

Abstract
A library of quinoxaline derivatives were prepared to target non-structural protein 1 of influenza A (NS1A) as a means to develop anti-influenza drug leads. An in vitro fluorescence polarization assay demonstrated that these compounds disrupted the dsRNA-NS1A interaction to varying extents. Changes of substituent at positions 2, 3 and 6 on the quinoxaline ring led to variance in responses. The most active compounds (35 and 44) had IC50 values in the range of low micromolar concentration without exhibiting significant dsRNA intercalation. Compound 44 was able to inhibit influenza A/Udorn/72 virus growth.
Keywords: Quinoxaline derivatives, NS1A protein, Influenza A virus, Fluorescence polarization
Influenza viruses cause a highly contagious respiratory disease in humans. They are RNA viruses composed of three general types: influenza A, influenza B, and influenza C.1 The type A viruses cause the most severe diseases, and as a result, are the most serious threat to human health.2 The influenza A virus can be further divided into different serotypes. H1N1 caused the 2009 flu pandemic,3 and H5N1 is a current pandemic threat.4 Therefore, the development of small molecule based anti-influenza therapeutics continues to capture significant attention.5,6
The NS1 protein,7 a highly conserved influenza virus encoded protein, has been identified as a potential target for antiviral development.8 Specifically, the double-stranded RNA (dsRNA) binding domain, comprising residues 1 – 73, is crucial for virus replication, and is the primary target of our work. Detailed biophysical and structural studies by high-resolution NMR and X-ray analysis revealed that the the N-terminal domain of the NS1A protein forms a homodimer with a unique six-helical chain fold.7 There is a deep cavity at the center of dsRNA-binding surface. If a small molecule can fit into this cavity, it can block dsRNA binding and thus inactivate the NS1 protein.
(−)-Epigallocatechin-3-gallate (EGCG)9 was identified to inhibit NS1A through high-throughput screening. EGCG and its derivatives10 display a broad range of biological activities.11 In an effort to design and synthesize structurally simple molecules targeting NS1A protein,
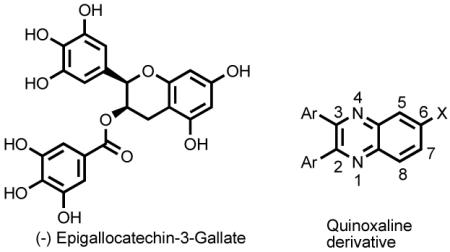 we turned our attention into the quinoxaline scaffold, which can be rapidly constructed. Quinoxalines, an important class of heterocycles, are components of several biologically active compounds.12 Quinoxaline and EGCG share structural similarities: a bicyclic ring and the potential for substitution with polar groups on the ring. Here, we report a structure-activity relationship (SAR) study with quinoxaline analogs targeting the NS1A protein.
we turned our attention into the quinoxaline scaffold, which can be rapidly constructed. Quinoxalines, an important class of heterocycles, are components of several biologically active compounds.12 Quinoxaline and EGCG share structural similarities: a bicyclic ring and the potential for substitution with polar groups on the ring. Here, we report a structure-activity relationship (SAR) study with quinoxaline analogs targeting the NS1A protein.
A library of 46 compounds were designed and synthesized. While keeping the quinoxaline core, various aromatic residues, such as 4-methoxyphenyl, 4-hydroxyphenyl, 2-furyl, and 2-pyridyl, were incorporated into positions 2 and 3, and different substituents were also placed in position 6. In general, 2,3-disubstituted quinoxalines were prepared by condensation of 1,2-diketones and o-phenylenediamine derivatives in refluxing EtOH or HOAc/NaOAc (eq 1).12
 |
(1) |
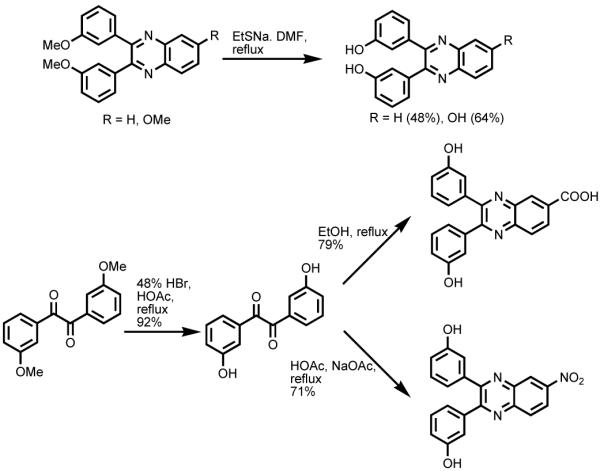
For demethylation of the methoxyphenyl substituted derivatives, many conditions were tested, including HBr/HOAc, BBr3/CH2Cl2, and EtSNa/DMF. For 3-methoxyphenyl and 4-methoxyphenyl substituted quinoxalines, treatment with EtSNa in refluxing DMF afforded the corresponding 3-hydroxyphenyl and 4-hydroxyphenyl derivatives when either H or OMe was in position 6. When electron-withdrawing groups, such as COOH and NO2, were in position 6 of quinoxalines, demethylation of 3,3′-dimethoxybenzil or 4,4′-dimethoxybenzil was achieved utilizing 48% HBr in HOAc under refluxing conditions, prior to condensation with o-phenylenediamine derivatives (Scheme 1).
Scheme 1.
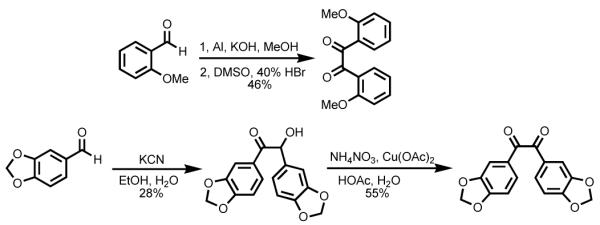
Several of the 1,2-diketones we used in eq 1 are not readily available. For example, 2,2′-dimethoxybenzil was prepared from o-anisaldehyde using Pinacol coupling followed by oxidation.13 Benzoin condensation of piperonal followed by oxidation afforded 3,4,3′,4′-bis(methylenedioxy)-benzil (Scheme 2). Condensation with these 1,2-phenylenediamines was done as described above. However, attempts to deprotect the catechol using either BBr3/CH2Cl2 or EtSNa/DMF afforded a complicated mixture.
Scheme 2.
In addition, 2,3-furyl-quinoxaline-6-carboxylic acid was coupled with various amines using PyBOP or TBTU as a coupling reagent and DIPEA as a base to afford a library of amide substituted quinoxaline analogs (eq 2).
 |
(2) |
In order to examine whether the quinoxaline analogues can disrupt the dsRNA binding to NS1A protein, an in vitro fluorescence polarization-based binding assay (FP assay)14 was employed. In this assay, a carboxyfluorescein-labeled dsRNA (FAM-dsRNA) was used as a signaling probe. In detail, when FAM-dsRNA binds to the NS1A protein, the mobility of the fluorophore (FAM) decreases and as a result, the fluorescence polarization increases. The addition of potential NS1A inhibitors targeting the dsRNA binding domain will displace the FAM-dsRNA from NS1A and lead to a decrease of fluorescence polarization. The data were reported as % binding at 50 μM, where a higher percentage represents stronger activity in breaking the dsRNA-NS1A interaction. A similar FP based assay to probe dsRNA intercalation of the quinoxaline derivatives was utilized as a control experiment, because targeting NS1A instead of dsRNA was desired. The data were reported as % intercalation at 50 μM, and (+) sign means intercalating to the dsRNA while (−) sign means denaturation of the dsRNA to ssRNAs. All assays were run in duplicates, and data were averaged. The compounds with high % binding at 50 μM and low % intercalation at 50 μM were subjected to further studies.
We first set out to explore SARs of 2,3,6-substituted quinoxaline derivatives, and the results are shown in Table 1. Substitution at positions 2 and 3 on the quinoxaline core had the most significant impact on the activity. Compounds with bis 2-furyl substitutions (27-30) were the most potent. Replacements of the furyl residue with methoxyphenyl (1-12), phenol (13-22), 3,4-(methylenedioxy)phenyl (23-25), or 2-pyridyl groups (31-33), reduced the activity. Compounds with phenol substitution (13-22) generally showed improved activity compared to the corresponding compounds with methoxyphenyl substitution (1-12). For bis 2-furyl substituted quinoxaline derivatives, substitution at position 6 also played an important role in the biological activity. Compounds 27, 28 and 29 exhibited significantly higher activity than 26, which has a hydrogen atom at position 6.
Table 1.
Activities of 2,3,6-substituted quinoxaline derivatives
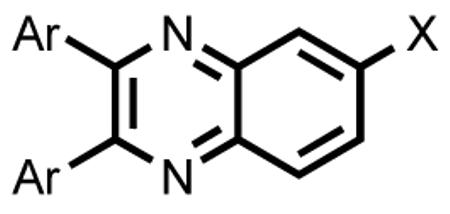
| Compound | Ar | X | % Binding at 50 μM |
% Intercalation at 50 μM |
|---|---|---|---|---|
| 1 | 2-OMe- Ph- |
H | 5.0 | −6.7 |
| 2 | 2-OMe- Ph- |
OMe | 4.4 | −5.0 |
| 3 | 2-OMe- Ph- |
COOH | 2.8 | −2.5 |
| 4 | 2-OMe- Ph- |
NO2 | 6.7 | −3.7 |
| 5 | 3-OMe- Ph- |
H | 1.4 | −6.0 |
| 6 | 3-OMe- Ph- |
OMe | 7.2 | −8.4 |
| 7 | 3-OMe- Ph- |
COOH | 9.1 | −4.4 |
| 8 | 3-OMe- Ph- |
NO2 | 1.5 | 1.9 |
| 9 | 4-OMe- Ph- |
H | 0.5 | 1.4 |
| 10 | 4-OMe- Ph- |
OMe | 1.8 | 5.7 |
| 11 | 4-OMe- Ph- |
COOH | 4.5 | −4.8 |
| 12 | 4-OMe- Ph- |
NO2 | −19.7 | 112.7 |
| 13 | 2-OH- Ph- |
H | −0.9 | −1.0 |
| 14 | 2-OH- Ph- |
OH | 10.9 | 0.9 |
| 15 | 3-OH- Ph- |
H | 10.9 | −4.8 |
| 16 | 3-OH- Ph- |
OH | 25.0 | 18.2 |
| 17 | 3-OH- Ph- |
COOH | 28.7 | −3.4 |
| 18 | 3-OH- Ph- |
NO2 | 42.8 | −15.8 |
| 19 | 4-OH- Ph- |
H | 10.2 | 8.5 |
| 20 | 4-OH- Ph- |
OH | 38.8 | 29.5 |
| 21 | 4-OH- Ph- |
COOH | 13.3 | −7.7 |
| 22 | 4-OH- Ph- |
NO2 | 19.5 | −32.0 |
| 23 15 | 3,4-O,O- CH2-Ph- |
H | 1.5 | 7.9 |
| 24 | 3,4-O,O- CH2-Ph- |
COOH | 23.2 | −4.8 |
| 25 | 3,4-O,O- CH2-Ph- |
NO2 | 56.6 | 24.4 |
| 26 | 2-Furyl- | H | 7.3 | −23.7 |
| 27 | 2-Furyl- | OMe | 54.3 | −3.2 |
| 28 | 2-Furyl- | COOH | 60.9 | 5.8 |
| 29 | 2-Furyl- | COOMe | 76.0 | −12.3 |
| 30 | 2-Furyl- | NO2 | 25.5 | 25.9 |
| 31 | 2- Pyridyl- |
H | −3.1 | 2.3 |
| 32 | 2- Pyridyl- |
COOH | 15.7 | 2.9 |
| 33 | 2- Pyridyl- |
NO2 | 7.8 | −5.6 |
Next, we prepared a library based on compound 28, and the results are listed in Table 2. Keeping 2-furyl at positions 2 and 3, phenyl groups with various substitution or heterocycles were incorporated into position 6 through an amide linker. The compounds showing the most potency were 35 and 44, with 3-methoxyphenyl and 2-furyl at position 6, respectively. Their percentage binding at 50 μM is 79.5 ± 1.5 and 74.0 ± 1.4 μM for 44 and 35 respectively, which is comparable to that of EGCG (89.0 ± 11.4 μM). Replacement of 3-methoxyphenyl with other methoxy substituted phenyl groups (34, 36-38), fluorophenyl (39-41), or phenyl 4-methyl ester (42) resulted in reduced activity. 2-pyridyl (43) substitution also decreased the biological activity. Replacement of 2-furyl with 2-thenyl (45) was not tolerated. An aliphatic 2-furyl mimic, glycine methyl ester derivative (46), showed decreased activity. The four compounds with the highest percentage binding at 50 μM were further studied in dose dependent FP assay. They have the following IC50 (μM): 74.8 (29), 6.2 (35), 43.3 (43) and 3.5 (44). None of these four compounds showed significant dsRNA interference.
Table 2.
Activities of amide derivatives of compound 28
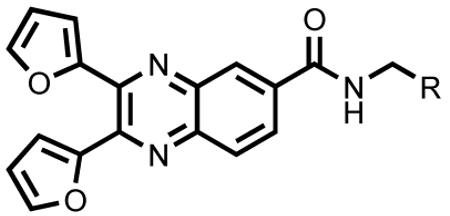
| Compound | R | % Binding at 50 μM |
% Intercalation at 50 μM |
|---|---|---|---|
| 34 | 2-OMe-Ph- | 29.5 | −17.7 |
| 35 | 3-OMe-Ph- | 74.0 | 4.5 |
| 36 | 4-OMe-Ph- | 44.0 | 7.6 |
| 37 | 3,4-(OMe)2- Ph- |
39.5 | 5.6 |
| 38 | 3,4,5- (OMe)3-Ph- |
53.6 | −4.7 |
| 39 | 2-F-Ph- | 11.6 | −11.3 |
| 40 | 3-F-Ph- | 13.6 | −11.6 |
| 41 | 4-F-Ph- | 49.0 | 6.3 |
| 42 | 4-COOMe- Ph- |
47.9 | 8.8 |
| 43 | 2-Pyridyl- | 64.7 | −9.6 |
| 44 | 2-Furyl- | 79.5 | 5.9 |
| 45 | 2-Thenyl- | 8.2 | −10.5 |
| 46 | COOMe | 26.7 | −27.9 |
We then set out to explore the biological activity of the most potent compound (44) identified from the in vitro assay described above. Viral growth was assayed using MDCK cells infected with influenza A/Udorn/72 virus. Figure 1 shows the single cycle growth curve at MOI (multiplicity of infection) 5 (top panel) and multiple cycles growth curve at MOI 0.001 (bottom panel). Following one hour infection, growth media containing compound 44 diluted in DMSO or control (0.1 % DMSO) was added. Compound 44 inhibited influenza A/Udorn/72 virus growth ~ 10 fold (top panel). The multiple cycles growth assay indicates that compound 44 appeared to lose effectiveness of inhibition after 37 h (bottom panel). Although these are qualitative experiments, they demonstrate the antiviral potential of those quinoxlaine derivatives.
Figure 1.

Single cycle virus growth curve (top panel) and multiple cycles virus growth curve (bottom panel) after compound 44 was incubated.
In conclusion, we have explored preliminary structure-activity relationships (SARs) for a library of quinoxaline derivatives targeting the NS1A protein for the development of anti-influenza therapeutics. Those quinoxline derivatives were designed to mimic (−)-Epigallocatechin-3-gallate, a hit identified by high-throughput screening. In vitro fluorescence polarization experiments showed that these compounds can bind to NS1A to disrupt the NS1A-dsRNA interaction. Substitution at positions 2 and 3 on the quinoxaline core played an important role, with 2-furyl being the best among those investigated. Substitution at position 6 was also crucial. Compounds 35 and 44 were the most potent, with an IC50 of 6.2 and 3.5 μM, respectively. The dsRNA intercalation experiments indicated that both 35 and 44 do not inhibit NS1A-dsRNA interactions by interfering with dsRNA, but likely function by binding to the NS1A dsRNA-binding domain itself. Results from a cell assay demonstrated that compound 44 was able to inhibit influenza A virus growth. Detailed structural analysis and optimization are currently ongoing.
Supplementary Material
Acknowledgement
We thank the National Institutes of Health (U01 AI074497) and The Texas Institute for Drug and Diagnostic Development for support of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Lagoja IM, De Clercq E. Med. Res. Rev. 2008;28:1. doi: 10.1002/med.20096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taubenberger JK, Morens DM. Annu. Rev. Pathol. Mech. Dis. 2008;3:499. doi: 10.1146/annurev.pathmechdis.3.121806.154316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bautista E, Chotpitayasunondh T, Gao Z, Harper SA, Shaw M, Uyeki TM, Zaki SR, Hayden FG, Hui DS, Kettner JD, Kumar A, Lim M, Shindo N, Penn C, Nicholson KG. N. Engl. J. Med. 2010;362:1708. doi: 10.1056/NEJMra1000449. [DOI] [PubMed] [Google Scholar]
- 4.Abdel-Ghafar A-N, Chotpitayasunondh T, Gao Z, Hayden FG, Hien ND, de Jong M, Naghdaliyev A, Peiris JSM, Shindo N, Soeroso S, Uyeki TM. N. Engl. J. Med. 2008;358:261. doi: 10.1056/NEJMra0707279. [DOI] [PubMed] [Google Scholar]
- 5.De Clercq E. Nat. Rev. Drug Discov. 2006;5:1015. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Das K, Aramini JM, Ma L-C, Krug RM, Arnold E. Nat. Struct. Mol. Biol. 2010;17:530. doi: 10.1038/nsmb.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7 (a).Chien C, Tejero R, Huang Y, Zimmerman DE, Rios CB, Krug RM, Montelione GT. Nature Struct. Biol. 1997;4:891. doi: 10.1038/nsb1197-891. [DOI] [PubMed] [Google Scholar]; (b) Liu J, Lynch PA, Chien C, Montelione GT, Krug RM, Berman HM. Nature Struct. Biol. 1997;4:896. doi: 10.1038/nsb1197-896. [DOI] [PubMed] [Google Scholar]; (c) Min J-Y, Krug RM. Proc. Natl. Acad. Sci. 2006;103:7100. doi: 10.1073/pnas.0602184103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hale BG, Randall RE, Ortin J, Jackson D. J. Gen. Virol. 2008;89:2359. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]; (e) Yin C, Khan JA, Swapna GVT, Ertekin A, Krug RM, Tong L, Montelione GT. J. Biol. Chem. 2007;282:20584. doi: 10.1074/jbc.M611619200. [DOI] [PubMed] [Google Scholar]
- 8 (a).Fernandez-Sesma A. Infect Disord. Drug Targets. 2007;7:336. doi: 10.2174/187152607783018754. [DOI] [PubMed] [Google Scholar]; (b) Krug RM, Aramini JM. Trends in Pharmacological Sciences. 2009;30:269. doi: 10.1016/j.tips.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Basu D, Walkiewicz MP, Frieman M, Baric RS, Auble DT, Engel DA. J. Virol. 2009;83:1881. doi: 10.1128/JVI.01805-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Walkiewicz MP, Basu D, Jablonski JJ, Geysen HM, Engel DA. J. Gen. Virol. 2010 doi: 10.1099/vir.0.025015-0. DOI: 10.1099/vir.0.025015-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9 (a).Yamamoto T, Juneja LR, Chu D-C, Kim M. Chemistry and Applications of Green Tea. CRC; Boca Raton: 1997. [Google Scholar]; (b) Li L, Chan TH. Org. Lett. 2001;3:739. doi: 10.1021/ol000394z. [DOI] [PubMed] [Google Scholar]
- 10 (a).Wan SB, Dou QP, Chan TH. Tetrahedron. 2006;62:5897. [Google Scholar]; (b) Tanaka H, Miyoshi H, Chuang Y-C, Ando Y, Takahashi T. Angew. Chem. Int. Ed. 2007;46:5934. doi: 10.1002/anie.200701276. [DOI] [PubMed] [Google Scholar]
- 11 (a).Park KD, Lee SG, Kim SU, Kim SH, Sun WS, Cho SJ, Jeong DH. Bioorg. Med. Chem. Lett. 2004;14:5189. doi: 10.1016/j.bmcl.2004.07.063. [DOI] [PubMed] [Google Scholar]; (b) Anderson JC, Headley C, Stapleton PD, Taylor PW. Bioorg. Med, Chem. Lett. 2005;15:2633. doi: 10.1016/j.bmcl.2005.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hayes CJ, Whittaker BP, Watson SA, Grabowska AM. J. Org. Chem. 2006;71:9701. doi: 10.1021/jo061740e. [DOI] [PubMed] [Google Scholar]; (d) Sánchez-del-Campo L, Otón F, Tárraga A, Cabezas-Herrera J, Chazarra S, Rodríguez-López JN. J. Med. Chem. 2008;51:2018. doi: 10.1021/jm701346h. [DOI] [PubMed] [Google Scholar]
- 12.For several recent examples, see: Hui X, Desrivot J, Bories C, Loiseau PM, Franck X, Hocquemiller R, Figadère B. Bioorg. Med. Chem. Lett. 2006;16:815. doi: 10.1016/j.bmcl.2005.11.025. Rong F, Chow S, Yan S, Larson G, Hong Z, Wu J. Bioorg. Med. Chem. Lett. 2007;17:1663. doi: 10.1016/j.bmcl.2006.12.103. Zhang L, Qiu B, Xiong B, Li X, Li J, Wang X, Li J, Shen J. Bioorg. Med. Chem. Lett. 2007;17:2118. doi: 10.1016/j.bmcl.2007.01.094. Porter J, Lumb S, Lecomte F, Reuberson J, Foley A, Calmiano M, Riche K, Edwards H, Delgado J, Franklin RJ, Gascon-Simorte JM, Maloney A, Meier C, Batchelor M. Bioorg. Med. Chem. Lett. 2009;19:397. doi: 10.1016/j.bmcl.2008.11.062. Romeiro NC, Aguirre G, Hernández P, González M, Cerecetto H, Aldana I, Pérez-Silanes S, Monge A, Barreiro EJ, Lima LM. Bioorg. Med. Chem. 2009;17:641. doi: 10.1016/j.bmc.2008.11.065.
- 13.Liu Y, Sandoval CA, Yamaguchi Y, Zhang X, Wang Z, Kato K, Ding K. J. Am. Chem. Soc. 2006;128:14212. doi: 10.1021/ja063350f. [DOI] [PubMed] [Google Scholar]
- 14 (a).Chien C, Xu Y, Xiao R, Aramini JM, Sahasrabudhe PV, Krug RM, Montelione GT. Biochemistry. 2004;43:1950. doi: 10.1021/bi030176o. [DOI] [PubMed] [Google Scholar]; (b) Rishi V, Potter T, Laudeman J, Reinhart R, Silvers T, Selby M, Stevenson T, Krosky P, Stephen AG, Acharya A, Moll J, Oh WJ, Scudiero D, Shoemaker RH, Vinson C. Anal. Biochem. 2005;340:259. doi: 10.1016/j.ab.2005.02.012. [DOI] [PubMed] [Google Scholar]
-
15.Ar (in 23, 24 and 25) =
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.