

Abstract
Objective
To describe the Alzheimer disease (AD)-like clinical and pathological features, including marked neurofibrillary tangle (NFT) pathology, of a familial prion disease due to a rare nonsense mutation of the prion gene (PRNP).
Methods
Longitudinal clinical assessments were available for the proband and her mother. After death, both underwent neuropathological evaluation. PRNP was sequenced after failure to find immunopositive Aβ deposits in the proband and the documentation of prion protein (PrP) immunopositive pathology.
Results
The proband presented at age 42 years with a 3-year history of progressive short-term memory impairment and depression. Neuropsychological testing found impaired memory performance, with relatively preserved attention and construction. She was diagnosed with AD and died at age 47 years. Neuropathologic evaluation revealed extensive limbic and neocortical NFT formation and neuritic plaques consistent with a Braak stage of VI. The NFTs were immunopositive, with multiple tau antibodies, and electron microscopy revealed paired helical filaments. However, the neuritic plaques were immunonegative for Aβ, whereas immunostaining for PrP was positive. The mother of the proband had a similar presentation, including depression, and had been diagnosed clinically and pathologically as AD. Reevaluation of her brain tissue confirmed similar tau and PrP immunostaining findings. Genetic analysis revealed that both the proband and her mother had a rare PRNP mutation (Q160X) that resulted in the production of truncated PrP.
Interpretation
We suggest that PRNP mutations that result in a truncation of PrP lead to a prolonged clinical course consistent with a clinical diagnosis of AD and severe AD-like NFTs.
Inherited prion diseases are a heterogeneous group of autosomal dominantly inherited neurodegenerative syndromes that were originally divided into Gerstmann-Sträussler-Scheinker syndrome (GSS), familial Creutzfeldt-Jacob disease (CJD), and fatal familial insomnia (FFI) based primarily on clinical and pathological characteristics. Mutations in the prion protein (PrP) gene (PRNP) were eventually found to be causative in all of these prion diseases, and since then it has become apparent that PRNP mutation-associated diseases manifest overlapping as well as distinctive clinical and pathological features.1,2
Here, we report a family with a rare PRNP mutation in which the clinical presentation, course, and initial neuropathological studies were strongly suggestive of Alzheimer disease (AD). A clinical diagnosis of early-onset AD was initially made for our proband. Ten years prior, the proband’s mother was also clinically and pathologically diagnosed with AD; the autopsy, carried out in 1987, revealed abundant neuritic plaques and neurofibrillary tangles (NFTs). After an 8-year course, consisting solely of cognitive decline, the proband expired. Her autopsy was also remarkable for abundant limbic and neocortical neuritic plaque-like structures and NFTs, consistent with a neuropathologic diagnosis of AD. However, immunohistochemical studies, which were unavailable at the time of her mother’s autopsy, demonstrated PrP, rather than Aβ, immunopositive deposits.
Subsequently, we found this family to have a nonsense substitution for glutamine (Q) at position 160 (Q160X) in PRNP that results in production of truncated PrP. This mutation has been previously described in one family, although with limited clinical description and no microscopic neuropathological characterization.3 Interestingly, a similar mutation, Y145X, has also been reported with a truncated PrP and severe neurofibrillary tangle pathology.4
Subjects and Methods
Subjects
Both the proband and her mother were subjects in the University of Washington Alzheimer’s Disease Research Center. Informed consent was obtained for longitudinal clinical evaluation, genetic studies, and autopsy at the time of death.
Neuropathology
The proband and her mother received a standard neuropathological workup, including gross and microscopic examinations. Histological evaluations included hematoxylin-eosin (H&E), modified Bielschowsky, and thioflavin S methods. In addition, immunostaining was performed for PrP (3F4, Chemicon International, Temecula, CA, 1:500; PrP 23-40, PrP 90-102, PrP 220-2315; PrP 1:1,7506), Aβ (6E10, Signet Laboratories, Dedham, MA, 1:400), tau (Tau-2, Sigma, St Louis, MO, 1:500, 1:500; PHF-1, generous gift of P. Davies, 1:10; AT8, Endogen, Woburn, MA, 1:250; RD3 and RD4, Upstate, Charlottesville, VA, 1:800 and 1:80, respectively), neurofilament (SMI-31, Sternberger Monoclonals, Covance, Princeton, NJ, 1:4,000), TDP-43 (Proteintech, Chicago, IL, 1:2,000), and alpha-synuclein (antibody LB509, generous gift of J. Q. Trojanowski, 1:400).
Genetics
DNA samples from the affected mother and daughter were extracted from frozen brain tissue and from Epstein-Barr virus-transformed lymphoblasts by a salting out method using Gentra Systems (Minneapolis, MN) Puregene reagents and protocols. The PrP coding sequence was amplified as a single 851bp fragment using previously published primers (JS8-5′ CCCTCAAGCTGGAAAAAAGA and JS9-5′ ACTCTGACGTTCTCCTCTTCA7) and 100ng of genomic DNA in a 50μl reaction volume. Polymerase chain reaction products were subjected to electrophoresis using 2% NuSieve/0.1X TAE gels, and the appropriate fragments were purified using a Gene-Clean kit (Bio 101). The purified fragments were sequenced in both directions using the above amplification primers and an internal primer (JS10- 5′ GTGGCACCCACAGTCAGTGG), Big Dye fluorescent dye terminators, and an ABI 373 DNA Sequencer (Applied Biosystems, Foster City, CA). Tau exons 1, 9, 10, 11, 12, and 13 were sequenced from III-6, and no mutations were found.
Biochemistry
To enrich the fraction of proteinase K (PK)-resistant PrP scrapie isoform (rPrPSc), 1ml of a 20% weight/volume brain homogenate was prepared in 0.01M sodium phosphate, pH 7.4, 10% N-lauryl sarcosinate, 1mM phenylmethylsulfonyl fluoride, 1mM N-ethylmaleimide, incubated 30 minutes at room temperature, then centrifuged at 22,000 × g for 30 minutes at 10°C. The supernatant (S1) was then centrifuged at 215,000 × g for 150 minutes at 10°C, and the pellet (P2) was resuspended in 100μl 0.6M potassium iodide, 6mM sodium thiosulfate, 1% N-lauroyl sarcosinate, and 10mM sodium phosphate pH 8.5. This (S2) fraction was centrifuged at 215,000 × g for 90 minutes at 10°C through a cushion of 400μl of 20% sucrose (sucrose/sample; 1:4 volume/volume), and the pellet (P3) was resuspended in 100μl of phosphate-buffered saline. Samples were digested with PK (20μg/ml) for 1 hour at 37°C.
PrP fractions were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on 14% polyacrylamide gels, then transferred to polyvinylidene difluoride membranes that were probed with anti-PrP monoclonal 3F4 antibody (gift from R. Kascsak, Staten Island, NY) at 1:5,000 dilution, followed by goat-antimouse immunoglobulin G secondary antibody (1:5,000; Santa Cruz Biotechnology, Santa Cruz, CA) to detect human PrP. The chemiluminescent signal was detected on a Chemi Doc XRS imager (Bio-Rad, Laboratories, Hercules, CA), using SuperSignal West Pico substrate (Pierce Biotechnology, Rockford, IL).
Results
Case Histories
PROBAND
The proband began demonstrating signs of cognitive impairment at age 39 years and presented to our dementia clinic at the age of 42 years. She was a teacher’s assistant with 14 years of education. She was married and had 2 children. Her husband reported that her cognitive and behavioral problems began after an incident at school in which a disabled student charged her and hit her in the jaw. She suffered no loss of consciousness but developed chronic jaw pain, ultimately treated with surgical intervention. Soon after the traumatic incident, the husband began to note occasional episodes of short-term memory loss that became more consistent and severe over time. In addition, she became increasingly emotionally labile and reported significant problems with sleep, appetite, feelings of hopelessness, and lowered energy levels. By the time of her presentation to a dementia clinic at age 42 years, she had been diagnosed with major depression and started on an antidepressant. Despite this treatment, she continued to exhibit significant memory loss, with associated loss of function at home and work. Family history was remarkable for a mother who died at age 67 years with autopsy-confirmed AD after an 8-year course of dementia. The maternal grandmother died at age 84 years of cancer and heart failure, and the maternal grandfather died of a stroke at age 60 years. Both reportedly had memory loss of unknown severity. There were 2 maternal uncles; 1 died at age 71 years from a myocardial infarction, but no medical information was available for the other maternal uncle. Laboratory testing, electroencephalography (EEG), and magnetic resonance imaging were unremarkable.
On examination, the proband was anxious and preoccupied with the traumatic incident at school and the resultant chronic jaw pain. Bedside neuropsychological testing revealed a Short Blessed score of 24 (missing month, time of day, 1 number on counting backwards, 2 on months backward, recalling 0/5 items).8 Neurological examination was unremarkable, with normal cranial nerve, motor, sensory, and gait evaluation. There was no evidence of myoclonus or ataxia.
Neuropsychological testing (Table) was notable for impairment on Mattis initiation and perseveration, conceptualization, and memory, with relative preservation of attention and construction.
TABLE.
Clinical and Neuropsychological Characteristics
| Characteristic | Proband | Mother |
|---|---|---|
| Age of onset, yr | 39 | 59 |
| Age at death, yr | 47 | 67 |
| Disease duration, yr | 8 | 8 |
| Age at neuropsychological testing, yr | 42 | 63 |
| MMSE | 14/30 | 15/30 |
| Mattis | ||
| Attention | 35/37 | 36/37 |
| Initiation and perseveration | 24/37 | 32/37 |
| Construction | 6/6 | 6/6 |
| Conceptualization | 30/39 | 38/39 |
| Memory | 9/25 | 10/25 |
| Total | 104/144 | 122/144 |
MMSE = Mini Mental State Examination.
Over the ensuing 5 years, the proband developed significant language impairment, mild parkinsonism, and loss of activities of daily living. Three years before her death, her neurological exam was significant for symmetrically brisk reflexes, and an essential-like tremor that was more prominent on the right.
She died at the age of 47 years, after an 8-year course.
PROBAND’S MOTHER
Ten years before the proband was evaluated in our clinic, her mother presented to our dementia center at the age of 63 years. She was reported then to have developed cognitive problems at the age of 59 years, with personality change and significant memory loss (“forgetful,” including misplacing objects and missing appointments). She also began to have difficulties with performance at her retail job. During this initial period, she also complained of depression and had a short trial of antidepressant therapy. At the time of her initial visit, she denied depressed mood, but noted some symptoms of depression (distress over her deficits, crying spells, and sleep disturbance). One year prior to the onset of symptoms, she had a modest head injury that required sutures, but was without loss of consciousness. Her only other medical history included weight loss and intermittent diarrhea. The postprandial diarrhea began approximately 2 years after onset of cognitive and behavioral changes and continued until her death. The chronic diarrhea was associated with substantial weight loss of 60 pounds and 1 hospitalization for dehydration. Laboratory evaluation, computed tomography of the head, and EEG were unremarkable.
On examination, she was alert and cooperative. Neuropsychological testing revealed impairments on Mattis initiation and preservation and memory, with relative preservation of attention, construction, and conceptualization (see Table). Neurologic examination was unremarkable with normal cranial nerve, motor, sensory, and gait evaluation. There was no myoclonus or ataxia.
Over the next 4 years she showed progressive decline in cognitive function, including the development of significant language impairment and loss of activities of daily living. She continued to have chronic diarrhea throughout her course, leading to severe weight loss.
She died at the age of 67 years after an 8-year clinical course.
Pathology
PROBAND
At autopsy the brain weight for the proband was 980g, with mild to moderate frontotemporal atrophy. No gross focal lesions were observed. Microscopic examination with H&E and Bielschowsky silver stains revealed severe NFTs and neuritic plaque-like pathologic changes in the hippocampus and neocortex, consistent with a Braak stage of VI and Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) plaque “frequent” score (Fig 1).9,10 However, an absence of Bielschowsky staining in the core of neuritic plaques (see Fig 1A, arrow), as would be expected in AD, led to immunostaining with antibodies to Aβ and PrP. The plaques were not immunopositive for Aβ peptide. However, extensive PrP immunopositive deposits were observed in the gray matter of the neocortex and limbic system, whereas the deposits in the cerebellum were more sparse (Fig 2A, B). The PrP deposits were predominantly in the plaque-like structures and vessels; there was no synaptic or perineuronal involvement. The prion deposits were also immunopositive using a PrP antibody to amino acids 90 to 102, but not to 220 to 231 (see Fig 2C, D). Histological staining with thioflavin S showed robust staining of NFTs; however, cortical and hippocampal PrP deposits were difficult to observe. In the cerebellum, there were no NFTs or tau immunopositive neurites, but light thioflavin S labeling of PrP deposits was observed. PrP immunopositive amyloid angiopathy was also observed (see Fig 2E), although not as severe as previously reported in another PRNP truncation mutation.4 Neocortical and hippocampal NFTs and neuritic plaquelike structures were immunopositive with all tau antibodies (AT8, PHF-1, Tau-2, RD3, RD4). No glial tau pathology was observed. Electron microscopy revealed paired helical filaments and prion deposits (Fig 3, 4).
FIGURE 1.
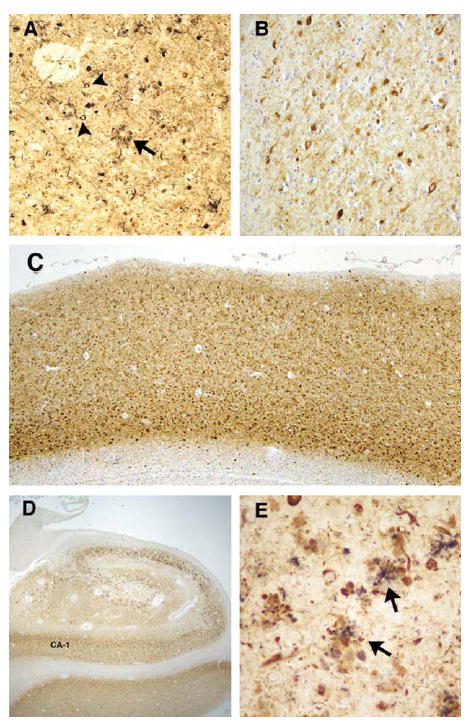
Silver stain and tau pathology in proband (A). Bielschowsky silver stained sections from the proband demonstrate significant neurofibrillary tangle (arrowheads) and neuritic plaque (arrow) formation in the frontal cortex. Note the coreless plaque (arrow). Tau immunohistochemistry revealed severe immunopositive staining of neurofibrillary pathology in the frontal cortex (B and C, PHF1 antibody) and hippocampus (D, PHF-1 antibody). (E) Double immunolabeling of plaquelike structures in the hippocampus with prion protein (3F4, purple) and tau (PHF-1, brown) antibodies revealed a close anatomic relationship between these pathologic changes (arrows). Original magnification, A and B, ×63; C, ×13; D, ×7; E, ×126.
FIGURE 2.
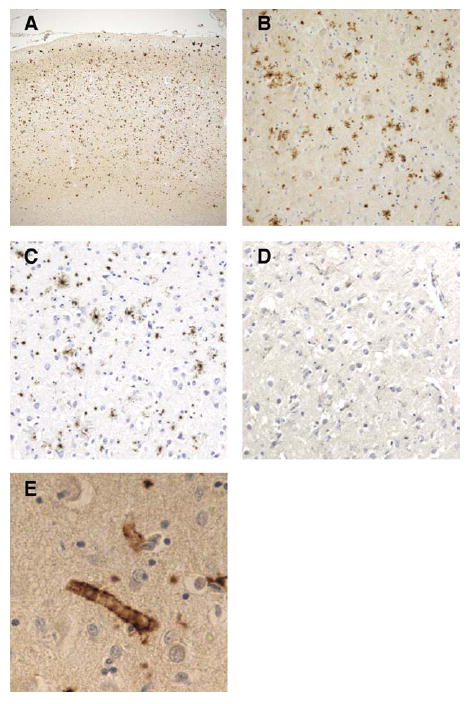
Prion protein (PrP) immunohistochemistry in frontal cortex of the proband. (A, B) PrP immunohistochemistry demonstrates widespread PrP deposition in frontal cortex. (C) Antibody specific to the amino acids 90 to 102 of PrP demonstrates immunopositive PrP deposits; (D) however, this was not observed with an antibody to amino acids 220 to 231 of the PrP. (E) PrP immunopositive angiopathy was also observed. Original magnification, A, ×13; B, C, and D, ×32; E, ×126.
FIGURE 3.
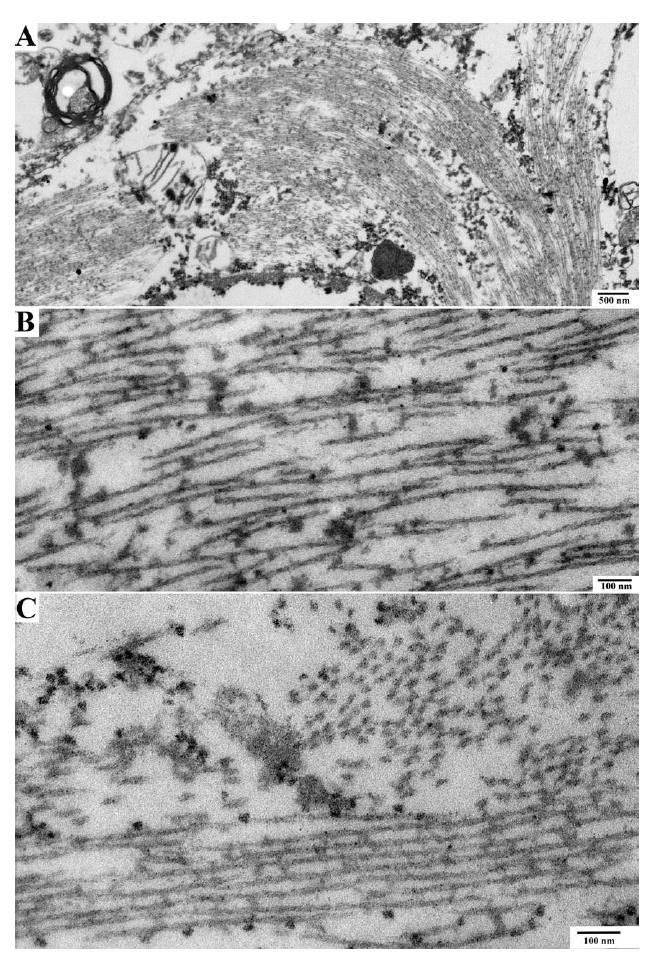
Electron microscopy of neurofibrillary tangles in proband. Electron microscopic pictures of neuronal cytoplasm show the presence of neurofibrillary tangles. (A) Cytoplasm of a nerve cell containing a neurofibrillary tangle. (B, C) High-power image of neurofibrillary tangle shows paired helical filaments seen longitudinally (lower portion of the image) and in cross-section (upper right portion of image).
FIGURE 4.
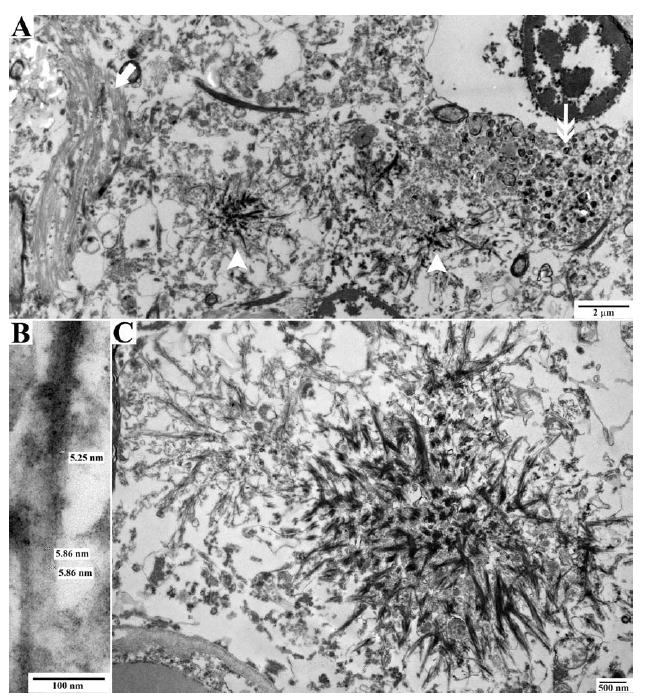
Electron microscopy of prion protein (PrP) deposition in proband. Electron microscopic pictures of neuropathologic lesions in the neuropil. (A) The neuropil shows a neurofibrillary tangle on the left (arrow), PrP amyloid deposits in the center (arrowheads), and a dystrophic neurite on the right (double arrow). (B) High-power image of filaments from a PrP amyloid plaque. (C) Starlike PrP amyloid plaques.
Neuritic plaque-like pathology in the limbic system and neocortex showed distended processes that were immunoreactive with tau and neurofilament antibodies. Double immunolabeling with tau (PHF-1) and PrP antibodies (3F4) revealed interdigitation of these distended tau and neurofilament immunopositive processes with the PrP deposits (see Fig 1E).
Alpha-synuclein immunohistochemistry revealed frequent neuronal inclusions and Lewy neurites in the amygdala and rare inclusions and neurites in the hippocampus and in the parahippocampal, cingulate, and superior frontal gyri. No inclusions or neurites were observed in the substantia nigra or medulla using immunohistochemistry for alpha-synuclein or with H&E.
Immunostaining for Aβ and TDP-43 was negative. Spongiform change was not observed, presumably due to the prolonged clinical course and associated slow rate of neuronal loss (relative to sporadic spongiform disease).
PROBAND’S MOTHER
At autopsy, the brain weight for the mother of the proband (II-2) was 1,435g, without significant gross lesions. Microscopic examination from the original neuropathologic evaluation utilized H&E, Holzer, and Holmes stains. The Holmes stain detected severe NFTs and neuritic plaque-like pathology in the neocortex and limbic system consistent with a Braak stage VI for NFTs and CERAD plaque stage “frequent” (Fig 5A and B).9,10 Amyloid angiopathy was mild in the gray matter of the neocortex and limbic system, whereas moderate to severe amyloid angiopathy was also observed in the occipital lobe and cerebellum. The predominant location of the amyloid angiopathy was gray matter capillaries and less frequently arterioles, whereas white matter and leptomeningeal vessels were only occasionally affected.
FIGURE 5.
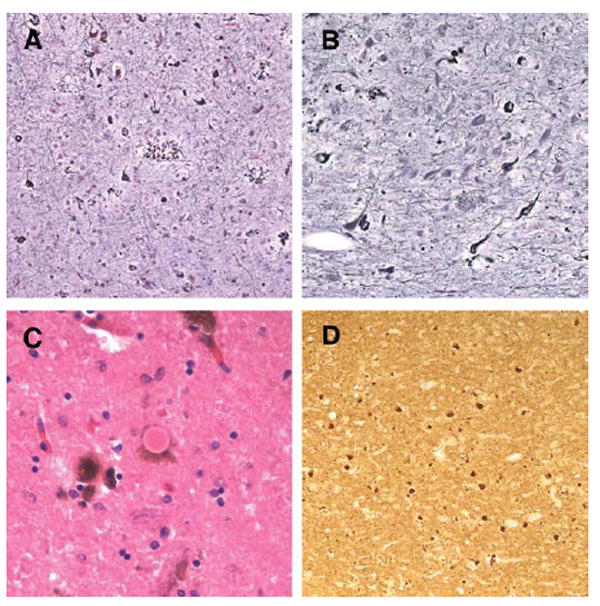
Histologic pathology in the proband’s mother. Holmes silver staining revealed severe neurofibrillary tangles and neuritic plaque pathology in the mother’s frontal cortex (A) and hippocampus (B). In addition, both classic Lewy bodies (C) and alpha-synuclein immunopositive inclusions and neurites (D) were observed. Original magnification, A and B, ×63; C, ×126; D, ×32.
Thirteen years after II-2’s original autopsy was performed, the diagnosis of prion disease was made in her daughter, and thus further diagnostic testing was performed to evaluate for PrP pathologic change. Immunohistochemistry revealed a remarkably similar staining pattern to the proband using antibodies to PrP and tau. Alpha-synuclein immunostaining revealed neuronal inclusions and Lewy neurites in the medulla, substantia nigra, amygdala, hippocampus, cingulate gyrus, and frontal cortex (see Fig 5D), consistent with neocortical predominant Lewy-related pathology.11 In addition, classic Lewy bodies were observed in the substantia nigra with H&E staining (see Fig 5C). Unlike the proband, Aβ immunostaining did detect modest diffuse plaque labeling in the temporal neocortex. TDP-43 immunostaining was negative.
The general autopsy showed no evidence of gastrointestinal pathology.
GENETICS
Two heterozygous variant sites were identified within the PrP coding sequence from the affected subjects. A silent A to G transition at nucleotide 400 of PRNP was identified in the proband (III-6) (based on numbering from Genbank accession number M13899); the mother (II-2) was homozygous A/A at this site. The second variant, a C to T transition at nucleotide 527, leading to premature truncation of the PrP protein (Q160Stop or Q160X), was identified in both patients (Fig 6). Our proband was found to be Met/Val at codon 129, whereas her mother was Met/Met. Apolipoprotein E genotyping in the proband revealed APOE E3/3.
FIGURE 6.
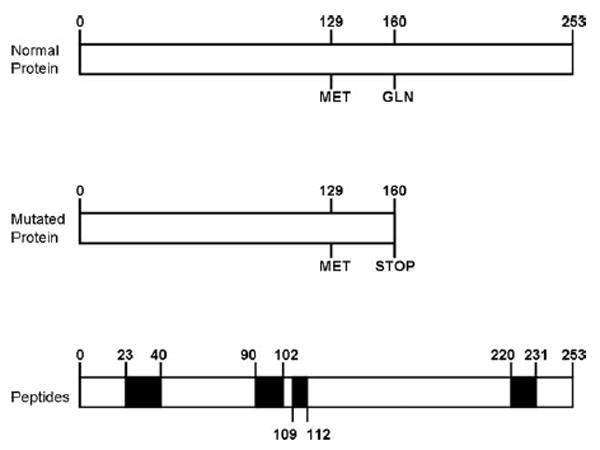
Schematic representation of normal prion protein and truncated prion protein in this family. The scheme of the prion protein highlights the regions that recognized the antibodies used in the present study.
BIOCHEMISTRY
The presence of PK-resistant PrP within a sample of frontal lobe from the proband was compared with that from a case of sporadic CJD (129VV2) by Western blot of the PrPSc-enriched fraction (see Subjects and Methods and Fig 7). Prior to PK digestion, in addition to full-length PrP of ~25-37kD, representing unglycosylated, monoglycosylated, and diglycosylated PrP, an additional smear of PrP ranging from ~11 to 18kDa was present, likely representing endogenously cleaved fragments of truncated PrP-Q160X. In contrast, only higher molecular weight PrP (~25–35kDa), in addition to a cleavage product of ~19kDa, was present in the sporadic CJD (129VV2). PK digestion typically cleaves PrPSc at residue ~90, leaving PrP 90-230 as the PK-resistant core. Thus, in CJD, 3 amino-truncated products are detected. In the Q160X case, however, a primary fragment of ~11kD was detected, in addition to a smear of higher molecular weight fragments, ranging from 13 to 18kDa and from ~21 to 35kDa. It is likely that the smaller range of fragments results from the mutated PrP that may be cleaved at 1 or more amino-terminal sites, whereas the higher molecular weight fragments might represent PrP-Q160X oligomers or full-length PrP that is converted to pathogenic PrP by mutated PrP. Further analysis and transmission studies may be necessary to confirm this possibility.
FIGURE 7.
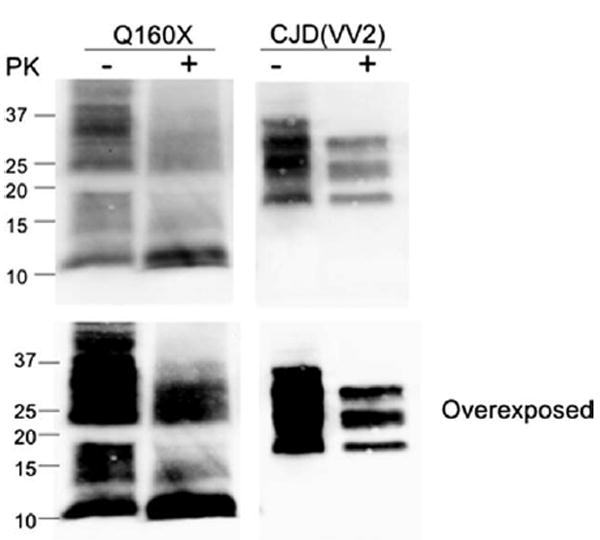
Western blot from proband brain tissue. Western blot analysis of enriched fractions of prion protein scrapie isoform (PrPSc) from brain samples of the PRNP-Q160X patient and a sporadic Creutzfeldt-Jakob disease (sCJD) subject with the 129VV genotype and type 2 proteinase K (PK)-resistant PrPSc (unglycosylated fragment ~19kDa) is shown. This enrichment reveals 2 major fractions of PrP that correspond to residual nonmutated (25–35kDa) PrP and full-length and cleaved (smear between 11 and 18kDa) mutated PrP. The predicted size of mature PrP-Q160X is ~17–18kDa, represented by the top of the second major fraction, with smaller fragments likely representing endogenously cleaved products of the PrP-Q160X mutated protein, the most prominent of which is ~11kDa. None of these smaller fractions is present in sCJD, even with overexposure, suggesting that they result directly from mutated PrP. Following PK treatment (+) the ~11kDa fragment remains most prominent. The smear of PK-resistant PrP that ranges from ~21 to 35kDa may represent PrP-Q160X oligomers or nonmutated PrP converted to PK-resistant fragments by PrP-Q160X. Blots were probed with antihuman PrP monoclonal antibody 3F4.
Discussion
We report a mother and daughter pair found to carry a nonsense mutation in PRNP (Q160X) leading to a truncated PrP. The clinical and initial pathological features in both patients were strongly suggestive of AD. Myoclonus and ataxia were not present in either the mother or daughter. The family’s first autopsy, in the mother, was remarkable for severe NFT and neuritic plaque formation in the limbic system and neocortex, using a Holmes silver stain, leading to a pathological diagnosis of AD. Not until coreless plaques were observed with Bielschowsky silver staining in the daughter, and specific Aβ and prion immunohistochemical labeling became available, was the diagnosis of prion disease established.
This specific mutation was previously reported in a study of mutations associated with early onset dementia.3 The 2 affected individuals were brothers who presented with early onset dementia at ages 32 and 48 years, both carrying Q160X in cis with M129. Their father was also reported to have died at age 60 years after a 12-year course of dementia. Of note, the wild-type allele for the younger onset brother was M129, whereas that of the later onset brother was V129. The authors suggested that this difference in the wild-type allele might have accounted for the difference in age of onset. Arguing against this hypothesis, the proband in our report had an age of onset that was 20 years earlier than her mother, but she carried the V129 allele, whereas her mother was M129 homozygous. Similar to the cases in the current report, other than dementia, neither brother demonstrated symptoms or signs of ataxia or other typical neurological sequelae of prion disease. Unfortunately, only the father underwent autopsy, and only the gross pathology was reported. To our knowledge, the current report is the first to describe the detailed neuropsychologic, neuropathologic, and biochemical characteristics associated with this rare PrP mutation.
Of particular relevance to the current report are the other reported cases of nonsense mutations in PRNP (Y145X, Y163X, Y226X, Q227X).4,12,13 Four of these cases had neuropathological assessment in addition to clinical history. Three had severe NFT formation and relatively long (>72 months) clinical disease duration. One case had short disease duration, 27 months, and very limited tau pathology at autopsy (Q227X).13 Ghetti et al4 originally commented on the severe cerebral amyloidosis in one of these nonsense mutation cases (Y145X). Others have also noted similar levels of cerebral amyloidosis in their nonsense mutation cases (Y163X, Y226X); however, our cases and one other (Q227X) had less severe or no vascular amyloidosis. The clinical presentation in our reported cases was consistent with a diagnosis of AD. One reported case (Y145X) also carried a clinical diagnosis of AD; however, the other reported nonsense mutation cases generally had early clinical characteristics (eg, hallucinations, extrapyramidal signs) that might suggest another diagnosis. The remarkable severity of the AD-like NFT pathology in most of these PRNP truncation mutation cases is intriguing and suggests that there may be an important association between these PrP truncation-associated mutations and the AD-like pathological phenotype. Unfortunately, the underlying pathophysiologic link for this association is not clear at this point.
NFT have certainly been described in prion diseases with morphologic and biochemical characteristics that are identical to those observed in AD.14,15 However, the strikingly high abundance of NFT observed in the current cases, and other nonsense mutation cases,4,12,13 distinguishes them from most, but not all, reported PRNP mutation cases. The few GSS cases associated with relatively severe NFT, specifically the mutations F198S and Q217R,14,16,17 are particularly interesting pathologically, as they also demonstrated coexistent neuritic plaques and Lewy body pathology. Our cases, and one of these GSS familial cases, also showed similar interdigitation of PrP deposits and neuritic plaque-like pathology,14 and Lewy body and alpha-synuclein pathology have been demonstrated previously in both sporadic and familial prion diseases.16,18-20 The majority of these cases also reported coexistent NFTs, suggesting a possible link between the presence of AD-like pathologic change and Lewy body pathology in certain prion diseases. Unfortunately, to date no systematic investigation of Lewy body pathology in all forms of prion disease has been performed, so this association is speculative.
Immunohistochemistry suggested that only the truncated PrP was present in the PrP deposits of both of our reported Q160X cases. The allelic origin of PrP found in amyloid plaques has been studied in GSS, CJD, and FFI mutations. Similar to this truncation mutation, amyloid formation in the Indiana (F198S), Swedish (Q217R), and A117V point mutations are also solely comprised of mutant PrP.21,22 Likewise, only the mutant PrP was shown to be detergent insoluble and protease resistant in cases of D178N linked to FFI and CJD.23 Interestingly, prion deposits in the patient carrying the Y145X truncation had PrP deposition that was immunopositive with both N-and C-terminus antibodies (PrP 220-231), indicating recruitment of the full-length protein.4
In conclusion, we report the clinical, neuropathologic, and biochemical characteristics of a mother and daughter with a rare PRNP mutation that results in the production of a truncated PrP. The AD-like clinical picture in both the proband and her mother is likely secondary to distribution and severity of the AD-like NFT pathology. The similar NFT pathology in another truncation mutation of PRNP, Y145X, suggests a link between the PrP truncation-associated mutations and some AD clinical and pathological characteristics. These cases also re-emphasize the remarkable clinical and pathological variability that can be observed in the prion diseases.
Acknowledgments
This work supported in part by the US Department of Veterans Affairs, Office of Research and Development Clinical R&D Program, and National Institutes of Health (NIH)/National Institute on Aging (NIA) 2P50AG005136-27, 5P50NS062684-02, 5R01NS051480-05, 5P30AG010133-20. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIA or the NIH.
We thank L. Greenup, C. Ulness, R. Small, C. Alyea, B. Dupree, and R. Richardson for their technical assistance and T. Sheehan and B. Glazier for assistance with the preparation of the manuscript.
Footnotes
Potential Conflicts of Interest T.D.B. has received speaking fees and royalties from Athena Diagnostics.
References
- 1.Ironside JW, Ghetti B, Head MW. Prions diseases. In: Love S, Louis DN, Ellison DW, editors. Greenfield’s neuropathology. London, UK: Hodder Arnold; 2008. pp. 1197–1274. [Google Scholar]
- 2.Kong Q, Suresicz WK, Petersen RB, et al. Inherited prion diseases. In: Prusiner SB, editor. Prion biology and diseases. 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2004. pp. 673–775. [Google Scholar]
- 3.Finckh U, Muller-Thomsen T, Mann U, et al. High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet. 2000;66:110–117. doi: 10.1086/302702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghetti B, Piccardo P, Spillantini MG, et al. Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP. Proc Natl Acad Sci U S A. 1996;93:744–748. doi: 10.1073/pnas.93.2.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piccardo P, Ghetti B, Dickson DW, et al. Gerstmann-Straussler-Scheinker disease (PRNP P102L): amyloid deposits are best recognized by antibodies directed to epitopes in PrP region 90-165. J Neuropathol Exp Neurol. 1995;54:790–801. doi: 10.1097/00005072-199511000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Kitamoto T, Ogomori K, Tateishi J, Prusiner SB. Formic acid pre-treatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest. 1987;57:230–236. [PubMed] [Google Scholar]
- 7.Collinge J, Harding AE, Owen F, et al. Diagnosis of Gerstmann-Straussler syndrome in familial dementia with prion protein gene analysis. Lancet. 1989;2:15–17. doi: 10.1016/s0140-6736(89)90256-0. [DOI] [PubMed] [Google Scholar]
- 8.Katzman R, Brown T, Fuld P, Peck A, Schechter R, Schimmel H. Validation of a short Orientation-Memory-Concentration Test of cognitive impairment. Am J Psychiatry. 1983;140:734–739. doi: 10.1176/ajp.140.6.734. [DOI] [PubMed] [Google Scholar]
- 9.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 10.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 11.Leverenz JB, Hamilton R, Tsuang DW, et al. Empiric refinement of the pathologic assessment of Lewy-related pathology in the dementia patient. Brain Pathol. 2008;18:220–224. doi: 10.1111/j.1750-3639.2007.00117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jansen C, Parchi P, Capellari S, et al. Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP. Acta Neuropathol. 2010;119:189–197. doi: 10.1007/s00401-009-0609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Revesz T, Holton JL, Lashley T, et al. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 2009;118:115–130. doi: 10.1007/s00401-009-0501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghetti B, Tagliavini F, Masters CL, et al. Gerstmann-Straussler-Scheinker disease. II. Neurofibrillary tangles and plaques with PrP-amyloid coexist in an affected family. Neurology. 1989;39:1453–1461. doi: 10.1212/wnl.39.11.1453. [DOI] [PubMed] [Google Scholar]
- 15.Tagliavini F, Giaccone G, Prelli F, et al. A68 is a component of paired helical filaments of Gerstmann-Straussler-Scheinker disease, Indiana kindred. Brain Res. 1993;616:325–329. doi: 10.1016/0006-8993(93)90226-d. [DOI] [PubMed] [Google Scholar]
- 16.Ghetti B, Tagliavini F, Giaccone G, et al. Familial Gerstmann-Straussler-Scheinker disease with neurofibrillary tangles. Mol Neurobiol. 1994;8:41–48. doi: 10.1007/BF02778006. [DOI] [PubMed] [Google Scholar]
- 17.Ghetti B, Bugiani O, Tagliavini F, Piccardo P. Gerstmann-Straussler-Scheinker disease. In: Dickson DW, editor. Neurodegeneration: the molecular pathology of dementia and movement disorders. Basel, Switzerland: ISN Neuropath Press; 2003. pp. 318–325. [Google Scholar]
- 18.Ghetti B, Gambetti P. Human prion disease. In: Mattson MP, editor. Advances in cell aging and gerontology volume 3: genetic aberrancies and neurodegenerative disorders. Greenwich, CT: Jai Press Inc; 1999. [Google Scholar]
- 19.Haraguchi T, Terada S, Ishizu H, et al. Coexistence of Creutzfeldt-Jakob disease, Lewy body disease, and Alzheimer’s disease pathology: an autopsy case showing typical clinical features of Creutzfeldt-Jakob disease. Neuropathology. 2009;29:454–459. doi: 10.1111/j.1440-1789.2008.00964.x. [DOI] [PubMed] [Google Scholar]
- 20.Vital A, Canron MH, Gil R, Hauw JJ, Vital C. A sporadic case of Creutzfeldt-Jakob disease with beta-amyloid deposits and alpha-synuclein inclusions. Neuropathology. 2007;27:273–277. doi: 10.1111/j.1440-1789.2007.00755.x. [DOI] [PubMed] [Google Scholar]
- 21.Tagliavini F, Lievens PM, Tranchant C, et al. A 7-kDa prion protein (PrP) fragment, an integral component of the PrP region required for infectivity, is the major amyloid protein in Gerstmann-Straussler-Scheinker disease A117V. J Biol Chem. 2001;276:6009–6015. doi: 10.1074/jbc.M007062200. [DOI] [PubMed] [Google Scholar]
- 22.Tagliavini F, Prelli F, Porro M, et al. Amyloid fibrils in Gerstmann-Straussler-Scheinker disease (Indiana and Swedish kindreds) express only PrP peptides encoded by the mutant allele. Cell. 1994;79:695–703. doi: 10.1016/0092-8674(94)90554-1. [DOI] [PubMed] [Google Scholar]
- 23.Chen SG, Parchi P, Brown P, et al. Allelic origin of the abnormal prion protein isoform in familial prion diseases. Nat Med. 1997;3:1009–1015. doi: 10.1038/nm0997-1009. [DOI] [PubMed] [Google Scholar]