

Abstract
Background & Aims
Hepatic ischemic reperfusion injury (IRI) is a major complication of liver transplantation and resectional hepatic surgeries. Natural killer T (NKT) cells predominate in liver, where they recognize lipid antigens bound to CD1d molecules. Type I NKT cells utilize a semi-invariant T-cell receptor and react with α-galactosylceramide; type II NKT cells use diverse T-cell receptors. Some type II NKT cells recognize the self-glycolipid sulfatide. It is not clear whether or how these distinct NKT cell subsets mediate hepatocellular damage following IRI.
Methods
We examined the roles of type I and type II NKT cells in mice with partial hepatic, warm ischemia and reperfusion injury.
Results
Mice that lack type I NKT cells (Jα18−/−) were protected from hepatic IRI, indicated by reduced hepatocellular necrosis and serum levels of alanine aminotransferase. Sulfatide-mediated activation of type II NKT cells reduced IFN-γ secretion by type I NKT cells and prevented IRI. Protection from hepatic IRI by sulfatide-mediated inactivation of type I NKT cells was associated with significant reductions in hepatic recruitment of myeloid cell subsets, especially the CD11b+Gr-1int, Gr-1−, and NK cells.
Conclusion
In mice, subsets of NKT cells have opposing roles in hepatic IRI: type I NKT cells promote injury whereas sulfatide-reactive type II NKT cells protect against injury. CD1d activation of NKT cells is conserved from mice to humans, so strategies to modify these processes might be developed to treat patients with hepatic reperfusion injury.
Keywords: Ischemia, liver injury, inflammation, CD1
Introduction
Liver injury following ischemia and reperfusion (IRI) is a major complication of liver transplantation, hepatic resections, trauma surgery, and shock. It is mediated by a biphasic inflammatory response. The initial phase (following 1–6 hrs of reperfusion) involves Kupffer cell activation, release of reactive oxygen species, CD4+ cell recruitment, and secretion of proinflammatory cytokines such as TNF-α and IFN-γ 1–5. In the later phase (following 6–48 hrs of reperfusion), accumulated neutrophil granulocytes release oxidants and proteases directly injuring hepatocytes 4, 6, 7. This results in elevated liver enzymes in serum, in the histological picture of hepatic necrosis, and can lead to organ dysfunction 4, 8, 9.
Due to the rapid development of hepatic ischemic reperfusion injury (IRI) not consistent with the timeframe required for conventional T cell responses 10 and due to the early recruitment of CD4+ T cells 1, 11 consisting of mostly Natural Killer T (NKT) cells in mice, recent studies in mice have focused on the role of NKT cells in hepatic IRI 10, 12, 13. NKT cells express both a T cell receptor and NK markers (such as NK1.1), are mainly CD4+ in the liver, and secrete cytokines rapidly upon stimulation 14–16. Most NKT cells recognize lipid antigens presented in the context of the monomorphic MHC class I-like molecule CD1d 14–16. CD1d-restricted NKT cells are categorized into type I and type II 14–17. Type I or invariant NKT cells express a semi-invariant TCR encoded by the Vα14-Jα18 gene segments in mice, are strongly reactive with the marine sponge-derived glycolipid α-galactosyl ceramide (αGalCer), and accordingly are identified by αGalCer/CD1d-tetramers in flow cytometry 14–16, 18. They also recognize bacterial-derived lipids and a self-glycolipid, isoglobotrihexosyl ceramide (iGb3) 19, 20. Type II NKT cells are less well studied. Our laboratory has shown earlier that a major subset of type II NKT cells is reactive to the self-glycolipid 3-sulfated β-galactosyl ceramide, called sulfatide 21. These sulfatide-reactive type II NKT cells are identified using sulfatide/CD1d-tetramers in flow cytometry 21. In experimental autoimmune diseases and cancer, activated sulfatide-reactive type II NKT cells exert a regulatory role on type I NKT cells, consequently mediating protection from autoimmune disease 22 and suppression of anti-tumor immunity 23. Thus in Concanavalin A-induced hepatitis, a murine model of autoimmune hepatitis, type I NKT cells mediate hepatitis, while activation of type II NKT cells protects from liver injury by inducing anergy in type I NKT cells 22.
In the murine model of hepatic IRI, a role for CD1d-restricted NKT cells has recently been suggested. It has been shown that liver injury is significantly decreased in CD1d−/− mice lacking these cells 12 or in WT mice following treatment with blocking anti-CD1d antibodies 10, 13. In both cases decreased levels of serum IFN-γ are found during IRI compared to untreated WT mice 10, 12. However a recent study indicates that experiments with “blocking” anti-CD1d Abs are difficult to interpret, since anti-CD1d Abs act also as an agonist in directly stimulating IL-12 production from APCs 24. Additionally, restoration of susceptibility to hepatic IRI upon adoptive transfer of CD4+NK1.1+ cells from WT mice into Rag-1−/− mice 10 further supports the involvement of NKT cells. However, distinct investigations on type I vs. type II NKT cells in hepatic IRI have not been carried out so far.
Since sulfatide is a self-glycolipid which is enriched in liver and has been shown by us to influence the activity of type I NKT cells through sulfatide-reactive type II NKT cells, we have examined the involvement of type II vs. type I NKT cells in hepatic IRI. Specifically, we investigated the development of IRI in the absence of type I NKT cells in Jα18−/− mice or following their inactivation subsequent to sulfatide-mediated activation of type II NKT cells. We found that Jα18−/− mice are significantly protected from hepatic IRI, as they develop less hepatocellular necrosis and reduced serum alanine aminotransferase (ALT) levels. Furthermore, decreased IFN-γ secretion by hepatic type I NKT cells and prevention of liver injury were found following activation of type II NKT cells with sulfatide. We found that in both cases prevention of liver injury was associated with decreased recruitment of myeloid cell subsets and NK cells into the liver. These findings clearly demonstrate a pathogenic role for type I NKT cells and a protective role for sulfatide-reactive type II NKT cells in liver injury following IR. Hence, sulfatide pretreatment may represent a novel therapeutic strategy for improving the outcome in liver transplantation and other surgeries associated with hepatic reperfusion.
Material and Methods
Animals
C57BL/6 mice were purchased from The Jackson Laboratory. C57BL/6-CD1d−/− and C57/B6-Jα18−/− mice were originally generated by Van Kaer 25 and Taniguchi 18, respectively. All mice were bred and maintained in specific pathogen free-conditions in the TPIMS animal facility. Treatment of animals was in compliance with federal and institutional guidelines and approved by the TPIMS Animal Care and Use Committee.
Lipids and tetramers
Purified bovine myelin-derived sulfatide (>90% pure) was purchased from Matreya Inc., Pleasant Gap, PA. Synthetic αGalCer was provided by Y. Koezuka (Kirin Brewery Co., Tokyo, Japan). All lipids were dissolved in vehicle (0.5% polysorbate-20 (Tween-20) and 0.9% NaCl solution) and diluted in PBS. Murine CD1d was made in a baculovirus expression system as described earlier 21. PE-labeled mCD1d-tetramers loaded with either sulfatide, αGalCer or PBS were generated as described earlier 21, 22.
Hepatic IRI model
The hepatic IRI model was established as described by Prof. Dr. J. W. Kupiec-Weglinski’s laboratory 26 with few modifications. Female mice (8–16 weeks, age-matched) were anesthetized by i.p. injection of 60 mg/kg sodium pentobarbital. After midline laparotomy, an atraumatic clip was applied to the hepatic triad (hepatic artery, portal vein, bile duct) of the 3 cephalad liver lobes. The caudal lobes retained intact blood circulation to prevent intestinal venous congestion. The peritoneum was closed and mice were placed on a heating pad (~37°C). Ambient temperature ranged between 25–26°C. After 90 min of partial hepatic warm ischemia, the clip was removed, initiating reperfusion, and the abdominal wall was sutured. Mice were euthanized after 6 or 24 hrs of reperfusion, and blood and cephalad liver lobes were collected. Sham controls underwent the same procedure but without vascular occlusion.
Blood and histological analyses
Blood was obtained by cardiac puncture and serum was isolated. Serum Alanine aminotransferase (ALT) levels were determined on Olympus 5400 chemistry analyzer (IDEXX Laboratories Inc., Westbrook, Maine). Liver tissues (cephalad lobes) were fixed in 10% formalin, embedded in paraffin and sections were stained with hematotoxylin and eosin for histological analysis (IDEXX Laboratories Inc., Westbrook, Maine).
Cell preparation
Leukocytes were isolated from murine cephalad liver lobes, using mechanical crushing followed by Percoll gradient separation and RBC lysis as described earlier 22.
Flow cytometry
Leukocytes were suspended in FACS buffer (PBS containing 0.02% NaN3 and 2% FCS), blocked (anti-mouse FcR-γ, BD Pharmingen, San Diego, CA) and stained with loaded mCD1d-tetramer-PE or PE-, FITC-, or PECy5-labeled anti-mouse antibodies (BD Pharmingen, San Diego, CA or eBioscience Inc., San Diego, CA) as indicated. Intracellular cytokine staining (ICCS) of liver mononuclear cells (MNCs) was carried out as described earlier 22. Analysis was performed on a FACSCalibur instrument using CellQuest software (version 4.0.2, BD, Franklin Lakes, NJ).
Statistics
Data are expressed as mean ± SEM for each group. Statistical differences between groups were evaluated by unpaired, one-tailed Student’s t test using GraphPad Prism software (version 5.0a, GraphPad Software Inc., La Jolla, CA).
Results
Significant reduction in hepatic IRI in mice deficient in type I NKT cells
To determine the role of type I NKT cells in hepatic IRI, the extent of liver injury was analyzed in groups of Jα18−/− mice lacking only type I NKT cells but harboring normal levels of type II NKT cells 21 and compared to WT mice. As shown in Figure 1, large necrotic areas were found in cephalad liver lobes of WT mice following 90 min of ischemia and 24 hrs of reperfusion, whereas necrotic areas in Jα18−/− mice were remarkably reduced. Sham controls of both WT and Jα18−/− mice did not show any necrotic areas.
Figure 1. Reduced hepatic necroses in the absence of type I NKT cells or following activation of type II NKT cells.

Untreated WT and Jα18−/− mice or mice treated with 20µg sulfatide/mouse i.p. 3hrs earlier (WT sulfatide or Jα18−/− sulfatide) were subjected to 90 min of hepatic ischemia followed by 24hrs of reperfusion (IRI) or sham surgery. Respective H&E staining of tissue from cephalad liver lobes is representative of at least 3 mice. 100 × magnification.
As a marker of hepatocellular damage, alanine aminotransferase (ALT) enzyme levels were assessed in serum of mice after 90 min of ischemia and 6 hrs of reperfusion. In Jα18−/− mice, serum ALT levels were decreased by ~51% compared to WT mice (1238.1 ± 178.4 U/l vs. 2502.0 ± 783.2 U/l, p < 0.05) (see Fig. 2). As reported earlier 12 serum ALT levels of CD1d−/− mice lacking both type I and type II NKT cells were also reduced following IRI induction (data not shown). Among sham controls serum ALT levels were quite similar, regardless of presence of type I NKT cells, and means ranged between 204.0–285.1 U/l.
Figure 2. Reduced levels of serum ALT in the absence of type I NKT cells or following activation of type II NKT cells.

Bar graphs depict serum ALT levels following 90 min of ischemia and 6 hrs of reperfusion (IRI) or sham surgery. WT or Jα18−/− mice (5–11 mice/group) were analyzed. Where indicated (+ Sulf.) mice were treated with sulfatide (20 µg/mouse) i.p. 3–48 hrs prior to surgeries. Values are mean ± SEM. * p < 0.05.
Thus liver injury following ischemia/reperfusion is significantly reduced in the absence of type I NKT cells, as assessed by histology and blood chemistry. These data clearly indicate a pathogenic role for type I NKT cells in mediating hepatic IRI.
Sulfatide-mediated activation of type II NKT cells provides significant protection from hepatic IRI
We have shown earlier 22 that activation of type II NKT cells by administration of sulfatide in vivo results in anergy induction or inactivation of type I NKT cells that is associated with a significant inhibition in their proliferation and cytokine secretion in response to αGalCer. To analyze the effect of sulfatide administration on IRI development, in which type I NKT cells are pathogenic (Fig. 1, Fig. 2), groups of BL/6 mice were treated with sulfatide (20 µg/mouse) intraperitoneally 3 to 48 hrs prior to ischemia induction or sham surgery. As shown in Figure 1 mice pretreated with sulfatide 3 hrs prior to ischemia induction followed by 24 hrs of reperfusion developed only minimal or no hepatic necrosis. Sham controls pretreated with sulfatide showed no necrosis.
Next mice were administered sulfatide 3, 16 and 48 hrs prior to surgical procedures and serum ALT levels were measured following 6 hrs of reperfusion or sham surgeries. No differences in ALT levels were seen between mice injected with sulfatide at the different time points (data not shown), so the groups were pooled in Figure 2. In parallel, IRI induction or sham surgeries were performed on mice injected with vehicle. Vehicle-treated mice and naïve mice did not differ in the ALT levels (data not shown). As shown in Figure 2, WT mice pretreated with sulfatide showed a significant decrease (~60%) in serum ALT levels compared to naïve WT mice (1008.0 ± 117.8 U/l vs. 2502.0 ± 783.2 U/l, p < 0.05). Sulfatide pretreatment of Jα18−/− mice did not further reduce the ALT-levels in injured mice (Fig. 2). These data clearly indicate that sulfatide-mediated inactivation of type I NKT cells accounts for the protection from hepatic IRI.
Since IFN-γ production by NKT cells seems to be involved in pathogenesis of hepatic IRI 10, we determined IFN-γ secretion by type I NKT (αGalCer/tetramer+TCRβ+) cells in cephalad liver lobes following 90 min of ischemia and 6 hrs of reperfusion. In Figure 3 increased IFN-γ production by type I NKT cells after IRI induction compared to sham surgery is shown. Administration of sulfatide 3 hrs prior to ischemia induction significantly reduced IFN-γ secretion by type I NKT cells (p < 0.01) (Fig. 3).
Figure 3. Sulfatide administration prior to IRI induction significantly inhibits IFN-γ secretion by type I NKT cells.
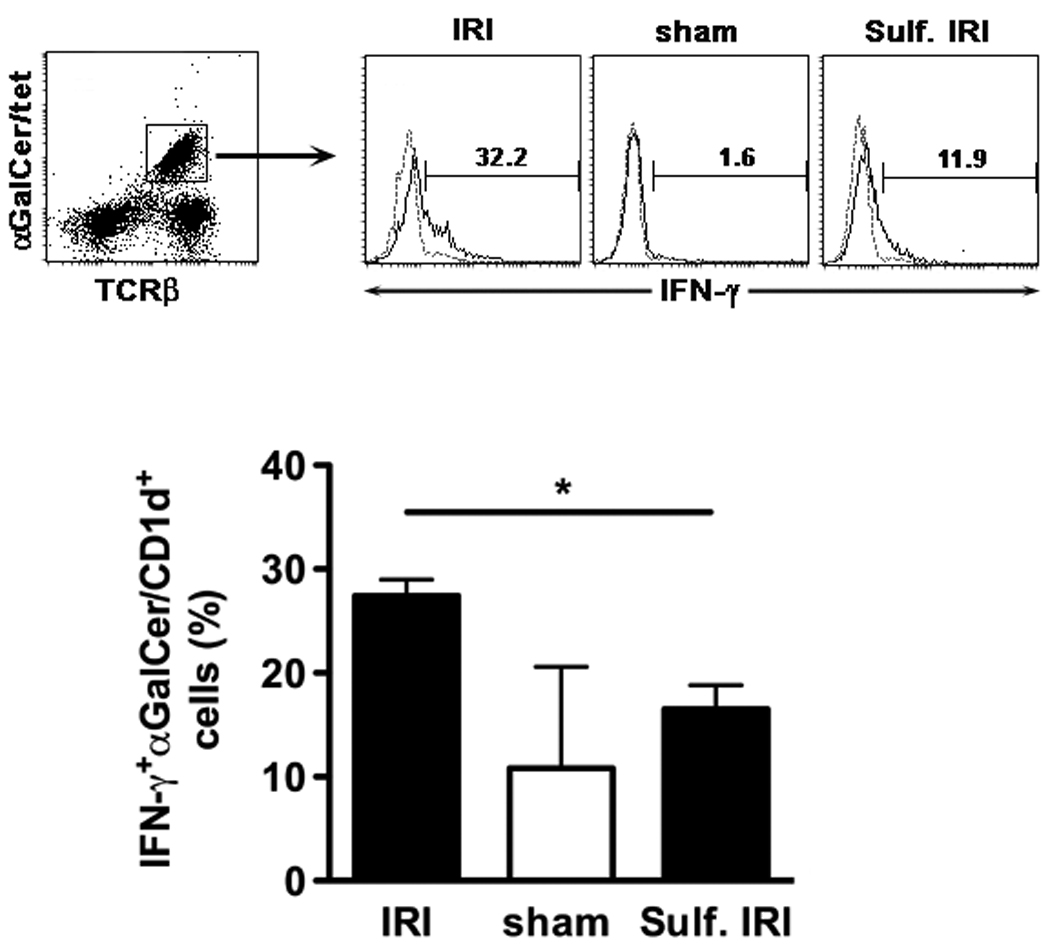
Intracellular cytokine staining of MNCs from cephalad liver lobes after 90 min of ischemia and 6 hrs of reperfusion (IRI) or sham surgery. One group of WT mice (n=3) was injected with sulfatide (20 µg/mouse i.p.) 3 hrs prior to ischemia induction (Sulf. IRI), the other groups (IRI (n=3), sham (n=2)) were not pretreated. (Upper panels) Tri-color flow cytometric analysis of IFN-γ+ cells in αGalCer/CD1d-tetramer+ cells. IFN-γ (black line) vs. isotype (dashed line). Numbers above brackets indicate percent positive cells. (Lower panel) Summarizing bar graphs. Values are mean ± SEM. * p < 0.01.
Thus activation of type II NKT cells with sulfatide results in protection from hepatic IRI to an extent similar to that observed in the absence of type I NKT cells. This is due to sulfatide-mediated inactivation of type I NKT cells and is associated with the reduction in IFN-γ secretion.
Alterations in hepatic NKT and T cell subsets in early IRI
To further analyze the involvement of distinct lymphocyte populations in the immune response in the reperfused liver, their numbers were determined by flow cytometry in wild type mice or in mice deficient in functional type I NKT cells. Since innate-like cellular interactions during the initial phase of IRI set the stage for the development of late phase hepatocelluar injury, we analyzed cellular profiles during the initial phase. Naïve WT and Jα18−/− mice or WT mice treated with 20 µg sulfatide/mouse i.p. 3 hrs earlier were subjected to 90 min of ischemia and 6 hrs of reperfusion or sham surgeries. Following staining of MNCs from cephalad liver lobes with mCD1d-tetramers or antibodies, the proportions and absolute numbers of NKT (NK1.1+TCRβ+) cells, type I NKT (αGalCer/tetramer+TCRβ+) cells, type II NKT (sulfatide/tetramer+TCRβ+) cells, CD4+ T cells, CD8+ T cells, and CD4−CD8− T cells were analyzed (Fig. 4). At this reperfusion time point hepatic NKT cell subsets, CD8+ T cells and CD4−CD8− T cells showed no significant changes in IRI-induced WT mice compared to sham controls (Fig. 4). However, a slight reduction in type I NKT and total NKT cells was observed. Consistently, CD4+ T cells were slightly but significantly decreased in livers from naïve WT and sulfatide-treated WT mice following 6 hrs of reperfusion.
Figure 4. Increase in CD11b+ cells in IRI is dependent on type I NKT cells and diminished by activation of type II NKT cells.

Flow cytometric analysis of indicated cell populations (depicted as bar graphs in % (left panels) and absolute numbers (right panels)) among MNCs or leukocytes (for CD11b+ cells) from cephalad liver lobes following 90 min of ischemia and 6 hrs of reperfusion (IRI) or sham surgery in WT or Jα18−/− mice (3–5 mice/group). Data from WT mice treated with sulfatide (20 µg /mouse i.p.) 3 hrs prior to surgeries (2–3 mice/group) (Sulfatide). Values are mean ± SEM. * p < 0.05. n.d. = not done.
The increase in hepatic NK cells during IRI is dependent on type I NKT cells and reduced by sulfatide pretreatment
The role of NK cells in hepatic IRI is unclear. Shimamura et al. suggested that NK cells can contribute to IRI to a certain extent 12. We therefore determined changes in the proportion and absolute number of hepatic NK (NK1.1+TCRβ−) cells during IRI in naïve WT mice, Jα18−/− mice, and sulfatide-treated WT mice. As shown in Figure 4 proportion of NK cells was significantly (p < 0.01) increased in livers of WT mice following 6 hrs of reperfusion compared to sham controls. In mice lacking type I NKT cells (Jα18−/−) however, no significant change in NK cells was observed (Fig. 4), indicating that the increase in hepatic NK cells is dependent on the presence of type I NKT cells. Furthermore in vivo activation of type II NKT cells by sulfatide prior to IRI induction reduced the increase in hepatic NK cells (Fig. 4). These data suggest that the recruitment of NK cells is dependent upon the activation of type I NKT cells during hepatic IRI and may contribute to tissue damage.
The hepatic recruitment of myeloid cell subsets during IRI is dependent on type I NKT cells and reduced by activation of type II NKT cells
The accumulation of neutrophil granulocytes in the liver and their subsequent injuring of hepatocytes has been reported following ischemia and reperfusion 1, 10, 27, 28. We analyzed CD11b+Gr-1+ cells, including neutrophil granulocytes, and CD11b+Gr-1− cells among liver leukocytes from naïve WT mice, Jα18−/− mice, and sulfatide-treated WT mice following 6 hrs of reperfusion or sham surgeries by flow cytometry (see Figure 4). A significant increase in hepatic CD11b+ cells was observed in IRI-induced WT mice compared to sham controls (37.8 ± 3.9% vs. 23.4 ± 1.8%, p < 0.01). This increase included Gr-1+ cells comprising granulocytes, monocytes and myeloid precursor cells, as well as Gr-1− cells mainly consisting of macrophages and myeloid dendritic cells but also of NK cells (Fig. 4). To further investigate the increase in the CD11b+Gr-1+ population, Gr-1high and Gr-1int subsets were analyzed separately. The Gr-1int subset, predominantly comprising monocytes and myeloid precursors, is increased around 3.5-fold (p < 0.005) in livers of WT mice following 6 hrs of reperfusion compared to sham surgeries (Fig. 5). In contrast the Gr-1high subset, mainly consisting of granulocytes, did not differ significantly between livers of sham controls and IRI-induced mice at this time point (Fig. 5).
Figure 5. Myeloid precursor cells are increased in IRI in the presence of type I NKT cells.

Flow cytometric analysis of CD11b+Gr-1+ subsets among leukocytes from cephalad liver lobes following 90 min of ischemia and 6 hrs of reperfusion (IRI) or sham surgery in WT or Jα18−/− mice (3–5 mice/group) and in WT mice treated with sulfatide 3 hrs earlier (2–3 mice/group). (Upper panels) Gr-1high and Gr-1int populations were gated. Numbers next to boxes indicate percent positive cells among liver leukocytes. (Lower panels) Summarizing bar graphs depict % (left) and absolute numbers (right) of the Gr-1high and Gr-1int populations. Values are mean ± SEM. * p < 0.005.
Notably, the hepatic recruitment of myeloid cell subsets during IRI is dependent on the presence of type I NKT cells, since it is not observed in Jα18−/− mice (Fig. 4 and Fig. 5). Furthermore in vivo activation of type II NKT cells by sulfatide 3hrs prior to IRI induction diminished hepatic recruitment of myeloid cells, as proportions of CD11b+Gr-1+ and CD11b+Gr-1− populations were not significantly altered between sulfatide-treated sham and IRI mice (Fig. 4). CD11b+Gr-1int cells were reduced by ~50% in sulfatide-treated mice compared to naïve mice (p < 0.05) (Fig. 5).
Collectively, we found a significant accumulation of myeloid cell subsets that are predominantly myeloid precursor cells/monocytes in reperfused livers. This myeloid cell accumulation is dependent on type I NKT cells and significantly inhibited following activation of type II NKT cells.
Discussion
In this study we show that type I NKT cells mediate hepatic ischemic reperfusion injury, since Jα18−/− mice lacking type I NKT cells but harboring normal levels of type II NKT cells are significantly protected from injury. In these mice only mild hepatitis with significantly reduced hepatic necrosis and decreased serum ALT levels developed. Increased intracellular IFN-γ expression in hepatic type I NKT cells during IRI of WT mice further substantiates a pathogenic role for type I NKT cells in reperfusion injury. In contrast, a protective role in hepatic IRI was found for a major subset of type II NKT cells, which are reactive to sulfatide. Activation of this subset by intraperitoneal sulfatide administration 3–48 hrs prior to ischemia induction prevents liver injury. Sulfatide-mediated protection is associated with inhibition of IFN-γ secretion by hepatic type I NKT cells and suppression of type I NKT cell-mediated recruitment of myeloid cells, especially the CD11b+Gr-1int and Gr-1− subsets, and NK cells into the liver. A mechanism involving different subsets of NKT cells in hepatic IRI has been proposed and is summarized in Figure 6.
Figure 6. Model of proposed immune mechanisms in hepatic IRI.

Sulfatide-mediated activation of type II NKT cells results in inactivation of type I NKT cells as exemplified by the inhibition of IFN-γ secretion. This results in reduced hepatic recruitment of myeloid cells and consequently, in reduced hepatocyte and endothelial cell (EC) injury. Collectively, proposed model suggest that appropriate targeting of NKT cells could lead to novel strategies for intervention in hepatic IRI.
It is clear from our data that hepatic type I NKT cells secrete IFN-γ during the early phase of IRI (Figure 3) and that this is significantly blunted following their inactivation leading to protection from liver injury. The crucial involvement of IFN-γ secretion by type I NKT cells in the pathogenesis of IRI reported here is consistent with an earlier study 10 showing increased IFN-γ secretion by type I NKT cells during early hepatic IRI and failure of adoptively transferred CD4+NK1.1+ cells from IFN-γ−/− mice to restore susceptibility to IRI in Rag-1−/− mice. Role of IFN-γ secretion by type I NKT cells may be a common feature of ischemic organ injury as kidney IRI is also attenuated in Jα18−/− mice29. Collectively these data suggest that during ischemic tissue injury type I NKT cells become promptly activated resulting in IFN-γ secretion followed by cellular recruitment into the liver. Consistent with the pathogenic role of type I NKT cells, we and others (data not shown and 30) have observed a significant increase in IRI upon αGalCer administration 6 or 16 hrs prior to liver ischemia. Moreover, αGalCer is not a physiological ligand for type I NKT cells and most mammalian glycolipids that are β-linked may behave differently. Results with αGalCer should be viewed with caution as background genes, dosage and timing of the administration of αGalCer can produce contradictory results in different experimental systems 30–33.
Interestingly, the extent of IRI prevention following treatment with sulfatide is strikingly similar to that found in the total absence of type I NKT cells (Jα18−/− mice). In both cases ALT levels were reduced by >50%, and only minimal or no hepatic necrotic areas were present. It is clear that sulfatide administration results in inactivation of type I NKT cells 22 and is associated with a significant inhibition of IFN-γ secretion in reperfused livers (Fig. 3). These data are consistent with a novel immunoregulatory pathway proposed earlier 22 in which activation of type II NKT cells by sulfatide in vivo mediates anergy induction in type I NKT cells that is dependent upon IL-12 and modification of hepatic DC subsets. In fact, adoptive transfer of hepatic CD11c+ cells from sulfatide-treated animals is sufficient to blunt liver damage in the recipients 22. Thus it is likely that the protective effect of sulfatide pretreatment in hepatic IRI results from inactivation of pathogenic type I NKT cells by CD11c+ myeloid cells modified through their interaction with sulfatide-activated type II NKT cells.
The development of hepatic ischemic reperfusion injury is divided into two phases: an initial period (following 1–6 hrs of reperfusion) dominated by Kupffer cell activation, release of reactive oxygen species, CD4+ cell recruitment and secretion of proinflammatory cytokines 1–5. This initial phase sets the stage for the later period (following 6–48 hrs of reperfusion) characterized by neutrophil accumulation and induction of hepatic necrosis 4, 6, 7. In this study we focused on the earlier phase, after 6 hrs of reperfusion, directly prior to the induction of liver damage. We have defined an important role for myeloid (CD11b+) cell subsets that are recruited into reperfused WT livers at this time point. They are mainly of the CD11b+Gr-1int subset, comprising myeloid precursor cells and monocytes, and of the CD11b+Gr-1− subset, containing macrophages and myeloid dendritic cells (Fig. 4 and 5). However we did not observe a significant increase in the CD11b+Gr-1high subset, consisting mainly of neutrophil granulocytes, in livers following 6 hrs of reperfusion. This is compatible with previous studies reporting hepatic neutrophil accumulation in the later phase, following 18–24 hrs of reperfusion 1, 10, 27, 28. Our observation concerning the recruitment of myeloid cells other than neutrophils, predominantly of the CD11b+Gr-1int subset, is consistent with a recent report of recruitment of inflammatory monocytes into reperfused livers enhancing the injury 34. Interestingly, Bamboat et al. further show inhibition of inflammatory monocyte function by IL-10 from conventional DCs 34.
Strikingly, the hepatic recruitment of myeloid cells is dependent on the presence of type I NKT cells. It is still not clear whether the myeloid cell recruitment is dependent upon the secretion of a specific cytokine, such as IFN-γ or osteopontin or other chemokines. Consequentially activation of type II NKT cells with sulfatide, resulting in inactivation of type I NKT cells, suppresses hepatic recruitment of myeloid cells and results in protection from liver injury, emphasizing the significant role of myeloid cells in the mediation of hepatic IRI.
A link between CD1d-dependent NKT cells and myeloid cells has been suggested previously 10, 12, 22. One group 10 reported that blocking of the CD1d pathway results in significantly reduced hepatic neutrophil accumulation following 24 hrs of reperfusion, suggesting NKT-cell dependent recruitment of neutrophil granulocytes in hepatic IRI. However another study 13 did not detect a significant CD1d-dependent change in neutrophil accumulation after 8 hrs of reperfusion. Both studies differ in the time point of assessment and in the respective protocol of anti-CD1d treatment. Anti-CD1d blocking experiments are problematic because anti-CD1d antibodies can have direct agonistic effect on APCs leading to the secretion of IL-12 24. Additionally data from Shimamura et al. indicates CD1d-dependent reduction of activation levels of granulocytes in hepatic IRI 12. A reduced number of hepatic myeloid cells following sulfatide administration in IRI model is consistent with earlier study showing their decreased accumulation following sulfatide administration in the ConA-induced hepatitis model 22. In contrast to these inflammatory liver injury models, type I NKT cells are considered to suppress hepatic neutrophil accumulation and activity in a mouse model of cholestatic liver damage 35.
Our data also support a role for NK cells in hepatic IRI, since they are recruited into reperfused livers in the presence of type I NKT cells, but not in their absence or following their inactivation. A role of type I NKT cells in the recruitment or activation of NK cells is consistent with the report of their activation following αGalCer administration 36. Similarly to our study, a tendency for an early increase in hepatic NK cells of IRI-induced WT mice has been shown by others 11, 12. Consistently, depletion of NK cells either with anti-asialoGM1 or with anti-NK1.1 antibodies has been shown to attenuate hepatic IRI 10, 12. However, these treatments eliminate both NK and NKT cells and hence cannot clearly identify the contribution of NK cells to liver injury.
At the end of the initial phase, following 6 hrs of reperfusion, we observed a slight reduction in hepatic NKT cells and the hepatic type I NKT cell subset. Consistently, a slight but significant decrease in CD4+ T cells was found in injured mice. These data are entirely compatible with studies showing a very early hepatic recruitment (after 1–4 hrs of reperfusion) of CD4+ T cells, including NKT (CD4+NK1.1+TCRβint) cells 1,2, 11. A later rebound decrease in hepatic T cells below the base line was observed in the IRI model of Shimamura et al. 12. They further reported a very early decrease (after 1h of reperfusion) in NKT (NK1.1+CD3int) cells, followed by an increase with a peak around 10–20 hrs 12. Thus the respective roles of type I and type II NKT cells were not reflected in a change of their hepatic proportions at the end of the initial IRI phase.
Our findings support the idea of a pivotal role for self-antigens in reperfusion injury 37. It is likely that ischemia-induced release of reactive oxygen species (ROS) results in lipid modifications and their release from disrupted membranes. These lipids act as self-antigens and bind to CD1d expressed by local cells such as dendritic cells, Kupffer cells, hepatic stellate cells and hepatocytes 38–40. Following uptake and presentation in the context of CD1d these self-lipids can activate NKT cells and initiate their rapid immune response in hepatic IRI. Our data clearly show that it is the type I NKT cell subset that mediates liver injury following ischemia and reperfusion. Activation of type II NKT cells can neutralize the pathogenic role of such activated type I NKT cells and provide protection from IRI. This is mediated by suppressing the effects of type I NKT cells, such as their IFN-γ secretion and the recruitment of myeloid and NK cells into the liver.
Collectively, data presented here in liver injury due to ischemia and reperfusion as well as our findings in Concanavalin A-induced hepatitis 22 are consistent with the idea of an immunoregulatory pathway in which type I NKT cells play a detrimental role and sulfatide-reactive type II NKT cells a protective role in hepatic inflammation. Since CD1d immune pathways are highly conserved between mice and human, our data have important implications for the development of immunotherapeutics based upon the usage of sulfatide or its analogs to ameliorate the outcome of surgeries associated with hepatic reperfusion such as liver transplantation and hepatic resection. Such novel strategies would allow more patients to undergo successful liver transplantation by expanding the donor pool to marginal organs that are currently highly susceptible to IRI and by diminishing the level of overall immune activation predisposing to graft rejections.
Acknowledgment
The authors thank Prof. Dr. Jerzy W. Kupiec-Weglinski and Dr. Xiu-Da Shen from University of California Los Angeles for generously demonstrating the hepatic IRI model. Authors thank Dr. Randle Ware for critical reading of the manuscript.
Grant Support: This work was supported by grants from the NIH (R01-CA100660), JDRF, MSNRC and DNRG (to VK) and the DFG (German Research Foundation, AR 645/1-1) and YAEL Foundation (to PA).
Abbreviations
- αGalCer
α-galactosyl ceramide
- DC
dendritic cell
- IRI
ischemic reperfusion injury
- NKT
Natural Killer T
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author’s Contributions:
PA: study design, acquisition, analysis and interpretation of data, writing manuscript; IM: technical support and data acquisition; VK: study concept and design, analysis and interpretation of data, writing manuscript.
References
- 1.Zwacka RM, Zhang Y, Halldorson J, et al. CD4(+) T-lymphocytes mediate ischemia/reperfusion-induced inflammatory responses in mouse liver. J Clin Invest. 1997;100:279–289. doi: 10.1172/JCI119533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caldwell CC, Tschoep J, Lentsch AB. Lymphocyte function during hepatic ischemia/reperfusion injury. J Leukoc Biol. 2007;82:457–464. doi: 10.1189/jlb.0107062. [DOI] [PubMed] [Google Scholar]
- 3.Jaeschke H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol. 2003;284:G15–G26. doi: 10.1152/ajpgi.00342.2002. [DOI] [PubMed] [Google Scholar]
- 4.Lentsch AB, Kato A, Yoshidome H, et al. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology. 2000;32:169–173. doi: 10.1053/jhep.2000.9323. [DOI] [PubMed] [Google Scholar]
- 5.Jaeschke H, Farhood A. Neutrophil and Kupffer cell-induced oxidant stress and ischemia-reperfusion injury in rat liver. Am J Physiol. 1991;260:G355–G362. doi: 10.1152/ajpgi.1991.260.3.G355. [DOI] [PubMed] [Google Scholar]
- 6.Jaeschke H, Smith CW. Mechanisms of neutrophil-induced parenchymal cell injury. J Leukoc Biol. 1997;61:647–653. doi: 10.1002/jlb.61.6.647. [DOI] [PubMed] [Google Scholar]
- 7.Jaeschke H. Mechanisms of Liver Injury II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1083–G1088. doi: 10.1152/ajpgi.00568.2005. [DOI] [PubMed] [Google Scholar]
- 8.Kupiec-Weglinski JW, Busuttil RW. Ischemia and reperfusion injury in liver transplantation. Transplant Proc. 2005;37:1653–1656. doi: 10.1016/j.transproceed.2005.03.134. [DOI] [PubMed] [Google Scholar]
- 9.Teoh NC, Farrell GC. Hepatic ischemia reperfusion injury: pathogenic mechanisms and basis for hepatoprotection. J Gastroenterol Hepatol. 2003;18:891–902. doi: 10.1046/j.1440-1746.2003.03056.x. [DOI] [PubMed] [Google Scholar]
- 10.Lappas CM, Day YJ, Marshall MA, et al. Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d-dependent NKT cell activation. J Exp Med. 2006;203:2639–2648. doi: 10.1084/jem.20061097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caldwell CC, Okaya T, Martignoni A, et al. Divergent functions of CD4+ T lymphocytes in acute liver inflammation and injury after ischemia-reperfusion. Am J Physiol Gastrointest Liver Physiol. 2005;289:G969–G976. doi: 10.1152/ajpgi.00223.2005. [DOI] [PubMed] [Google Scholar]
- 12.Shimamura K, Kawamura H, Nagura T, et al. Association of NKT cells and granulocytes with liver injury after reperfusion of the portal vein. Cell Immunol. 2005;234:31–38. doi: 10.1016/j.cellimm.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 13.Kuboki S, Sakai N, Tschop J, et al. Distinct contributions of CD4+ T cell subsets in hepatic ischemia/reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1054–G1059. doi: 10.1152/ajpgi.90464.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- 15.Kronenberg M, Gapin L. The unconventional lifestyle of NKT cells. Nat Rev Immunol. 2002;2:557–568. doi: 10.1038/nri854. [DOI] [PubMed] [Google Scholar]
- 16.Van Kaer L. NKT cells: T lymphocytes with innate effector functions. Curr Opin Immunol. 2007;19:354–364. doi: 10.1016/j.coi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 17.Arrenberg P, Halder R, Kumar V. Cross-regulation between distinct natural killer T cell subsets influences immune response to self and foreign antigens. J Cell Physiol. 2009;218:246–250. doi: 10.1002/jcp.21597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawano T, Cui J, Koezuka Y, et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science. 1997;278:1626–1629. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- 19.Zhou D, Mattner J, Cantu C, 3rd, et al. Lysosomal glycosphingolipid recognition by NKT cells. Science. 2004;306:1786–1789. doi: 10.1126/science.1103440. [DOI] [PubMed] [Google Scholar]
- 20.Kinjo Y, Wu D, Kim G, et al. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature. 2005;434:520–525. doi: 10.1038/nature03407. [DOI] [PubMed] [Google Scholar]
- 21.Jahng A, Maricic I, Aguilera C, et al. Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J Exp Med. 2004;199:947–957. doi: 10.1084/jem.20031389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halder RC, Aguilera C, Maricic I, et al. Type II NKT cell-mediated anergy induction in type I NKT cells prevents inflammatory liver disease. J Clin Invest. 2007;117:2302–2312. doi: 10.1172/JCI31602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ambrosino E, Terabe M, Halder RC, et al. Cross-regulation between type I and type II NKT cells in regulating tumor immunity: a new immunoregulatory axis. J Immunol. 2007;179:5126–5136. doi: 10.4049/jimmunol.179.8.5126. [DOI] [PubMed] [Google Scholar]
- 24.Yue SC, Nowak M, Shaulov-Kask A, et al. Direct CD1d-mediated stimulation of APC IL-12 production and protective immune response to virus infection in vivo. J Immunol. 184:268–276. doi: 10.4049/jimmunol.0800924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hong S, Scherer DC, Singh N, et al. Lipid antigen presentation in the immune system: lessons learned from CD1d knockout mice. Immunol Rev. 1999;169:31–44. doi: 10.1111/j.1600-065x.1999.tb01304.x. [DOI] [PubMed] [Google Scholar]
- 26.Shen XD, Ke B, Zhai Y, et al. CD154-CD40 T-cell costimulation pathway is required in the mechanism of hepatic ischemia/reperfusion injury, and its blockade facilitates and depends on heme oxygenase-1 mediated cytoprotection. Transplantation. 2002;74:315–319. doi: 10.1097/00007890-200208150-00005. [DOI] [PubMed] [Google Scholar]
- 27.Jaeschke H, Farhood A, Smith CW. Neutrophils contribute to ischemia/reperfusion injury in rat liver in vivo. Faseb J. 1990;4:3355–3359. [PubMed] [Google Scholar]
- 28.Jaeschke H, Farhood A, Bautista AP, et al. Functional inactivation of neutrophils with a Mac-1 (CD11b/CD18) monoclonal antibody protects against ischemia-reperfusion injury in rat liver. Hepatology. 1993;17:915–923. [PubMed] [Google Scholar]
- 29.Li L, Huang L, Sung SS, et al. NKT cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J Immunol. 2007;178:5899–5911. doi: 10.4049/jimmunol.178.9.5899. [DOI] [PubMed] [Google Scholar]
- 30.Cao Z, Yuan Y, Jeyabalan G, et al. Preactivation of NKT cells with alpha-GalCer protects against hepatic ischemia-reperfusion injury in mouse by a mechanism involving IL-13 and adenosine A2A receptor. Am J Physiol Gastrointest Liver Physiol. 2009;297:G249–G258. doi: 10.1152/ajpgi.00041.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jahng AW, Maricic I, Pedersen B, et al. Activation of natural killer T cells potentiates or prevents experimental autoimmune encephalomyelitis. J Exp Med. 2001;194:1789–1799. doi: 10.1084/jem.194.12.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Kaer L. Natural killer T cells as targets for immunotherapy of autoimmune diseases. Immunol Cell Biol. 2004;82:315–322. doi: 10.1111/j.0818-9641.2004.01252.x. [DOI] [PubMed] [Google Scholar]
- 33.Sireci G, La Manna MP, Di Sano C, et al. Pivotal advance: alpha-galactosylceramide induces protection against lipopolysaccharide-induced shock. J Leukoc Biol. 2007;81:607–622. doi: 10.1189/jlb.0506298. [DOI] [PubMed] [Google Scholar]
- 34.Bamboat ZM, Ocuin LM, Balachandran VP, et al. Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion. J Clin Invest. 120:559–569. doi: 10.1172/JCI40008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wintermeyer P, Cheng CW, Gehring S, et al. Invariant natural killer T cells suppress the neutrophil inflammatory response in a mouse model of cholestatic liver damage. Gastroenterology. 2009;136:1048–1059. doi: 10.1053/j.gastro.2008.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carnaud C, Lee D, Donnars O, et al. Cutting edge: Cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J Immunol. 1999;163:4647–4650. [PubMed] [Google Scholar]
- 37.Zhang M, Alicot EM, Chiu I, et al. Identification of the target self-antigens in reperfusion injury. J Exp Med. 2006;203:141–152. doi: 10.1084/jem.20050390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mandal M, Chen XR, Alegre ML, et al. Tissue distribution, regulation and intracellular localization of murine CD1 molecules. Mol Immunol. 1998;35:525–536. doi: 10.1016/s0161-5890(98)00055-8. [DOI] [PubMed] [Google Scholar]
- 39.Winau F, Hegasy G, Weiskirchen R, et al. Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity. 2007;26:117–129. doi: 10.1016/j.immuni.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 40.Brigl M, Brenner MB. CD1: antigen presentation and T cell function. Annu Rev Immunol. 2004;22:817–890. doi: 10.1146/annurev.immunol.22.012703.104608. [DOI] [PubMed] [Google Scholar]