

Abstract
Stem cells generate the differentiated cell types within many organs throughout the lifespan of an organism and are thus ultimately responsible for the longevity of multicellular organisms. Therefore, senescence of stem cells must be prevented. Bmi1 is required for the maintenance of adult stem cells in some tissues partly because it represses genes that induce cellular senescence and cell death.
Many tissues are maintained throughout the lifespan of an organism by a small number of adult stem cells. These cells are unique in that they have both the ability to give rise to new stem cells via a process called self-renewal and the ability to differentiate into the mature cells of a tissue. To maintain tissue homeostasis, stem cells have developed strict regulatory mechanisms to self-renew, differentiate, and prevent premature senescence and apoptosis (see review, ref. 1). The recent observation that Bmi1, a Polycomb group repressor, is essential for the self-renewal of adult murine hematopoietic stem cells (HSCs) and neuronal stem cells, in part via repression of genes involved in senescence, suggests that stem cells have evolved specific mechanisms to repress senescence and to prolong their capacity to proliferate. In this Perspective, we discuss the possible role of Bmi1 in the prevention of senescence in stem cells.
What makes a cell a stem cell?
HSCs are among the best-characterized stem cells. The existence of these cells was proven using clonal assays and retroviral marking (2, 3). Flow cytometry was then used to isolate HSCs based on cell-surface marker expression (4, 5). Subsequently, other types of somatic stem cells such as neuronal stem cells from the peripheral and central nervous systems have been identified (6, 7).
Stem cells possess three fundamental properties (1). First, they must self-renew, allowing the maintenance of the original stem cell population. Self-renewal is a cell division in which one or both of the daughter cells are stem cells that retain the same developmental potential as the mother cell. In contrast, proliferation is a more general term that refers to all types of mitosis, whether they yield stem cells, restricted progenitors, or terminally differentiated cells. Second, stem cells must be able to differentiate into multiple types of mature cells in order to replace the mature cells that turn over in adult tissues. Third, the total number of stem cells is strictly regulated via both extrinsic and intrinsic mechanisms, resulting in the stability of a stable stem cell pool (8–11).
Stem cells and senescence
Senescence is a state in which a cell no longer has the ability to proliferate. Since stem cells maintain many tissues during the lifetime of an animal, it follows that stem cell senescence must be prevented to maintain an organ throughout life. Several studies suggest that cellular senescence is accompanied by changes in gene expression, which might be regulated by epigenetic mechanisms. In support of this hypothesis, histone deacetylase inhibitors, which decondense chromatin and activate the transcription of some genes, can induce a senescence-like state in human fibroblasts (12), suggesting that conversion of some heterochromatin to euchromatin may be a feature of replicative senescence (13, 14). Other studies suggest that chromatin condensation and subsequent downregulation of certain genes might regulate senescence. Senescence accompanies changes in nuclear morphology and formation of a distinct chromatin structure, called senescence-associated heterochromatic foci (SAHF) (15). SAHF do not contain active transcription sites, and they recruit heterochromatin proteins to the genes that are to be stably repressed during senescence. It was shown that SAHF contained the retinoblastoma protein (pRB) in the E2F-responsive promoters, such as cyclin A and proliferating cell nuclear antigen promoters, and silenced the expression of E2F-responsive genes during senescence but not during quiescence (15). Formation of SAHF and silencing require an intact pRB pathway, since inhibition of p16Ink4a prevents SAHF formation and leads to DNA replication. These results provide a molecular mechanism for the maintenance of the senescent state and demonstrate the importance of pRB as a tumor suppressor.
HSCs have an impressive regenerative potential, as demonstrated by transplantation experiments using limited numbers of cells. In mice, serial transplantation is possible for four to six passages, suggesting that individual HSCs are capable of extensive self-renewal but may not be immortal. Even though HSCs express telomerase (16, 17), it is not sufficient to completely prevent telomere erosion during aging (18). Overexpression of the catalytic subunit of the telomerase enzyme in hematopoietic cells prevents telomeres from shortening during serial transplantation of bone marrow. However, even HSCs overexpressing telomerase could be serially transplanted no more than four times, as is the case with wild-type HSCs; this suggests that a telomere-independent mechanism regulates replicative senescence of mouse HSCs during serial transplantation (19). On the other hand, telomerase-deficient HSCs can be serially transplanted only twice, accompanied by an increased rate of telomere shortening, indicating that telomerase is nonetheless needed to prevent premature loss of telomere function during serial transplantation (20, 21).
Role of Bmi1 in stem cell self-renewal
Since epigenetic events such as histone modification have been implicated in senescence, it follows that genes involved in chromatin remodeling and gene expression, such as members of the Polycomb and Trithorax families, might be directly involved in decisions that affect stem cell fate, including self-renewal, senescence, and possibly aging. Polycomb and Trithorax proteins form large multimeric structures, which can lead to repression or activation of gene expression, respectively, via a concerted process of chromatin modifications (22, 23).
Both HSCs and neuronal stem cells express high levels of Bmi1 (24–26), a member of the Polycomb group of transcription repressors that was initially identified as an oncogene cooperating with c-myc in a murine model of lymphoma (27, 28). Bmi1 has a RING finger at the amino-terminus and a central helix-turn-helix domain. The RING finger domain is required for the generation of lymphoma in Eμ-Bmi1 transgenic mice (29, 30). Postnatal mice lacking Bmi1 exhibit defects in hematopoiesis, skeletal patterning, neurological functions, and development of the cerebellum (31).
It has recently been shown that Bmi1 is necessary for efficient self-renewing cell divisions of adult HSCs as well as adult peripheral and central nervous system neural stem cells, but that it is less critical for the generation of differentiated progeny (25, 26). Transplantation of Bmi1–/– fetal liver cells resulted in only transient hematopoietic cell reconstitution, suggesting that the transplanted mutant fetal liver HSCs failed to generate more HSCs but gave rise to multipotent progenitors that could sustain hematopoiesis for up to 4–8 weeks. Similarly, Bmi1 is needed for the maintenance of neural stem cells found in both the central and peripheral nervous systems. As with HSCs, the reduced self-renewal of Bmi1-deficient neural stem cells led to their postnatal depletion in vivo, but the proliferation and survival of committed progenitor cells were essentially normal (26). Given the broad ranges of phenotypic changes in Bmi1-deficient mice, including posterior transformation and neurological abnormalities (31), and its broad tissue distribution (32), it is likely that Bmi1 regulates the self-renewal of other types of somatic stem cells.
Bmi1
may also play a key role in some types of cancer (33–35). In approximately 11% of cases of mantle cell lymphoma, the malignant cells have a three- to sevenfold amplification of Bmi1 DNA and express high levels of the protein, implicating this gene in this invariably lethal form of lymphoma. In a mouse model of leukemia, Bmi1 was essential for the maintenance of leukemic cells (36). Enforced expression of Hoxa9/Meis-1 in both normal and Bmi1-deficient mouse fetal liver cells, followed by transplantation, initially resulted in infiltration of the bone marrow by cells that looked like acute myeloid leukemia (AML) blasts, and mice developed a bone marrow infiltrate that resembled AML. However, only Bmi1 wild-type AML could be serially transplanted. Taken together with the detection of high levels of Bmi1 in human AML stem cells (25), these results suggest that Bmi1 is also required for the self-renewal of leukemic stem cells.
Bmi1 and senescence
In WI-38 human fetal lung fibroblasts, Bmi1 is downregulated when the cells undergo replicative senescence, but not when they are quiescent. Additionally, Bmi1 extends replicative lifespan but does not induce immortalization when overexpressed (37). In the absence of Bmi1, both the p16Ink4a and the p19Arf genes from the Ink4a locus are expressed (38). Lifespan extension by Bmi1 is mediated in part by suppression of the p16Ink4a-dependent senescence pathway and requires an intact pRB pathway, but not the p53 tumor-suppressor protein. The RING finger and helix-turn-helix domains of Bmi1 were required for lifespan extension and p16Ink4a suppression. Furthermore, a RING finger deletion mutant acted as a dominant negative, inducing p16Ink4a and premature senescence (37).
Normal mouse embryonic fibroblasts (MEFs) reach replicative senescence after seven passages in culture, whereas MEFs from Bmi1–/– mice show a premature-senescence phenotype at the third passage. This was correlated with increased expression of p16Ink4a. Re-expression of Bmi1 in Bmi1–/– MEFs prevented premature senescence (28). Overexpression of Bmi1 gave a proliferative advantage and extended MEF lifespan. Furthermore, unlike human fibroblasts, Bmi1 could immortalize MEFs.
Downstream targets of Bmi1
Gene-profiling studies suggest that Bmi1 modulates HSC self-renewal through the regulation of genes important for stem cell fate decisions, as well as survival genes, antiproliferative genes, and stem cell–associated genes (Figure 1) (25). The previously mentioned Bmi1 target, the Ink4a locus (28), encodes p16Ink4a and p19Arf using different promoters (38). Enforced expression of p16Ink4a and p19Arf in HSCs led to senescence and apoptosis, respectively (25). In neural stem cells, p16Ink4a deficiency partially restored the ability of Bmi1-deficient stem cells to self-renew (26). Figure 2 illustrates regulation of the cell cycle and senescence by p16Ink4a and p19Arf. During the cell cycle, pRB is hyperphosphorylated by the cyclin D/cyclin-dependent kinases 4 and 6 (cyclin D/Cdk4/6) complex (39). The hyperphosphorylated pRB is unable to bind and inhibit E2F transcription factor, allowing transcription of E2F target genes that are important for the G1/S transition, such as DNA polymerase II, cyclin E, p19, myb, and dihydrofolate reductase (40). This allows cell cycle progression. In the absence of Bmi1, p16Ink4a is upregulated and prevents binding of Cdk4/6 to cyclin D, inhibiting the kinase activity. This results in hypophosphorylated pRB, which then binds E2F and inhibits E2F-mediated transcription, leading to cell cycle arrest and senescence (39). p19Arf sequesters mouse double minute 2 (MDM2) and inhibits p53 degradation, resulting in p53-mediated cell cycle arrest and apoptosis (41, 42). Point mutations and deletion of p16Ink4a and p19Arf are frequently found in many types of human cancers, which implicates them as key regulators of immortalization and/or senescence checkpoints.
Figure 1.
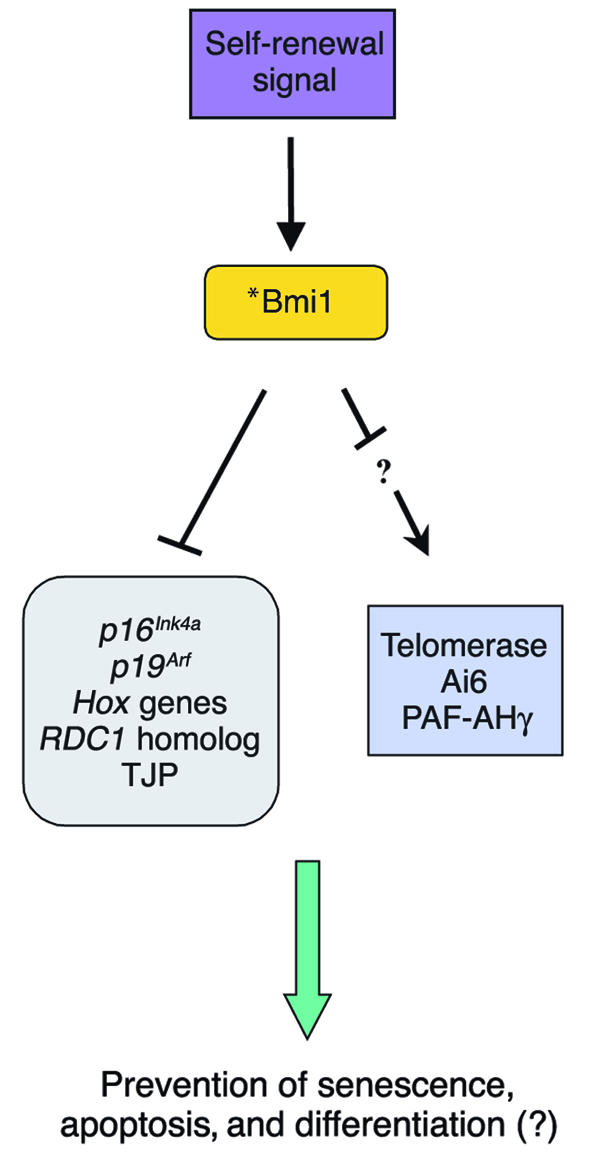
Postulated Bmi1 targets. Extrinsic signals for a stem cell to self-renew result in elevation of the Bmi1 level in stem cells. This allows repression of various genes including the Ink4a locus genes, p16Ink4a and p19Arf, and possibly activation, via indirect mechanisms, of some genes including telomerase, apoptosis inhibitor-6 (Ai6), and platelet-activating factor acetylhydrolase (PAF-AHγ). These genes are likely play a role in stem cell fate decisions including self-renewal and differentiation. *Sites of frequent mutations associated with cancer. TJP, tight junction protein. RDC1, chemokine orphan receptor 1.
Figure 2.

Regulation of cell cycle, apoptosis, and senescence by Bmi1. In normal stem cells, p16Ink4a and p19Arf genes are repressed in a Bmi1-dependent manner. In the absence of p16Ink4a, the cyclin D/Cdk4/6 complex can phosphorylate pRB, allowing the E2F-dependent transcription that leads to cell cycle progression and DNA synthesis. In addition, MDM2-mediated p53 degradation causes low p53 levels in the absence of p19Arf, thus preventing cell cycle arrest and apoptosis. The absence of Bmi1 relieves the repression of the Ink4a locus, resulting in the expression of p16Ink4a and p19Arf. p16Ink4a inhibits binding of cyclin D to Cdk4/6, resulting in inhibition of the kinase activity. This leads to a hypophosphorylated pRB, which then can bind E2F and inhibit E2F-dependent transcription, resulting in cell cycle arrest and senescence. p19Arf inhibits MDM2, which mediates ubiquitin-dependent degradation of p53, thus leading to accumulation of p53 protein in the cell. This leads to induction of various p53 target genes involved in cell cycle arrest and apoptosis. Proteins affected by high and low levels of Bmi1 are shown by black and red arrows, respectively. *Sites of frequent mutations associated with cancer.
Mice lacking Bmi1 showed induction of both p16Ink4a and p19Arf in various hematopoietic and neuronal tissues (25). Overexpression of p16Ink4a and p19Arf in adult HSCs induced cell cycle arrest and apoptosis via the pRB and the p53-dependent pathway, respectively. Double deletion of the Bmi1 and p16Ink4a/p19Arf genes partially rescued the phenotypes observed in Bmi1-deficient mice (28), suggesting that p16Ink4a, p19Arf, and p53 are downstream effectors of Bmi1 that are involved in the control of the proliferation and survival of HSCs during self-renewing cell divisions (Figure 2). Therefore, Bmi1 maintains the HSC pool in part by repressing genes involved in cellular senescence. Increased expression of the p53 target gene Wig1 in Bmi1–/– bone marrow suggests that the p19Arf pathway may have been activated in Bmi1–/– hematopoietic cells. Wig1 is a double-stranded RNA-binding protein and inhibits tumor growth in vitro, suggesting that it may function in stress-induced p53 responses (43). The observation that p53-deficient mice have increased numbers of stem cells is consistent with the notion that p53 might be a downstream effector of Bmi1 (44). In addition, some of the Hox9 family of genes are also affected in Bmi1-deficient hematopoietic tissues and neurospheres (25, 26). Determination of the relative contribution of each of these pathways to the regulation of HSC self-renewal will require careful analysis of the HSCs from double- or triple-knockout mice.
There is evidence that Bmi1 might regulate telomerase expression in human mammary epithelial cells (MECs) and might play a role in the development of human breast cancer. Bmi1 is overexpressed in several breast cancer cell lines and postselection human MECs immortalized with human papilloma virus E6 oncogene, which abrogates the p53/p21waf pathway (45), suggesting that Bmi1 might be involved in immortalization. Postselection MECs can be obtained by regular feeding of a heterogeneous population of MECs from primary mammary tissue. During this process, the p16Ink4a gene is progressively silenced and not expressed in postselection MECs (46, 47). Overexpression of Bmi1 in postselection MECs bypasses senescence, extending replicative lifespan and immortalizing MECs. This is associated with human telomerase reverse transcriptase (hTERT) expression, which leads to induction of telomerase activity. Although hTERT is a direct target of c-Myc–induced transcription in MECs (48, 49), Bmi1 appeared to act independently of c-Myc. Since Bmi1 is a transcription repressor, induction of telomerase is probably mediated by an indirect mechanism. Deletion analysis of the Bmi1 protein suggested that the RING finger, as well as the conserved helix-turn-helix domain, was required for its ability to induce telomerase and immortalization. These data suggest that Bmi1 directly or indirectly regulates telomerase expression in MECs and might play a role in the development of human breast cancer. However, Bmi1 induction of telomerase is cell type specific; Bmi1 fails to induce telomerase in fibroblasts (45). This is consistent with the observation that Bmi1 overexpression did not immortalize human fibroblasts (37). It is not known whether Bmi1 is involved in telomere function in normal breast stem cells.
Future directions
Bmi1 maintains the stem cell pool by preventing premature senescence, either through repression of genes involved in senescence or perhaps through induction of telomerase to prevent telomere shortening. It is very likely that Bmi1 is important for maintenance of multiple types of somatic stem cells, since it is widely expressed and Bmi1-deficient mice have developmental defects in other organs. Bmi1 is also important for maintenance of leukemic stem cells and perhaps other tumorigenic stem cells; therefore, Bmi1 could be used as a molecular target to induce senescence in cancer stem cells (50).
Since Bmi1 maintains the HSC pool size and regulates key genes implicated in senescence and aging, it is of interest to determine whether expression of Bmi1 and its target genes changes during stem cell transplantation and/or aging. Whether stem cells undergo senescence during aging is controversial (51–53). In C57BL mice, in which most HSC studies have been performed, HSC numbers increase with age without losing overall function (54–56). However, HSC senescence might occur during aging in certain other strains of mice (57, 58). The number of times that HSCs can reconstitute the bone marrow of lethally irradiated mice is limited in serial-transplantation experiments. This observation might be either a result of an intrinsic stem cell aging program that occurs only when stem cell proliferation far exceeds that seen during normal aging, or a result of damage to the stem cells that is secondary to the stress of the transplant. In either model, it is possible that the loss of stem cell activity is mediated by Bmi1 or its downstream targets.
References
- 1.Reya T, Morrison S, Clarke M, Weissman I. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 2.Till J, McCulloch E. A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat. Res. 1961;14:1419–1430. [PubMed] [Google Scholar]
- 3.Williams D, Lemischka I, Nathan D, Mulligan R. Introduction of new genetic material into pluripotent haematopoietic stem cells of the mouse. Nature. 1984;310:476–480. doi: 10.1038/310476a0. [DOI] [PubMed] [Google Scholar]
- 4.Spangrude GJ, Heimfeld S, Weissman IL. Purification and characteristics of mouse hematopoietic stem cells. Science. 1988;241:58–62. doi: 10.1126/science.2898810. [DOI] [PubMed] [Google Scholar]
- 5.Morrison SJ, Weissman IL. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity. 1994;1:661–673. doi: 10.1016/1074-7613(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 6.Stemple D, Anderson D. Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell. 1992;71:973–985. doi: 10.1016/0092-8674(92)90393-q. [DOI] [PubMed] [Google Scholar]
- 7.Davis A, Temple S. A self-renewing multipotential stem cell in embryonic rat cerebral cortex. Nature. 1994;372:263–266. doi: 10.1038/372263a0. [DOI] [PubMed] [Google Scholar]
- 8.Domen J, Cheshier SH, Weissman IL. The role of apoptosis in the regulation of hematopoietic stem cells: overexpression of BCL-2 increases both their number and repopulation potential. J. Exp. Med. 2000;191:253–264. doi: 10.1084/jem.191.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lemischka IR, Moore KA. Stem cells: interactive niches. Nature. 2003;425:778–779. doi: 10.1038/425778a. [DOI] [PubMed] [Google Scholar]
- 10.Calvi LM, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- 12.Ogryzko V, Hirai T, Russanova V, Barbie D, Howard B. Human fibroblast commitment to a senescence-like state in response to histone deacetylase inhibitors is cell cycle dependent. Mol. Cell. Biol. 1996;16:5210–5218. doi: 10.1128/mcb.16.9.5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howard B. Replicative senescence: considerations relating to the stability of heterochromatin domains. Exp. Gerontol. 1996;31:281–293. doi: 10.1016/0531-5565(95)00022-4. [DOI] [PubMed] [Google Scholar]
- 14.Villeponteau B. The heterochromatin loss model of aging. Exp. Gerontol. 1997;32:383–394. doi: 10.1016/s0531-5565(96)00155-6. [DOI] [PubMed] [Google Scholar]
- 15.Narita M, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 16.Pathak S. Organ- and tissue-specific stem cells and carcinogenesis. Anticancer Res. 2002;22:1353–1356. [PubMed] [Google Scholar]
- 17.Morrison S, Prowse K, Ho P, Weissman I. Telomerase activity in hematopoietic cells is associated with self-renewal potential. Immunity. 1996;5:207–216. doi: 10.1016/s1074-7613(00)80316-7. [DOI] [PubMed] [Google Scholar]
- 18.Yui J, Chiu C-P, Lansdorp PM. Telomerase activity in candidate stem cells from fetal liver and adult bone marrow. Blood. 1998;91:3255–3262. [PubMed] [Google Scholar]
- 19.Allsopp RC, Weissman IL. Replicative senescence of hematopoietic stem cells during serial transplantation: does telomere shortening play a role? Oncogene. 2002;21:3270–3273. doi: 10.1038/sj.onc.1205314. [DOI] [PubMed] [Google Scholar]
- 20.Samper E, et al. Long-term repopulating ability of telomerase-deficient murine hematopoietic stem cells. Blood. 2002;99:2767–2775. doi: 10.1182/blood.v99.8.2767. [DOI] [PubMed] [Google Scholar]
- 21.Allsopp RC, Morin GB, DePinho R, Harley CB, Weissman IL. Telomerase is required to slow telomere shortening and extend replicative lifespan of HSCs during serial transplantation. Blood. 2003;102:517–520. doi: 10.1182/blood-2002-07-2334. [DOI] [PubMed] [Google Scholar]
- 22.Simon J, Tamkun J. Programming off and on states in chromatin: mechanisms of Polycomb and trithorax group complexes. Curr. Opin. Genet. Dev. 2002;12:210–218. doi: 10.1016/s0959-437x(02)00288-5. [DOI] [PubMed] [Google Scholar]
- 23.Orlando V. Polycomb, epigenomes, and control of cell identity. Cell. 2003;112:599–606. doi: 10.1016/s0092-8674(03)00157-0. [DOI] [PubMed] [Google Scholar]
- 24.Lessard J, Baban S, Sauvageau G. Stage-specific expression of Polycomb group genes in human bone marrow cells. Blood. 1999;91:1216–1224. [PubMed] [Google Scholar]
- 25.Park I-K, et al. Bmi1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 26.Molofsky AV, et al. Bmi1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425:962–967. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haupt Y, Bath M, Harris A, Adams J. Bmi1 transgene induces lymphomas and collaborates with myc in tumorigenesis. Oncogene. 1993;8:316–314. [PubMed] [Google Scholar]
- 28.Jacob J, Kieboom K, Marino S, Depinho R, van Lohuizen M. The oncogene and Polycomb-group gene Bmi1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 29.van Lohuizen M, et al. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell. 1991;65:737–752. doi: 10.1016/0092-8674(91)90382-9. [DOI] [PubMed] [Google Scholar]
- 30.Alkema M, Jacobs H, van Lohuizen M, Berns A. Pertubation of B and T cell development and predisposition to lymphomagenesis in Emu Bmi1 transgenic mice require the Bmi1 RING finger. Oncogene. 1997;15:899–910. doi: 10.1038/sj.onc.1201262. [DOI] [PubMed] [Google Scholar]
- 31.van der Lugt NM, et al. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the Bmi1 proto-oncogene. Genes Dev. 1994;8:757–769. doi: 10.1101/gad.8.7.757. [DOI] [PubMed] [Google Scholar]
- 32.Haupt Y, Alexander W, Barri G, Klinken S, Adams J. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell. 1991;65:753–763. doi: 10.1016/0092-8674(91)90383-a. [DOI] [PubMed] [Google Scholar]
- 33.Bea S, et al. BMI1 gene amplification and overexpression in hematological malignancies occur mainly in mantle cell lymphomas. Cancer Res. 2001;61:2409–2412. [PubMed] [Google Scholar]
- 34.van Kemenade FJ, et al. Coexpression of BMI1 and EZH2 polycomb-group proteins is associated with cycling cells and degree of malignancy in B-cell non-Hodgkin lymphoma. Blood. 2001;97:3896–3901. doi: 10.1182/blood.v97.12.3896. [DOI] [PubMed] [Google Scholar]
- 35.Vonlanthen S, et al. The Bmi1 oncoprotein is differentially expressed in non-small cell lung cancer and correlates with INK4A-ARF locus expression. Br. J. Cancer. 2001;84:1372–1376. doi: 10.1054/bjoc.2001.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lessard J, Sauvageau G. Bmi1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 37.Itahana K, et al. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi1. Mol. Cell. Biol. 2003;23:389–401. doi: 10.1128/MCB.23.1.389-401.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;84:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 39.Sharpless N, DePinho R. The INK4A/ARF locus and its two gene products. Curr. Opin. Genet. Dev. 1999;9:22–30. doi: 10.1016/s0959-437x(99)80004-5. [DOI] [PubMed] [Google Scholar]
- 40.Vernell R, Helin K, Muller H. Identification of target genes of the p16INK4A-pRB-E2F pathway. J. Biol. Chem. 2003;278:46124–46137. doi: 10.1074/jbc.M304930200. [DOI] [PubMed] [Google Scholar]
- 41.Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat. Cell Biol. 1999;1:20–26. doi: 10.1038/8991. [DOI] [PubMed] [Google Scholar]
- 42.Honda R, Yasuda H. Association of p19ARF with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 1999;18:22–27. doi: 10.1093/emboj/18.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mendez-Vidal C, Wilhelm MT, Hellborg F, Qian W, Wiman KG. The p53-induced mouse zinc finger protein wig-1 binds double-stranded RNA with high affinity. Nucleic Acids Res. 2002;30:1991–1996. doi: 10.1093/nar/30.9.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.TeKippe M, Harrison DE, Chen J. Expansion of hematopoietic stem cell phenotype and activity in Trp53-null mice. Exp. Hematol. 2003;31:521–527. doi: 10.1016/s0301-472x(03)00072-9. [DOI] [PubMed] [Google Scholar]
- 45.Dimri GP, et al. The Bmi1 oncogene induces telomerase activity and immortalizes human mammary epithelial cells. Cancer Res. 2002;62:4736–4745. [PubMed] [Google Scholar]
- 46.Brenner A, Stampfer M, Aldaz C. Increased p16 expression with first senescence arrest in human mammary epithelial cells and extended growth capacity with p16 inactivation. Oncogene. 1998;17:199–205. doi: 10.1038/sj.onc.1201919. [DOI] [PubMed] [Google Scholar]
- 47.Wong D, Foster S, Galloway D, Reid B. Progressive region-specific de novo methylation of the p16 CpG island in primary human mammary epithelial cell strains during escape from M(0) growth arrest. Mol. Biol. Cell. 1999;19:5642–5651. doi: 10.1128/mcb.19.8.5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu K, et al. Direct activation of TERT transcription by c-MYC. Nat. Genet. 1999;21:220–224. doi: 10.1038/6010. [DOI] [PubMed] [Google Scholar]
- 49.Greenberg R, et al. Telomerase reverse transcriptase gene is a direct target of c-Myc but is not functionally equivalent in cellular transformation. Oncogene. 1999;18:1219–1226. doi: 10.1038/sj.onc.1202669. [DOI] [PubMed] [Google Scholar]
- 50.Schmitt C. Senescence, apoptosis and therapy: cutting the lifelines of cancer. Nat. Rev. Cancer. 2003;3:286–295. doi: 10.1038/nrc1044. [DOI] [PubMed] [Google Scholar]
- 51.Van Zant G, Liang Y. The role of stem cells in aging. Exp. Hematol. 2003;31:659–672. doi: 10.1016/s0301-472x(03)00088-2. [DOI] [PubMed] [Google Scholar]
- 52.Liang Y, Van Zant G. Genetic control of stem-cell properties and stem cells in aging. Curr. Opin. Hematol. 2003;10:195–202. doi: 10.1097/00062752-200305000-00001. [DOI] [PubMed] [Google Scholar]
- 53.Geiger H, True JM, de Haan G, Van Zant G. Age- and stage-specific regulation patterns in the hematopoietic stem cell hierarchy. Blood. 2001;98:2966–2972. doi: 10.1182/blood.v98.10.2966. [DOI] [PubMed] [Google Scholar]
- 54.Morrison S, Prowse K, Ho P, Weissman I. The aging of hematopoietic stem cells. Nat. Med. 1996;2:1011–1016. doi: 10.1038/nm0996-1011. [DOI] [PubMed] [Google Scholar]
- 55.Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. J. Exp. Med. 2000;192:1273–1280. doi: 10.1084/jem.192.9.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim M, Moon H-B, Spangrude GJ. Major age-related changes of mouse hematopoietic stem/progenitor cells. Ann N. Y. Acad. Sci. 2003;996:195–208. doi: 10.1111/j.1749-6632.2003.tb03247.x. [DOI] [PubMed] [Google Scholar]
- 57.Van Zant G, Holland B, Eldridge P, Chen J. Genotype-restricted growth and aging patterns in hematopoietic stem cell populations of allophenic mice. J. Exp. Med. 1990;171:1547–1565. doi: 10.1084/jem.171.5.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen J, Astle CM, Harrison DE. Genetic regulation of primitive hematopoietic stem cell senescence. Exp. Hematol. 2000;28:442–450. doi: 10.1016/s0301-472x(99)00157-5. [DOI] [PubMed] [Google Scholar]