20mg/mL; Sigma, Deisenhofen, Germany), washed again and fixed in 1% formaldehyde for a time period of 3h. Bacteria were fixed in order to i) ascertain synchronization of bacteria in the late exponential phase, which has been shown in previous studies to be optimal for the in vivo interaction of bacteria with the endothelium (Kerdudou et al., 2006), and ii) to exclude surface adhesin-independent effects on endothelial and blood cells, which might be mediated by a panoply of factors released from viable organisms. The fixed FITC-labeled staphylococci were then washed three times in PBS and the inoculum was adjusted to 109 cells/mL sterile PBS / 1% BSA (extrapolated from the number of viable counts prior to fixation). The sample was stored for 2–3 days at 4°C in the dark p rior to application, and 1 mL of this suspension was injected into each animal.
Intravital fluorescence microscopy
For the in vivo microscopic analysis of the microcirculation, the animals were anesthetized and a fine polyethylene catheter (PE10, 0.28mm internal diameter) was inserted into the A. carotis for application of bacteria and fluorescent dyes. Subsequently, the hamsters were put in lateral decubital position on a Plexiglas pad and the dorsal skinfold chamber preparation was attached to the microscopic stage. Intravital fluorescence microscopy was performed after i.a. injection of 109 FITC-labeled bacteria. At the end of the experiment, i.e. after the analysis of intravascular bacterial adherence, 0.1mL 5% FITC-labeled dextran 150,000 were additionally injected for contrast enhancement by staining of blood plasma in order to determine microhemodynamic parameters (Fig. 1C) and 0.1mL 0.1% rhodamine 6G (Sigma) for direct staining of white blood cells.
The microscope used for our study was a Zeiss Axiotech microscope (Zeiss; Oberkochen, Germany) with a 100W mercury lamp attached to an epi-illumination filter block for blue (450–490nm excitation; >520nm emission wavelength), green (530– 560nm; >580nm) and ultraviolet (330– 390nm; >430nm) light. The microscopic images were recorded by a charge-coupled device video camera (FK6990; Pieper, Schwerte, Germany) and transferred to a DVD system for off-line evaluation (Panasonic AG-7350-SVHS; Matsushita, Tokyo, Japan). By means of 5x (numerical aperture (n.a.) = 0.15), 20x (n.a. = 0.4) and 50x (n.a. = 0.55) objectives, magnifications of ×115, ×430 and ×1150 were achieved on a 14- inch video screen (KV-14CT1E; Sony, Tokyo, Japan).
Microcirculatory analysis
Quantitative off-line analysis of the DVDs was performed using the computer-assisted image analysis system CapImage (Zeintl; Heidelberg, Germany). In each animal, analyses were performed in 8–10 postcapillary or collecting venules. To exclude that differences in bacteria-endothelial cell interaction between MAb 74.5.2-treated animals and respective controls were caused by changes in local microhemodynamics, we additionally analyzed the diameters, centerline velocity, volumetric blood flow, and wall-shear rate of these vessels as described previously (Laschke et al., 2005a).
Adherent staphylococci were defined as bacteria which did not move or detach from the endothelial lining of venules during an observation period of 20s and are given as the number of adherent cocci/mm2 of endothelial surface. Leukocytes were classified according to their interaction with the microvascular endothelium as rolling or adherent cells (Laschke et al., 2009). Rolling leukocytes were defined as cells moving with a velocity less than two-fifths of the centerline velocity, and are given as number of cells/min, passing a reference point within the microvessel. Adherent leukocytes were defined as cells that did not move or detach from the endothelial lining within a 20s observation period, and are given as number of cells/mm² of endothelial surface.
In addition, macromolecular leakage as a parameter of microvascular permeability and, thus, endothelial integrity was assessed after intravenous injection of the macromolecular fluorescent dye FITC-labeled dextran 150,000 by determining densitometrically grey levels in the tissue directly adjacent to the venular vessel wall (E1), as well as in the marginal cell free plasma layer within the vessel (E2). Extravasation (E) was then calculated as E = E1/E2 (Laschke et al., 2005b).
Experimental protocol
A dorsal skinfold chamber was prepared in 25 Syrian golden hamsters. After 48h, animals received either an i.p. injection of 100mg/kg MAb 74.5.2 (1.6 mL, n = 13), which was kindly provided by the Cell Culture/Hybridoma Facility, Department of Molecular Genetics and Microbiology, Stony Brook University (Stony Brook, NY, USA), or serum free medium control (1.6 mL, n = 12) according to prior studies (Peerschke et al., 2006). Then, 48h later, the tissue of the dorsal skinfold chamber was exposed to tumor necrosis factor (TNF)-α (topical application for 30 min, 2000 U dissolved in 100μl PBS; Boehringer Mannheim, Germany) in order to induce inflammation. In a second set of experiments, the chamber tissue was solely exposed to 100μl PBS (topical application for 30 min). Subsequently, intravital fluorescence microscopy was performed at 5, 10, 20, 30 and 60 min after application of 109 FITC-labeled bacteria. At the end of the experiment, each animal received 5% FITC-labeled dextran 150,000 and 0.1% rhodamine 6G i.a. for the quantitative assessment of microhemodynamic parameters, macromolecular leakage, and leukocyte-endothelial cell interaction.
Enzyme-linked immunosorbent assay (ELISA)
To quantify levels of anti-gC1qR antibodies in the hamster serum, an ELISA was used as described previously (Peerschke et al., 2006). In brief, soluble, recombinant gC1qR was immobilized on 96-well plates. After blocking the wells, standard concentrations of anti-gC1qR antibodies (0–10 μg/mL) were prepared in naïve hamster serum diluted 1/10 with 0.3M NaCl and then added to the wells. Test hamster serum was diluted 1/10 in 0.3M NaCl and added to separate wells. Bound antibody was detected spectrophotometrically (OD405nm), using alkaline phosphatase-conjugated goat anti-mouse immunoglobulin G (IgG; Sigma) and p-NPP substrate. The reactivity of test plasmas was compared to that of standard anti-gC1qR antibody solutions. Each serum sample was evaluated in duplicate for data reproducibility. Results were expressed in μg/mL serum.
In vitro adherence assay
Mock or TNF-α-pretreated EA.hy endothelial cells (Edgell et al., 1983) were detached by trypsin treatment and subsequently exposed to FITC-labeled bacteria. Samples were harvested at the indicated times and attachment of bacteria was analyzed by using flow-cytometry (FACS Calibur and CellQuest Pro Software; Becton Dickinson, Heidelberg, Germany). Experiments were performed with MAb 74.5.2 (500μg/mL in serum-free medium + 10% FCS), respective controls (serum free medium + 10% FCS) and with as well as without TNF-α stimulation. The results were confirmed in five independent experiments.
Statistics
Differences between the groups were tested separately at each time point by the unpaired Student’s t-test and Mann-Whitney U test. To test for time effects within individual groups, multivariate ANOVA for repeated measures was applied. This was followed by the paired Student’s t-test, including correction of the α-error according to Bonferroni probabilities for repeated measurements (SigmaStat; Jandel Corporation, San Rafael, CA, USA). All values are expressed as means ± SEM. Statistical significance was accepted for a value of p<0.05.
Results
Serum concentration of MAb 74.5.2
In contrast to control animals (gC1qR MAb titer: 0±0 μg/mL), significantly high sustainable concentrations of MAb 74.5.2 were measured 48h following i.p. administration of 100mg/kg MAb 74.5.2 (gC1qR MAb titer: 109±23 μg/mL).
Effects of gC1qR blockade during TNF-α-induced inflammation
TNF-α-induced inflammation resulted in the adherence of many late exponential phase staphylococci to the endothelium of postcapillary and collecting venules in dorsal skinfold chambers of control animals (Figs. 2A and C). Highest numbers of adherent bacteria (~850mm−2) could be observed 5 min after S. aureus administration. This was followed by a progressive decline in bacterial adherence to ~510mm−2 at time point 60 min (Fig. 2C). In contrast, blockade of gC1qR by application of MAb 74.5.2 effectively reduced S. aureus adherence (Figs. 2B and C). Throughout the entire experiment, antibody-treated animals presented with significantly decreased numbers of adherent bacteria of ~260–480mm−2 when compared to controls (Fig. 2C; 5min: p = 0.038; 10min: p = 0.011; 20min: p = 0.011; 30min: p = 0.002; 60min: p = 0.021).
Figure 2.

A, B: Intravital fluorescence microscopy of FITC-labeled Staphylococcus aureus in postcapillary and collecting venules of the dorsal skinfold chamber (5 min after intra-arterial injection of bacteria) after local TNF-α-exposure. Note that blockade of gC1qR by application of MAb 74.5.2 (B) results in a significant reduction of bacterial adherence when compared to controls (A). Blue light epi-illumination; scale bars: 100μm. C: Adherent bacteria (mm−2) in venules of TNF-α-exposed dorsal skin-fold chambers of Syrian golden hamsters, which were treated with MAb 74.5.2 (black bars; n = 7) or vehicle (control, white bars; n = 7), as assessed by intravital fluorescence microscopy and computer-assisted image analysis. Measurements were performed at 5, 10, 20, 30, and 60 min after intra-arterial injection of the bacteria. Values are given as means ± SEM; *p<0.05 vs. control.
The analysis of microhemodynamic parameters, i.e. microvessel diameter, centerline velocity, volumetric blood flow and wall-shear rate, did not show any significant differences between antibody-treated animals and respective controls (Tab. 1). Furthermore, leukocyte-endothelial cell interaction remained unaffected under administration of the gC1qR antibody. In both groups, animals exhibited numbers of rolling and adherent leukocytes of ~6–7min−1 and ~800mm−2 (Figs. 3A and B). However, treatment with MAb 74.5.2 resulted in a significantly decreased macromolecular leakage (0.40 ± 0.03) when compared to controls (0.54 ± 0.02; p = 0.002) (Fig. 3C).
Table 1.
Diameter, centerline velocity, volumetric blood flow and wall shear rate of postcapillary and collecting venules within the dorsal skinfold chamber of antibody-treated (MAb) and vehicle-treated (Con) Syrian golden hamsters. Chambers were pretreated with TNF-α (MAb: n = 7; Con: n = 7) or PBS (MAb: n = 6; Con: n = 5). Parameters were assessed by intravital fluorescence microscopy and computer-assisted image analysis. Values are given as means ± SEM; data did not significantly differ between the groups.
| TNF-α | PBS | ||
|---|---|---|---|
| Diameter (μm) | TNF-α vs. PBS | ||
| Vehicle | 26.4 ± 2.1 | 24.6 ± 0.5 | p = 0.509 |
| MAb | 25.6 ± 0.6 | 24.3 ± 2.2 | p = 0.138 |
| Vehicle vs. MAb | p = 0.902 | p = 0.177 | |
| Centerline velocity (μm/s) | |||
| Vehicle | 145.9 ± 23.1 | 153.5 ± 32.2 | p = 0.849 |
| MAb | 169.6 ± 31.6 | 193.3 ± 60.2 | p = 0.722 |
| Vehicle vs. MAb | p = 0.557 | p = 0.596 | |
| Volumetric blood flow (pL/s) | |||
| Vehicle | 52.9 ± 12.6 | 46.3 ± 10.7 | p = 0.719 |
| MAb | 55.0 ± 10.6 | 58.1 ± 17.4 | p = 0.600 |
| Vehicle vs. MAb | p = 0.532 | p = 0.558 | |
| Wall shear rate (s−1) | |||
| Vehicle | 45.2 ± 7.2 | 49.6 ± 9.9 | p = 0.715 |
| MAb | 53.0 ± 9.7 | 65.1 ± 21.5 | p = 0.879 |
| Vehicle vs. MAb | p = 0.900 |
Figure 3.
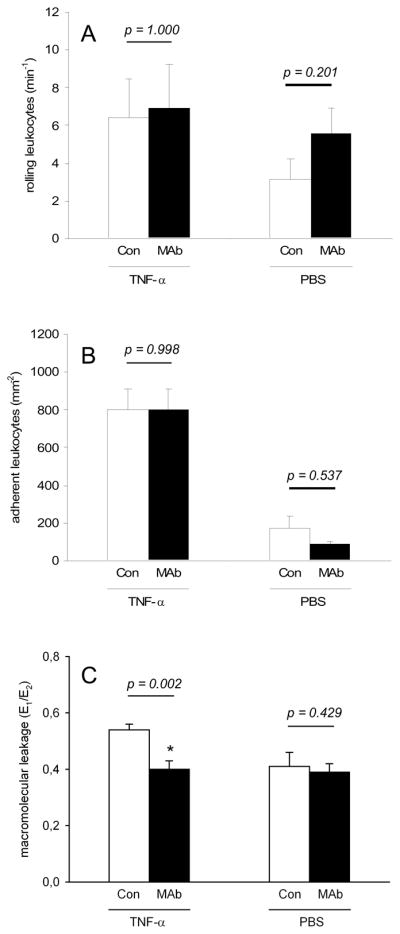
Rolling leukocytes (min−1) (A), adherent leukocytes (mm−2) (B) and macro-molecular leakage (E1/E2) (C) in venules of TNF-α- or PBS-exposed dorsal skinfold chambers of Syrian golden hamsters, which were treated with MAb 74.5.2 (MAb, black bars; TNF-α: n = 7, PBS: n = 6) or vehicle (Con, white bars; TNF-α: n = 7, PBS: n = 5), as assessed by intravital fluorescence microscopy and computer-assisted image analysis. Values are given as means ± SEM; *p<0.05 vs. control.
Effects of gC1qR blockade under baseline conditions
To determine whether the observed reduction of S. aureus adherence by gC1qR blockade was based on the interference with TNF-α-induced pathways, we further examined the effects of gC1qR blockade in dorsal skinfold chambers, which were not exposed to TNF-α. As expected, bacterial adherence in postcapillary and collecting venules was distinctly reduced under these baseline conditions when compared to TNF-α-treated tissue (Figs. 4A and C). Numbers of adherent bacteria ranged between ~200–335mm−2 in vehicle-treated animals throughout the observation period of 60 min. Nonetheless, treatment with the anti-gC1qR antibody again significantly decreased S. aureus adherence. In fact, antibody-treated animals presented with numbers of adherent bacteria of only ~70–135mm−2, according to a ~60% reduction when compared to controls (Figs. 4B and C; 5min: p = 0.052; 10min: p = 0.052; 20min: p = 0.018; 30min: p = 0.009; 60min: p = 0.030). These findings clearly indicate that bacterial adherence to the microvascular endothelium is directly affected by gC1qR blockade, independently from TNF-α-induced pathways. According to our first set of experiments, analysis of microhemodynamic parameters and leukocyte-endothelial cell interaction did not show any significant differences between antibody-treated and control animals (Tab. 1, Figs. 3A and B). Moreover, macromolecular leakage remained unaffected under treatment with MAb 74.5.2 (Fig. 3C).
Figure 4.

A, B: Intravital fluorescence microscopy of FITC-labeled Staphylococcus aureus in postcapillary and collecting venules of the dorsal skinfold chamber (5 min after intra-arterial injection of bacteria) under physiological conditions. Note that blockade of gC1qR by application of MAb 74.5.2 (B) results in a significant reduction of bacterial adherence when compared to controls (A). Blue light epi-illumination; scale bars: 100μm. C: Adherent bacteria (mm−2) in venules of PBS-exposed dorsal skinfold chambers of Syrian golden hamsters, which were treated with MAb 74.5.2 (black bars; n = 6) or vehicle (control, white bars; n = 5), as assessed by intravital fluorescence microscopy and computer-assisted image analysis. Measurements were performed at 5, 10, 20, 30, and 60min after intra-arterial injection of the bacteria. Values are given as means ± SEM; *p<0.05 vs. control.
Effects of gC1qR blockade on S. aureus adherence to human endothelial cells in vitro
In line with our in vivo experiments, we could also demonstrate in vitro the tendency that gC1qR blockade reduced the adherence of S. aureus to human endothelial cells, which had been exposed to TNF-α (Fig. 5A; 5min: p = 0.761; 10min: p = 0.217; 20min: p = 0.171; 30min: p = 0.220; 60min: p = 0.686). Moreover, bacterial adherence was also decreased, when cells were not pretreated with TNF-α (Fig. 5B; 5min: p = 0.529; 10min: p = 0.204; 20min: p = 0.180; 30min: p = 0.363; 60min: p = 0.486). However, the results of the in vitro experiments were not proven to be statistically significant.
Figure 5.

In vitro adherence of Staphylococcus aureus to endothelial cells. FITC-labeled bacteria were incubated with endothelial cells and adherence of Staphylococcus aureus to endothelial cells was measured using FACS analysis in the presence of MAb 74.5.2 (black bars; n = 5) and in respective controls (serum free medium + 10% FCS; white bars; n = 5). The experiments were performed with (A) and without TNF-α treatment (B). Values are given as means ± SEM.
Discussion
The present study demonstrates that blockade of gC1qR by administration of MAb 74.5.2 significantly reduces the adherence of S. aureus to the venular endothelium under in vivo conditions. For this purpose, we used the dorsal skinfold chamber model in combination with the technique of intravital fluorescence microscopy. This approach is unique as it allows for the real-time observation of staphylococcal interaction with the microvascular endothelium.
In a first set of experiments we studied the effects of MAb 74.5.2 administration on the adherence of S.aureus to the venular endothelium activated via TNF-α. TNF-α exerts a variety of different inflammatory effects on the endothelium (Bradley, 2008; Pober et al., 1998). Accordingly, we have previously shown that TNF-α markedly increases the number of vessel wall-adhering staphylococci (Laschke et al., 2005a). This was also the case in the present study, however, only under in vivo but not under in vitro conditions, indicating that the observed TNF-α-stimulated bacterial adherence may involve mechanisms additional to the expression of endothelial binding molecules on in situ or cultured endothelial cells. In fact, the endovascular system is characterized by various humoral and cellular factors contained in whole blood, many of which are also prone to TNF-α-mediated activation. These include binding of fibrinogen to endothelial cells or activation of circulating cells such as leukocytes (Vaday et al., 2001), which in turn may affect the binding of bacteria to the vessel wall.
In the present study, we now demonstrate that the TNF-α-induced adherence of S. aureus to the venular endothelium is significantly inhibited by blockade of gC1qR. On the one hand, this effect may be explained by the blockade of TNF-α-induced endothelial adherence of activated gC1qR-expressing platelets or leukocytes, which can serve as binding sites for free flowing bacteria in the microcirculation (Peerschke and Ghebrehiwet, 2007). However, in unactivated tissue we observed comparable results. Therefore, we suggest that administration of MAb 74.5.2 also directly affects the gC1qR-dependent binding of S. aureus to the microvascular endothelium. In line with this assumption, we could demonstrate in our in vitro assay excluding interfering platelets and leukocytes that S. aureus binding to human endothelial cells was inhibited by gC1qR-blockade albeit to a reduced extent when compared to our in vivo findings. Thereby, it was not surprising that in our in vitro and in vivo experiments not all adhesion of staphylococcus was inhibited by the anti-gC1qR antibody. In fact, there are various surface adhesins and ligands promoting the adhesion of S. aureus to the vascular endothelium (Moreillon et al., 2002). Particularly the interaction of vWF with its staphylococcal receptors protein A (Hartleib et al., 2000) or with the vWF binding protein (Bjerketorp et al., 2002) may mediate this interaction. Accordingly, it could not be expected that the MAb directed against gC1qR would completely abrogate this interaction, yet our study proves it to be a relevant and novel interaction partner in this system.
gC1qR is recognized by protein A on the surface of S. aureus (Nguyen et al., 2000). Although gC1qR interacts with protein A at or near the IgG binding site (Nguyen et al., 2000), we believe that the observed effects of our MAb 74.5.2 administration are based on specific interactions of the MAb rather than on unspecific IgG binding to protein A, because physiological concentrations of circulating IgG are high and application of low amounts of IgG as performed in this study is unlikely to have a significant effect.
As it was previously demonstrated that soluble gC1qR binds to S. aureus, it is conceivable that S. aureus decorated with gC1qR might interact with additional ligand molecules recognizing gC1qR. Candidate ligands include the fibrinogen D domain, which is a known binding partner for gC1qR. In fact, since fibrinogen/fibrin is an important component of endovascular lesions (Archer, 1998; Moreillon and Que, 2004; Peerschke et al., 2006), gC1qR-decoration may enhance S. aureus’ ability to interact with sites of local fibrinogen/fibrin exposure, e.g. sites of subendothelial exposure. Moreover, gC1qR has been shown to interact with various components of the human contact system, also known as the kallikrein-kinin cascade or intrinsic pathway of coagulation such as factor XII or high molecular-weight kininogen (HMW-kininogen) (Ghebrehiwet and Peerschke, 1998). The surface of S. aureus activates this contact system, resulting in the release of short-lived bradykinin (Mattsson et al., 2001) and a subsequent pro-inflammatory response mediated by conversion of bradykinin to the bradykinin 1 receptor (B1R) agonist desArg(9)bradykinin, which leads to a further up-regulation of B1R, and thereby ensues a chronic inflammatory response (Bengtson et al., 2006). Considering our findings, demonstrating a reduced interaction of S. aureus with the vessel wall upon blockade of gC1qR, it may be speculated that this interaction may also interfere with the S. aureus-triggered local contact system activation, resulting in a decreased inflammatory leukocyte and endothelial cell activation.
For these reasons, we also assessed the leukocyte-endothelial cell interaction in venules of the dorsal skinfold chamber. We found that exposure of the tissue to TNF-α resulted in a strong inflammation with significantly increased numbers of adherent leukocytes when compared to PBS-treated tissue. Meanwhile, it is well known that the interaction of leukocytes with the endothelium is regulated by specific ligand-receptor interactions. On activation, leukocytes roll along the venular endothelium mediated by the selectin family of adhesion molecules (Klintman et al., 2004; Laschke et al., 2008; Springer, 1994). This leukocyte rolling is a major prerequisite for the subsequent firm adhesion of leukocytes, which is mediated by integrins expressed on leukocytes, in particular lymphocyte function antigen-1 (LFA-1) and macrophage antigen-1 (MAC-1), which interact with molecules of the immunoglobulin supergene family expressed on endothelial cells, such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) (Carlos and Harlan, 1994; Springer, 1994). Interestingly, we found that blockade of gC1qR did not affect this well coordinated process, indicating that gC1qR blockade may selectively inhibit the adherence of S. aureus to the endothelium without interfering with the interaction of leukocytes and endothelial cells.
Furthermore, we could observe in our study a significantly decreased macromolecular leakage in TNF-α-exposed tissue of animals, which were treated with anti-gC1qR antibodies. Because we did not detect any effects of gC1qR blockade on leukocyte-endothelial cell interaction, we propose that this observation may be due to a direct inhibition of the gC1qR-mediated activation of the kallikrein-kinin cascade and, thus, the formation of bradykinin (Joseph et al., 1999). In fact, bradykinin has been shown to be a potent direct inducer of vascular permeability during infection (Matsumoto et al., 1986; Maeda et al., 1999). Accordingly, blockade of gC1qR may also prevent transvascular spread of bacteria.
Taken together, we could demonstrate in the present study that gC1qR blockade effectively inhibits the adherence of S. aureus to the microvascular endothelium. In light of the fact that gC1qR plays a pivotal role in the pathogenesis of S. aureus-associated endovascular disease and that the resistance of S. aureus to antimicrobial therapy is increasing (Chastre, 2008), immunological blockade of gC1qR represents a promising novel therapeutic strategy in the treatment of S. aureus-associated endovascular diseases.
Research highlights.
Intravital fluorescence microscopy for the analysis of bacteria-endothelial cell interaction
Blockade of gC1qR/p33 by administration of monoclonal antibodies
gC1qR/p33 blockade inhibits adherence of S. aureus to the microvascular endothelium
gC1qR/p33 blockade does not affect leukocyte-endothelial cell interaction
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) SPP 1130, He1870/7-2 (to M.H.), the Medical Faculty of the University of Saarland (HOMFOR) (to M.H. and M.D.M.) and the National Institutes of Health HL67211 (to E.I.P.) and A1060866 (to B.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Archer GL. Staphylococcus aureus: a well-armed pathogen. Clin Infect Dis. 1998;26:1179–1181. doi: 10.1086/520289. [DOI] [PubMed] [Google Scholar]
- Bengtson SH, Phagoo SB, Norrby-Teglund A, Påhlman L, Mörgelin M, Zuraw BL, Leeb-Lundberg LM, Herwald H. Kinin receptor expression during Staphylococcus aureus infection. Blood. 2006;108:2055–2063. doi: 10.1182/blood-2006-04-016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerketorp J, Nilsson M, Ljungh A, Flock JI, Jacobsson K, Frykberg L. A novel von Willebrand factor binding protein expressed by Staphylococcus aureus. Microbiology. 2002;148:2037–2044. doi: 10.1099/00221287-148-7-2037. [DOI] [PubMed] [Google Scholar]
- Boyle-Vavra S, Daum RS. Community-acquired methicillin-resistant Staphylococcus aureus: the role of Panton-Valentine leukocidin. Lab Invest. 2007;87:3–9. doi: 10.1038/labinvest.3700501. [DOI] [PubMed] [Google Scholar]
- Bradley JR. TNF-mediated inflammatory disease. J Pathol. 2008;214:149–160. doi: 10.1002/path.2287. [DOI] [PubMed] [Google Scholar]
- Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- Chastre J. Evolving problems with resistant pathogens. Clin Microbiol Infect. 2008;14:3–14. doi: 10.1111/j.1469-0691.2008.01958.x. [DOI] [PubMed] [Google Scholar]
- Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci USA. 1983;80:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster TJ, Hook M. Surface protein adhesins of Staphylococcus aureus. Trends Microbiol. 1998;6:484–488. doi: 10.1016/s0966-842x(98)01400-0. [DOI] [PubMed] [Google Scholar]
- Foster TJ, McDevitt D. Surface-associated proteins of Staphylococcus aureus: their possible roles in virulence. FEMS Microbiol Lett. 1994;118:199–205. doi: 10.1111/j.1574-6968.1994.tb06828.x. [DOI] [PubMed] [Google Scholar]
- Fridkin SK, Hageman JC, Morrison M, Sanza LT, Como-Sabetti K, Jernigan JA, Harriman K, Harrison LH, Lynfield R, Farley MM. Methicillin-resistant Staphylococcus aureus disease in three communities. N Engl J Med. 2005;352:1436–1444. doi: 10.1056/NEJMoa043252. [DOI] [PubMed] [Google Scholar]
- Ghebrehiwet B, Peerschke EI. Structure and function of gC1q-R: a multiligand binding cellular protein. Immunobiology. 1998;199:225–238. doi: 10.1016/S0171-2985(98)80029-6. [DOI] [PubMed] [Google Scholar]
- Guo WX, Ghebrehiwet B, Weksler B, Schweitzer K, Peerschke EI. Up-regulation of endothelial cell binding proteins/receptors for complement component C1q by inflammatory cytokines. J Lab Clin Med. 1999;133:541–550. doi: 10.1016/s0022-2143(99)90183-x. [DOI] [PubMed] [Google Scholar]
- Hartleib J, Kohler N, Dickinson RB, Chhatwal GS, Sixma JJ, Hartford OM, Foster TJ, Peters G, Kehrel BE, Herrmann M. Protein A is the von Willebrand factor binding protein on Staphylococcus aureus. Blood. 2000;96:2149–2156. [PubMed] [Google Scholar]
- Hasan AA, Zisman T, Schmaier AH. Identification of cytokeratin 1 as a binding protein and presentation receptor for kininogens on endothelial cells. Proc Natl Acad Sci USA. 1998;95:3615–3620. doi: 10.1073/pnas.95.7.3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann M, Vaudaux PE, Pittet D, Auckenthaler R, Lew PD, Schumacher-Perdreau F, Peters G, Waldvogel FA. Fibronectin, fibrinogen, and laminin act as mediators of adherence of clinical staphylococcal isolates to foreign material. J Infect Dis. 1988;158:693–701. doi: 10.1093/infdis/158.4.693. [DOI] [PubMed] [Google Scholar]
- Joseph K, Ghebrehiwet B, Kaplan AP. Cytokeratin 1 and gC1qR mediate high molecular weight kininogen binding to endothelial cells. Clin Immunol. 1999;92:246–255. doi: 10.1006/clim.1999.4753. [DOI] [PubMed] [Google Scholar]
- Kerdudou S, Laschke MW, Sinha B, Preissner KT, Menger MD, Herrmann M. Fibronectin binding proteins contribute to the adherence of Staphylococcus aureus to intact endothelium in vivo. Thromb Haemost. 2006;96:183–189. [PubMed] [Google Scholar]
- Klintman D, Li X, Thorlacius H. Important role of P-selectin for leukocyte recruitment, hepatocellular injury, and apoptosis in endotoxemic mice. Clin Diagn Lab Immunol. 2004;11:56–62. doi: 10.1128/CDLI.11.1.56-62.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laschke MW, Dold S, Menger MD, Jeppsson B, Thorlacius H. Platelet-dependent accumulation of leukocytes in sinusoids mediates hepatocellular damage in bile duct ligation-induced cholestasis. Br J Pharmacol. 2008;153:148–156. doi: 10.1038/sj.bjp.0707578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laschke MW, Kerdudou S, Herrmann M, Menger MD. Intravital fluorescence microscopy: a novel tool for the study of the interaction of Staphylococcus aureus with the microvascular endothelium in vivo. J Infect Dis. 2005a;191:435–443. doi: 10.1086/427193. [DOI] [PubMed] [Google Scholar]
- Laschke MW, Häufel JM, Roller J, Schorr H, Menger MD. Rapamycin, but not cyclosporine A, inhibits vascularization and incorporation of implanted surgical meshes. Transpl Int. 2009;22:654–662. doi: 10.1111/j.1432-2277.2009.00841.x. [DOI] [PubMed] [Google Scholar]
- Laschke MW, Häufel JM, Thorlacius H, Menger MD. New experimental approach to study host tissue response to surgical mesh materials in vivo. J Biomed Mater Res A. 2005b;74:696–704. doi: 10.1002/jbm.a.30371. [DOI] [PubMed] [Google Scholar]
- Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- Madani TA. Epidemiology and clinical features of methicillin-resistant Staphylococcus aureus in the University Hospital, Jeddah, Saudi Arabia. Can J Infect Dis. 2002;13:245–250. doi: 10.1155/2002/235213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda H, Wu J, Okamoto T, Maruo K, Akaike T. Kallikrein-kinin in infection and cancer. Immunopharmacology. 1999;43:115–128. doi: 10.1016/s0162-3109(99)00104-6. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Yamamoto T, Kamata R, Maeda H. Enhancement of vascular permeability upon serratial infection: activation of Hageman factor-kallikrein-kinin cascade. Adv Exp Med Biol. 1986;198:71–78. doi: 10.1007/978-1-4757-0154-8_9. [DOI] [PubMed] [Google Scholar]
- Mattsson E, Herwald H, Cramer H, Persson K, Sjöbring U, Björck L. Staphylococcus aureus induces release of bradykinin in human plasma. Infect Immun. 2001;69:3877–3882. doi: 10.1128/IAI.69.6.3877-3882.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDevitt D, Francois P, Vaudaux P, Foster TJ. Molecular characterization of the clumping factor (fibrinogen receptor) of Staphylococcus aureus. Mol Microbiol. 1994;11:237–248. doi: 10.1111/j.1365-2958.1994.tb00304.x. [DOI] [PubMed] [Google Scholar]
- Menger MD, Laschke MW, Vollmar B. Viewing the microcirculation through the window: some twenty years experience with the hamster dorsal skin-fold chamber. Eur Surg Res. 2002;34:83–91. doi: 10.1159/000048893. [DOI] [PubMed] [Google Scholar]
- Moreillon P, Que YA. Infective endocarditis. Lancet. 2004;363:139–149. doi: 10.1016/S0140-6736(03)15266-X. [DOI] [PubMed] [Google Scholar]
- Moreillon P, Que YA, Bayer AS. Pathogenesis of streptococcal and staphylococcal endocarditis. Infect Dis Clin North Am. 2002;16:297–318. doi: 10.1016/s0891-5520(01)00009-5. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Ghebrehiwet B, Peerschke EI. Staphylococcus aureus protein A recognizes platelet gC1qR/p33: a novel mechanism for staphylococcal interactions with platelets. Infect Immun. 2000;68:2061–2068. doi: 10.1128/iai.68.4.2061-2068.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni ED, Perkins S, Francois P, Vaudaux P, Höök M, Foster TJ. Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin of Staphylococcus aureus. Mol Microbiol. 1998;30:245–257. doi: 10.1046/j.1365-2958.1998.01050.x. [DOI] [PubMed] [Google Scholar]
- Peerschke EI, Bayer AS, Ghebrehiwet B, Xiong YQ. gC1qR/p33 blockade reduces Staphylococcus aureus colonization of target tissues in an animal model of infective endocarditis. Infect Immun. 2006;74:4418–4423. doi: 10.1128/IAI.01794-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peerschke EI, Ghebrehiwet B. The contribution of gC1qR/p33 in infection and inflammation. Immunobiology. 2007;212:333–342. doi: 10.1016/j.imbio.2006.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peerschke EI, Murphy TK, Ghebrehiwet B. Activation-dependent surface expression of gC1qR/p33 on human blood platelets. Thromb Haemost. 2003;89:331–339. [PubMed] [Google Scholar]
- Peterson KL, Zhang W, Lu PD, Keilbaugh SA, Peerschke EI, Ghebrehiwet B. The C1q-binding cell membrane proteins cC1q-R and gC1q-R are released from activated cells: subcellular distribution and immunochemical characterization. Clin Immunol Immunopathol. 1997;84:17–26. doi: 10.1006/clin.1997.4374. [DOI] [PubMed] [Google Scholar]
- Pober JS. Activation and injury of endothelial cells by cytokines. Pathol Biol (Paris) 1998;46:159–163. [PubMed] [Google Scholar]
- Roller J, Laschke MW, Sethi S, Herrmann M, Menger MD. Prolene-Monocryl-composite meshes do not increase microvascular Staphylococcus aureus adherence and do not sensitize for leukocytic inflammation. Langenbecks Arch Surg. 2008;393:349–357. doi: 10.1007/s00423-008-0295-5. [DOI] [PubMed] [Google Scholar]
- Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- Stam-Bolink EM, Mithoe D, Baas WH, Arends JP, Möller AV. Spread of a methicillin-resistant Staphylococcus aureus ST80 strain in the community of the northern Netherlands. Eur J Clin Microbiol Infect Dis. 2007;26:723–727. doi: 10.1007/s10096-007-0352-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaday GG, Franitza S, Schor H, Hecht I, Brill A, Cahalon L, Hershkoviz R, Lider O. Combinatorial signals by inflammatory cytokines and chemokines mediate leukocyte interactions with extracellular matrix. J Leukoc Biol. 2001;69:885–892. [PubMed] [Google Scholar]
- van den Berg RH, Prins F, Faber-Krol MC, Lynch NJ, Schwaeble W, van Es LA, Daha MR. Intracellular localization of the human receptor for the globular domains of C1q. J Immunol. 1997;158:3909–3916. [PubMed] [Google Scholar]
- Wann ER, Gurusiddappa S, Hook M. The fibronectin-binding MSCRAMM FnbpA of Staphylococcus aureus is a bifunctional protein that also binds to fibrinogen. J Biol Chem. 2000;275:13863–13871. doi: 10.1074/jbc.275.18.13863. [DOI] [PubMed] [Google Scholar]
- Westh H, Boye K, Bartels MD, Kristoffersen K, Bergen L, Havstreym J, Bagersted J, Petersen IS, Lester A, Lisby JG, Friis-Møller A, Knudsen JD, Slotsbjerg TD, Lundgren B. Epidemic increase in methicillin-resistant Staphylococcus aureus in Copenhagen. Ugeskr Laeger. 2006;168:671–673. [PubMed] [Google Scholar]
- Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis. 2004;39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- Yeaman MR, Bayer AS. Staphylococcus aureus, Platelets, and the Heart. Curr Infect Dis Rep. 2000;2:281–298. doi: 10.1007/s11908-000-0005-0. [DOI] [PubMed] [Google Scholar]