

Abstract
BACKGROUND & AIMS
Nonalcoholic steatohepatitis (NASH) is a disorder that consists of steatosis and hepatic inflammation. It is not known why only some people with steatosis develop NASH. Recently, we identified dietary cholesterol as a factor that directly leads to hepatic inflammation and hepatic foam cell formation. We propose a mechanism by which Kupffer cells (KCs) take up modified cholesterol-rich lipoproteins via scavenger receptors (SRs). KCs thereby accumulate cholesterol, become activated, and may then trigger an inflammatory reaction. Scavenging of modified lipoproteins mainly depends on CD36 and macrophage scavenger receptor 1.
METHODS
To evaluate the involvement of SR-mediated uptake of modified lipoproteins by KCs in the development of diet-induced NASH, female low-density lipoprotein receptor-deficient (Ldlr−/−) mice were lethally irradiated and transplanted with bone marrow from Msr1+/+/Cd36+/+ or Msr1−/−/Cd36−/− mice and fed a Western diet.
RESULTS
Macrophage and neutrophil infiltration revealed that hepatic inflammation was substantially reduced by approximately 30% in Msr1−/−/Cd36−/−-transplanted mice compared with control mice. Consistent with this, the expression levels of well-known inflammatory mediators were reduced. Apoptotis and fibro-sis were less pronounced in Msr1−/−/Cd36−/−-transplanted mice, in addition to the protective phenotype of natural antibodies against oxidized low-density lipoprotein in the plasma. Surprisingly, the effect on hepatic inflammation was independent of foam cell formation.
CONCLUSIONS
Targeted inactivation of SR pathways reduces the hepatic inflammation and tissue destruction associated with NASH, independent of hepatic foam cell formation.
Keywords: Fatty Liver, Inflammation, Scavenger Receptors, Kupffer Cells
Nonalcoholic steatohepatitis (NASH) is considered to be the hepatic event of the metabolic syndrome. It is characterized by hepatic lipid accumulation (steatosis) combined with inflammation. Estimates of the general United States population indicate that approximately 2%–3% of all adults have NASH and that this prevalence is expected to rise rapidly because of the increasing prevalence of obesity.1 The current view on the pathogenesis of diet-induced liver inflammation is that hepatic steatosis is a critical prerequisite for the development of inflammation. Whereas steatosis itself is generally considered benign and reversible, the presence of inflammation can lead to further progression of NASH, resulting in liver fibrosis, cirrhosis, and, eventually, liver failure and hepatocellular carcinoma.2 Thus, the progression toward hepatic inflammation represents a key step in NASH development. It is not yet known why only a small percentage of people with steatosis develop NASH. Although several processes have been identified that participate in the development of hepatic inflammation,3–7 the actual trigger for the inflammatory response remains uncertain.
We and other researchers have recently shown that dietary cholesterol is an important risk factor for the development of hepatic inflammation.8 –10 Interestingly, hyperlipidemic mice fed a high-fat, high-cholesterol (HFC) diet develop early hepatic inflammation, which is associated with bloated Kupffer cells (KC), resembling foam cells in atherosclerosis. These KCs with a foamy appearance have also been identified in other studies in the sinusoidal space of the liver.11,12 Strikingly, omitting cholesterol from the diet in these mice resulted in a dramatic inhibition of hepatic inflammation and foam cell formation.8 An attractive hypothesis is that, because of the decreased lipoprotein uptake by hepatocytes in hyperlipidemic mice, lipoproteins have a longer residence time in the plasma and are therefore more prone to be modified and subsequently scavenged by KCs. This process ultimately results in KC activation, which may be responsible for triggering the hepatic inflammation and KC cholesterol accumulation.
Like typical macrophages, KCs express scavenger receptors (SRs)13 and are thus capable of taking up modified lipoproteins. In vitro, foam cell formation has been shown to depend mainly on 2 different SRs: SR-A (macrophage scavenger receptor 1 [MSR1]) and CD36.14 MSR1 was found to account for the majority (80%) of macrophage uptake of acetylated low-density lipoprotein (LDL) (acLDL) but has a lower affinity for oxidized LDL (oxLDL).14,15 CD36 binds moderately oxLDL, rather than acLDL,16 and was found necessary for oxLDL-induced c-Jun-N-terminal kinase activation.17 Despite numerous studies addressing the role of these 2 SRs in atherosclerosis, their roles remain uncertain. Whereas clear effects of MSR1 and CD36 on cholesterol uptake and foam cell formation have been found in vitro,14 the results in vivo are less consistent. Findings in mouse models of atherosclerosis lacking one or both of these SRs have ranged from reduced foam cell formation and atherosclerosis to no effect or to greater foam cell formation and increased atherosclerosis.16,18,19 Whereas the reasons for these discrepancies are unclear, there is a growing appreciation of the numerous functions that SRs can play in addition to lipoprotein uptake, including their roles in inflammatory signal transduction and tissue homeostasis.
To investigate the contribution of Msr1 and Cd36 expression by macrophages on hepatic modified lipoprotein uptake and inflammation, we performed a bone marrow transplantation of donor bone marrow from mice with targeted deletions for Msr1 and Cd36 into lethally irradiated low-density lipoprotein receptor-deficient (Ldlr−/−) recipient mice. We hypothesized that Msr1−/−/Cd36−/−-transplanted (-tp) mice on a high-fat diet would develop reduced levels of hepatic inflammation compared with mice with normal hematopoietic cells because of a decreased uptake of modified lipoproteins by KCs. Surprisingly, whereas deletion of Msr1 and Cd36 in hematopoietic cells failed to block cholesterol accumulation in KCs, these transplanted mice showed reduced markers for NASH, including less inflammation, apoptosis, and fibrosis after high-fat feeding. These data suggest that SR expression by KCs in the liver increases the risk for NASH in the presence of high levels of plasma-modified lipoproteins.
Materials and Methods
Experimental Setup
Female Ldlr−/− mice were lethally irradiated and transplanted with Msr+/+/Cd36+/+ and Msr1−/−/Cd36−/− bone marrow. After a recovery period of 9 weeks, the mice were given an HFC diet for 7 days (n = 8 in both Msr+/+/Cd36+/+-tp and Msr1−/−/Cd36−/−-tp mice) and 3 months (n = 7 in Msr+/+/Cd36+/+-tp group, n = 8 in Msr−/−/Cd36−/−-tp group). Chow fed mice, killed after 9 weeks’ recovery, were used as the control group (n = 7 in both Msr+/+/Cd36+/+-tp and Msr1−/−/Cd36−/−-tp mice).
Collection of blood, specimens, lipid analysis in plasma and liver, liver histology, RNA isolation, complementary DNA (cDNA) synthesis and qualitative polymerase chain reaction, determination of chimerism, aminotransferases, oxysterols, and autoantibody titers against modified LDL are described extensively in Supplementary Materials and Methods.
Statistical Analysis
Data were statistically analyzed by performing 2-tailed nonpaired t tests for comparing Msr+/+/Cd36+/+-tp and Msr1−/−/Cd36−/−-tp mice for each diet group. One-way analysis of variance test was used for comparing the different time points of high-fat feeding within the same acceptor mice. Data were expressed as the mean ± standard error of mean and considered significant at *P < .05 (*P < .05, **P < .01, and ***P < .001, respectively).
Results
Plasma and Liver Lipid Levels in Msr1+/+/Cd36+/+ -and Msr1−/−/Cd36−/− Transplanted Mice
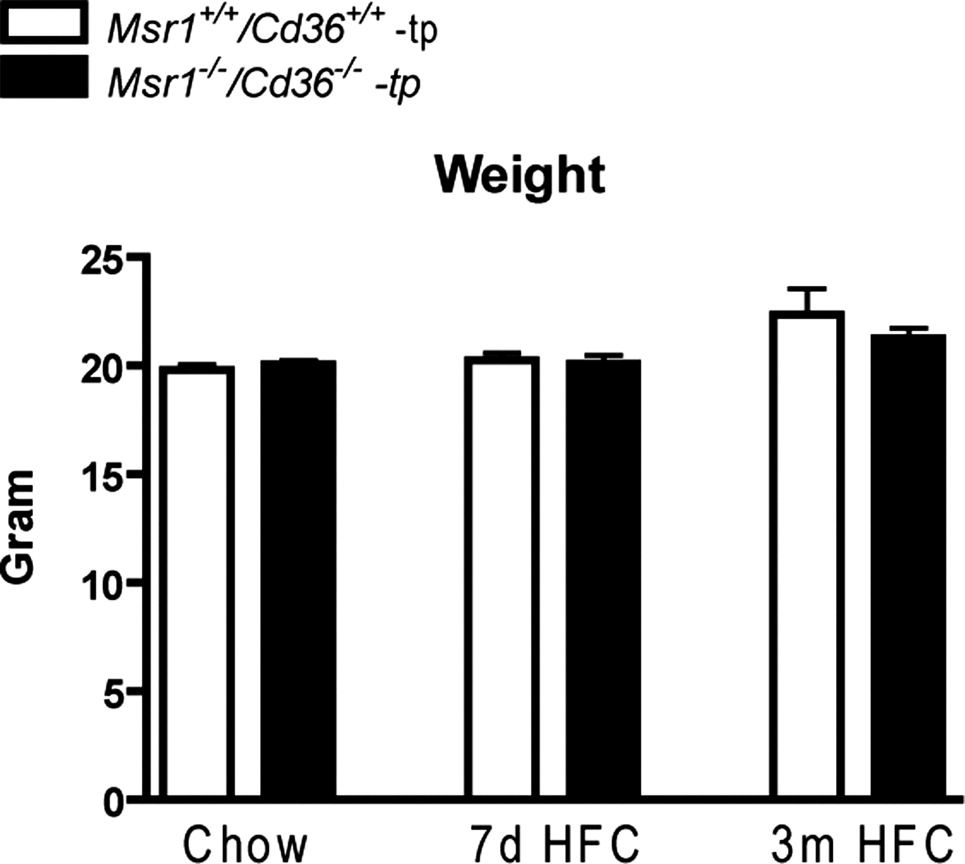
To establish the role of Msr1 and Cd36 on diet-induced NASH, Ldlr−/− mice were transplanted with Msr1+/+/Cd36+/+ and Msr1−/−/Cd36−/− bone marrow. Chimerism determination revealed an engraftment efficiency of 99%. After a recovery period of 9 weeks, mice received an HFC diet for either 7 days or 3 months. Chow fed mice that were killed after 9 weeks recovery served as control groups. The body weights were not significantly different between all the groups (Supplementary Figure 1). As expected, HFC feeding increased plasma triglycerides (TG) and total cholesterol (TC) levels in a time-dependent manner in both transplanted groups after periods of 7 days and 3 months (Figure 1A and B) (TG: P = .0003 for Msr1+/+/Cd36+/+-tp mice and P = .0007 for Msr1−/−/Cd36−/−-tp mice; TC: P < .0001 for both Msr1+/+/Cd36+/+-tp and Msr1−/−/Cd36−/−-tp mice). Surprisingly, Msr1−/−/Cd36−/−-tp mice displayed lower TG levels after 3 months of an HFC diet than Msr1+/+/Cd36+/+-tp mice (P = .009). A similar trend was observed for plasma cholesterol after 3 months of HFC diet; however, this did not reach statistical significance (P = .08). No effect on free fatty acid (FFA) levels was detected (Figure 1C).
Figure 1.

Plasma and liver lipid levels. (A–C) Plasma total triglycerides (TG), total cholesterol (TC), and free fatty acids (FFA) after chow, 7 days, and 3 months on an HFC diet. (D–F) Liver TG, TC, and FFA after chow and 7 days and 3 months on an HFC diet. *Significantly different from the age-matched Msr1+/+/Cd36+/+-tp group.
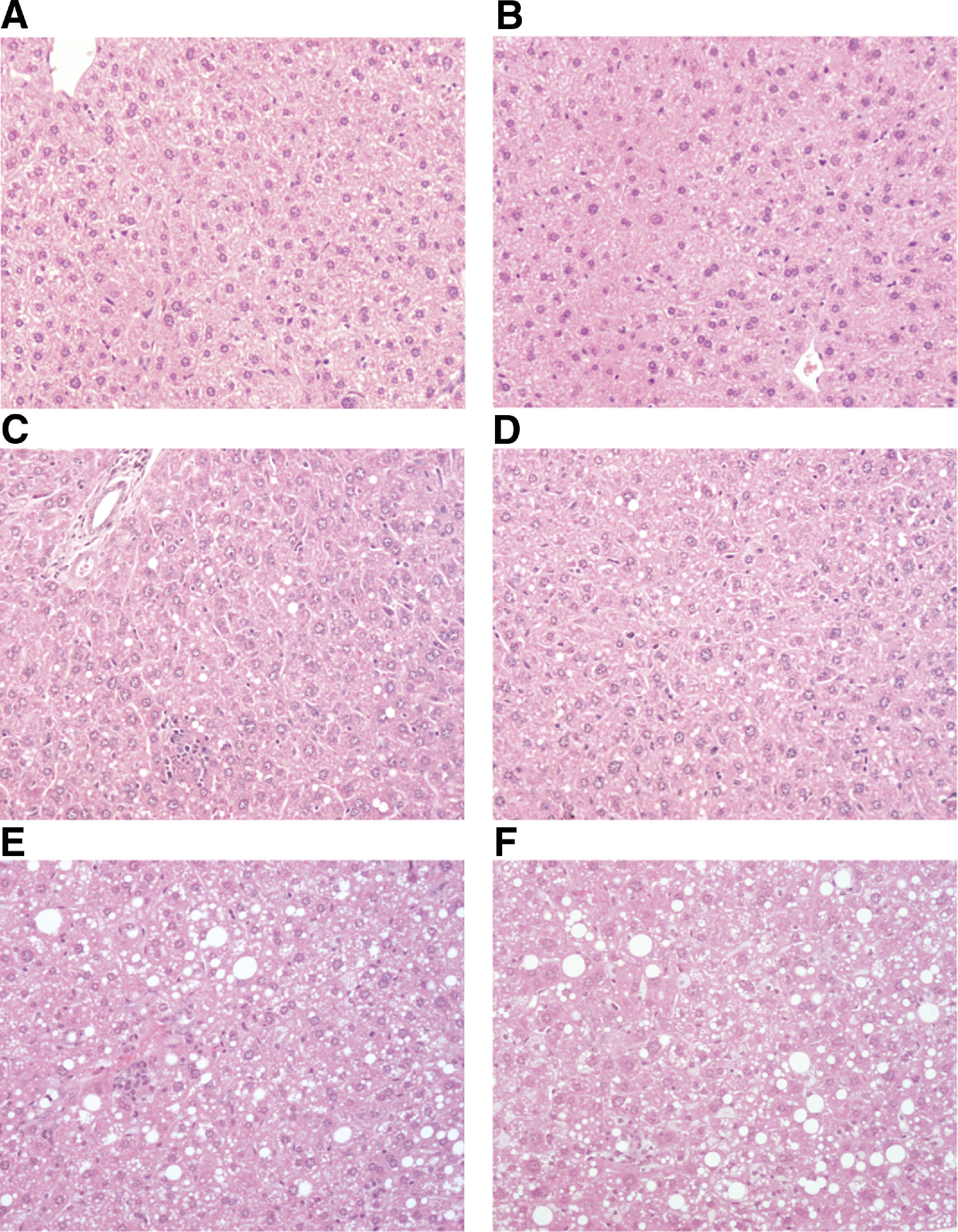
Both short- and long-term HFC feeding resulted in the development of equal levels of steatosis in the 2 transplanted groups, with increasing levels after 7 days and 3 months of HFC diet compared with chow fed mice (time effect of TG, TC, and FFA: P < .0001 for both Msr1+/+/Cd36+/+-tp and Msr1−/−/Cd36−/−-tp mice). Neither hepatic TG levels nor hepatic TC and FFA differed between the 2 transplanted groups (Figure 1D–F). Scoring of HE-and oil red O-stained liver sections by a trained pathologist confirmed this observation (Supplementary Figures 2 and 3).
Lower Levels of Hepatic Inflammation and Apoptosis in the Msr1−/−/Cd36−/− Transplanted Mice
To determine whether combined deletion of Msr1 and Cd36 on bone marrow cells affected the level of hepatic inflammation, liver sections were stained for inflammatory cell markers. For both short- and long-term HFC feeding, counting of Mac-1-positive cells (infiltrated macrophages and neutrophils) and nogo-interacting-mitochondrial protein (NIMP)-positive cells (specifically neutrophils) revealed that hepatic inflammation was increased compared with chow fed mice (P < .0001 for both Mac1 and NIMP in both Msr1+/+/Cd36+/+-tp and Msr1−/−/Cd36−/−-tp mice) but was significantly lower in the Msr1−/−/Cd36−/−-tp mice compared with Msr1+/+/Cd36+/+-tp mice (Mac1: P = .009 [7 days], P = .05 [3 months]; Nimp: P = .02 [7 days], P = .007 [3 months]) (Figure 2A and B). Moreover, when present, the positive cells were more clustered in the Msr1+/+/Cd36+/+-tp mice (Figure 2D and E). The number of CD3+ cells (T cells) was also elevated upon HFC diet (P < .0001 for both Msr1+/+/Cd36+/+-tp and Msr1−/−/Cd36−/−-tp mice), and Msr1−/−/Cd36−/−-tp mice showed less positive T cells after 3 months of HFC diet compared with Msr1+/+/Cd36+/+-tp mice (P = .0008) (Figure 2C).
Figure 2.

Parameters of hepatic inflammation and apoptosis. (A–C) Liver sections were stained for infiltrated macrophages and neutrophils (Mac-1), neutrophils (NIMP), and T cells (CD3+), respectively, and counted. (D and E) Representative pictures of Mac-1 staining (original magnification, 200×) after 7 days of HFC feeding in Msr1+/+/Cd36+/+-tp and Msr1−/−/Cd36−/−-tp mice, respectively. (F) Apoptosis was quantified by cleaved caspase 3 staining. *Significantly different from age-matched Msr1+/+/Cd36+/+-tp group. *P < .05, **P < .01, and ***P < .001, respectively.
To define further the differences in inflammation between the 2 groups, we determined the expression of several genes known to be involved in inflammation. Table 1 shows that expression of tumor necrosis factor (Tnf) was significantly lower in the Msr1−/−/Cd36−/−-tp mice after 3 months on an HFC diet compared with Msr1+/+/Cd36+/+-tp mice (P = .02), confirming the histologic data. Despite a trend toward reduced expression of the proinflammatory cytokine interleukin 6 (Il-6), it was not significantly changed. The expression of both toll-like receptors (TLR) 2 and 4 (Tlr-2 and Tlr-4), 2 well-known receptors involved in NASH, was lower in the Msr1−/−/Cd36−/−-tp mice after 7 days on an HFC diet compared with Msr1+/+/Cd36+/+-tp mice (P = .03; P = .004, respectively), but not after 3 months. Moreover, the expression levels of the anti-inflammatory nuclear receptors peroxisome proliferator activated receptor (Ppar)-α and -γ were higher in the Msr1−/−/Cd36−/−-tp mice after 3 months of an HFC diet than in mice with Msr1+/+/Cd36+/+ bone marrow (P = .001; P = .03, respectively). Together, these data are consistent with a reduction in inflammation in mice lacking hematopoietic expression of CD36 and SR-A.
Table 1.
Gene Expression Analysis of Inflammatory Related Genes
| Gene | Chow
|
7 Days’ HFC
|
3 Months’ HFC
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Msr1+/+/CD3+/+ | Msr1−/−/CD36−/− | P value | Msr1+/+/CD3+/+ | Msr1−/−/CD36−/− | P value | Msr1+/+/CD3+/+ | Msr1−/−/CD36−/− | P value | |
| Tnf | 1 (±0.12) | 1.01 (±0.15) | .97 | 1.36 (±0.29) | 0.86 (±0.11) | .12 | 6.44 (±0.97) | 3.71 (±0.42) | .02* |
| Il-6 | 1 (±0.17) | 0.89 (±0.13) | .61 | 1.80 (±0.31) | 1.29 (±0.19) | .17 | 0.49 (±0.11) | 0.44 (±0.08) | .10 |
| Tlr-2 | 1 (±0.10) | 0.86 (±0.09) | .31 | 1.38 (±0.24) | 0.77 (±0.07) | .03* | 2.12 (±0.26) | 1.95 (±0.23) | .64 |
| Tlr-4 | 1 (±0.08) | 0.78 (±0.05) | .04* | 1.23 (±0.08) | 0.91 (±0.02) | .004** | 2.30 (±0.16) | 1.92 (±0.02) | .07 |
| Ppar-α | 1 (±0.07) | 0.78 (±0.06) | .06 | 1.23 (±0.06) | 1.15 (±0.06) | .37 | 1.19 (±0.06) | 1.49 (±0.04) | .001*** |
| Ppar-γ | 1 (±0.25) | 0.99 (±0.20) | .97 | 3.85 (±0.65) | 2.58 (±0.48) | .13 | 11.15 (±1.09) | 18.97 (±1.98) | .007** |
NOTE. Gene expression analysis of 4 well-known inflammatory markers: tumor necrosis factor (Tnf), interleukin 6 (Il-6), toll-like receptors 2 and 4 (Tlr-2 and -4), and 2 anti-inflammatory markers, peroxisome proliferator-activated receptors α and γ (Ppar-α and -γ), respectively. Data were set relative to the Msr1+/+/Cd36+/+-tp group on chow diet.
P < .05.
P < .01.
P < .001.
The death of KCs and hepatocytes by apoptosis is thought to give rise to larger regions of liver damage. To test whether there was a relationship between inflammation and apoptosis in these mice, liver sections were stained for cleaved caspase 3 staining. After 3 months of HFC feeding, a significant decrease in cleaved caspase 3-positive inflammatory cells and, to a lesser extent, in hepatocytes was observed in livers of Msr1−/−/Cd36−/−-tp mice (P = .04) (Figure 2F).
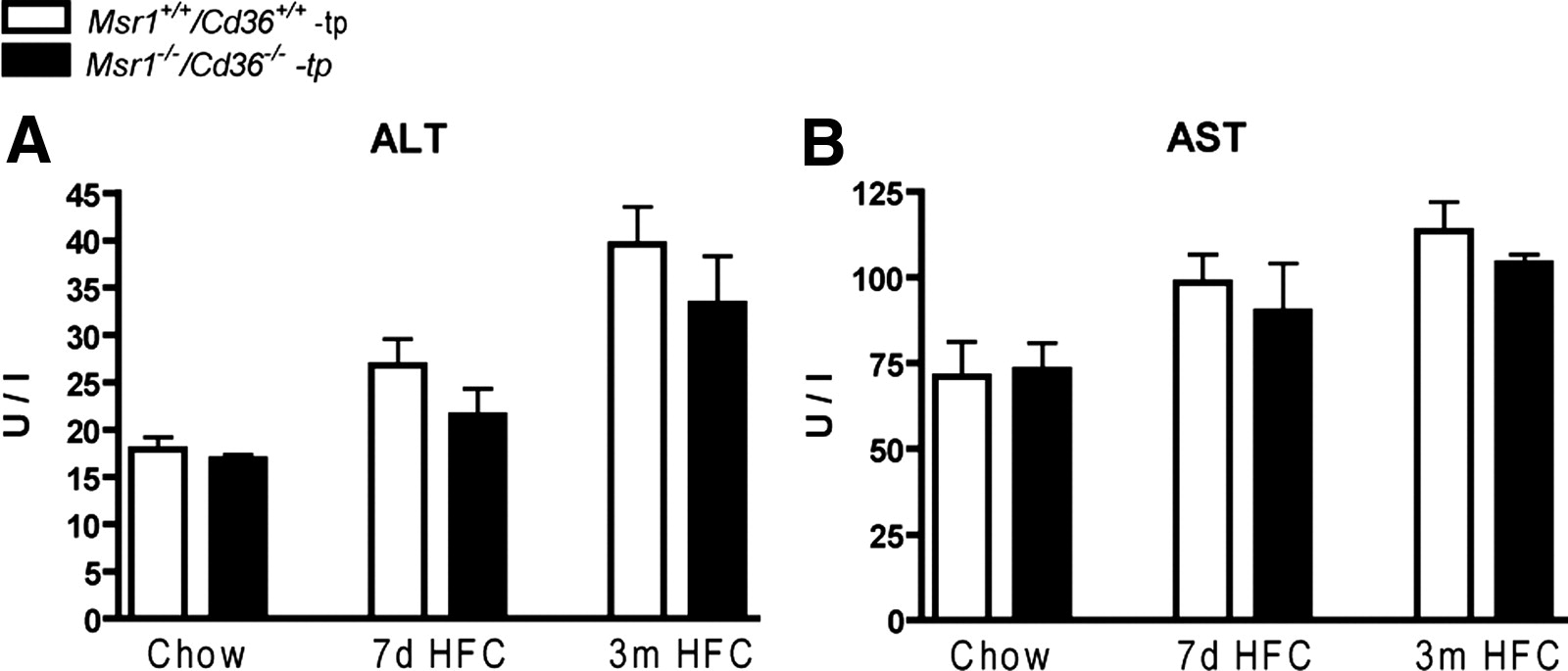
The presence of elevated transaminases like alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in plasma is considered to be a sensitive indicator of liver damage. In the present study, the plasma levels of these liver enzyme levels increased with the length of time of HFC feeding (ALT: P = .0017 for Msr1+/+/Cd36+/+-tp mice and P = .019 for Msr1−/−/Cd36−/−-tp mice; AST: P = .024 for Msr1+/+/Cd36+/+-tp mice and P = .12 for Msr1−/−/Cd36−/−-tp mice). However, neither ALT nor AST levels were significantly different between Msr1+/+/Cd36+/+-tp and Msr1−/−/Cd36−/−-tp mice (Supplementary Figure 4).
Hepatic Fibrosis Is Less Pronounced in Msr1−/−/Cd36−/− Transplanted Mice Compared With Msr1+/+/Cd36+/+ Transplanted Mice
Hepatic fibrosis is viewed as one of the advanced consequences of NASH. After 3 months of HFC diet, Msr1−/−/Cd36−/−-tp mice showed less hepatic fibrosis than Msr1+/+/CD3+/+-tp mice as evidenced by collagen staining with Sirius red (Figure 3A–C). Collagen content was lower in livers of Msr1−/−/Cd36−/−-tp mice and was primarily localized near vessels of periportal and centrolobular regions. Gene expression of 4 well-known fibrogenic-related genes in liver confirmed the findings of the Sirius red staining. After 3 months of HFC diet, expression levels of these genes were increased compared with chow and 7 days of HFC diet. Comparing Msr1−/−/Cd36−/−-tp mice with Msr1+/+/Cd36+/+-tp mice after 3 months of HFC feeding revealed a significant decrease in fibrosis (ie, collagen type 1α1 [Col1a1] [P = .005], matrix metalloprotease 13 [Mmp-13] [P = .05], tissue inhibitor of metalloproteinase 1 [Timp-1] [P = .002], and transforming growth factor β [Tgf-β] [P = .01]) (Figure 3D).
Figure 3.

Parameters of hepatic fibrosis. (A–C) Representative pictures (original magnification, 200×) of Sirius red-positive sections after 3 months of HFC diet in Msr1+/+/Cd36+/+-tp (A) and Msr1−/−/Cd36−/−-tp mice (B). (C) Quantification of Sirius red staining after 3 months of HFC diet. Livers were quantified as minimal, mild, or moderate positive for collagen. (D) Gene expression analysis of Col1α1, Mmp-13, Timp-1, and Tgf-β. Data were set relative to the Msr1+/+/Cd36+/+-tp mice on chow diet. *Significantly different from age-matched Msr1+/+/Cd36+/+-tp group. *P < .05 and **P < .01, respectively.
No Difference in Foamy Appearance of KCs Between the Models
Despite the absence of the 2 major modified lipoprotein receptors in macrophages, scoring of HE-stained (data not shown) and CD68 (macrophage marker that stains KCs)-positive sections did not reveal a reduction in the size or presence of foamy KCs in Msr1−/−/Cd36−/−-tp mice (Figure 4A–C). Moreover, a fluorescent double staining with CD68 and filipin (cholesterol marker) showed that the cholesterol content inside KCs increased with the size of the KCs upon HFC diet and that there were no differences in cholesterol content between KCs of Msr1+/+/Cd36+/+-tp and Msr1−/−/Cd36−/−-tp mice (Supplementary Figure 5A–G).
Figure 4.

Foamy appearance of Kupffer cells. (A–D) Representative pictures (original magnification, 200×) after chow, (A) 7 days (B) and 3 months (C) of HFC diet with Msr1+/+/Cd36+/+-tp and Msr1−/−/Cd36−/−-tp mice, respectively. (D) Gene expression analysis of macrophage and Kupffer cell marker Cd68 and ATP-binding cassette transporters Abca1 and Abcg1. Data were set relative to the Msr1+/+/Cd36+/+-tp mice on chow diet. *Significantly different from age-matched Msr1+/+/Cd36+/+-tp group. *P < .05, **P < .01, and ***P < .001, respectively.
Cd68 expression was comparable in livers of both groups after HFC feeding (Figure 4D). Notably, the expression levels of the adenosine triphosphate (ATP)-binding cassette transporter A1 (Abca1) in liver revealed a significant decrease after 3 months on an HFC diet in the Msr1−/−/Cd36−/−-tp mice compared with Msr1+/+/Cd36+/+-tp mice (P = .02). The ATP-binding cassette transporter G1 (Abcg1) showed the same trend after long-term HFC feeding (Figure 4D). Moreover, there were already basal differences between Msr1+/+/Cd36+/+-tp and Msr1−/−/Cd36−/−-tp mice for expression of Cd68, Abca1, and Abcg1 in total liver. Furthermore, gene expression analysis in whole liver revealed no compensation by other scavenger receptors (Supplementary Figure 6).
Msr1−/−/Cd36−/− Transplanted Mice Show Less Lipid Oxidation Compared With Msr1+/+/Cd36+/+ Transplanted Mice
To obtain an indirect measure of the degree of lipid oxidation, immunoglobulin (Ig) M and IgG auto-antibodies against malondialdehyde (MDA)-LDL and copper-oxidized (Cu-Ox) LDL were measured in plasma of transplanted animals. For each time point, IgM auto-antibody levels to MDA-LDL and Cu-OxLDL, which are thought to be to a large extent natural antibodies20 and to be inversely correlated with cardiovascular diseases,21,22 showed significant higher levels in Msr1−/−/Cd36−/−-tp mice compared with Msr1+/+/Cd36+/+-tp mice (MDA-IgM: P = .001 [chow], P = .01 [7 days], P < .0001 [3 months]; Cu-Ox-IgM: P = .006 [chow], P = .02 [7 days], P = .0008 [3 months]) (Figure 5A and B). IgG autoantibody levels to MDA-LDL are significantly decreased in the Msr1−/−/Cd36−/−-tp mice after HFC feeding, indicating a less pronounced oxidative stress-mediated immune response (P = .09 [chow], P = .006 [7 days], P = .03 [3 months]) (Figure 5C). The IgG levels against Cu-OxLDL were not significantly different after HFC diet but showed a basal difference between Msr1+/+/Cd36+/+-tp and Msr1−/−/Cd36−/−-tp mice (Figure 5D). In line with the protective pattern of autoantibodies against oxLDL, 2 well-known oxysterols in plasma, 27- and 24S-hydroxy-cholesterol, were significantly lower after 3 months HFC diet in Msr1−/−/Cd36−/−-tp mice compared with Msr1+/+/Cd36+/+-tp mice (P = .02, P = .008, respectively), indicating less oxidation of cholesterol in these animals (Figure 5E). Similar to the decreased oxidation processes in these mice, the number of myeloperoxidase (MPO)-positive cells, which can generate a variety of reactive oxygen species, was significantly lower in the livers of Msr1−/−/Cd36−/−-tp mice compared with Msr1+/+/Cd36+/+-tp mice (P = .003 [7 days], P = .05 [3 months]) (Figure 5F).
Figure 5.

Parameters of oxidative damage. (A–D) IgG and IgM autoantibody titers to MDA-LDL and CuOx-LDL in plasma, respectively. (E) Oxysterols in plasma: 27- and 24S-hydroxycholesterol. (F) Scoring of MPO-positive cells in liver sections. *Significantly different (P < .05) from aged matched Msr1+/+/Cd36+/+-tp group.
Discussion
Our study shows for the first time the involvement of macrophage SRs in early and advanced stages of NASH. Reconstitution of Ldlr−/− mice with bone marrow from mice lacking both Msr1 and Cd36 significantly reduced hepatic inflammation, lipid oxidation, and fibrosis without affecting steatosis. These novel observations support an analogy between mechanisms involved in the progression of liver inflammation and atherosclerosis.
KCs Initiate Early Hepatic Inflammation by Scavenging Modified Lipoproteins
Currently, the risk factors that drive hepatic inflammation during the progression to NASH are largely unknown. We have previously shown that hyperlipidemic mice are more sensitive to developing early, diet-induced NASH.8 In the current study, we demonstrate that the increased sensitivity of these mice to develop NASH is linked to expression of SR-A and CD36 on Kupffer cells.
The role of macrophage SRs CD36 and SR-A in atherogenesis has been intensively investigated. Uptake and internalization of modified LDL by SRs is believed to constitute one of the major pathways for foam cell formation in vivo. However, recent studies have revealed a wide spectrum of SR functions, including the activation of signal transduction pathways regulating inflammation, apoptotic cell clearance, chemoattraction, and angiogenesis, which may also contribute to atherogenesis.23–25 The current paradigm suggests that SR-mediated uptake of oxidized lipoproteins by macrophages sets off a cascade of proinflammatory events leading to the initiation of the inflammatory response. In line with this view, we have observed that, upon targeted deletion of Cd36 and Msr1 in macrophages, the hepatic inflammatory response was dramatically decreased in a mouse model of NASH, as indicated by a decreased number of inflammatory cells and Tnf. In line with this observation, deletion of both Msr1 and Cd36 in mice lacking Apoe was shown to reduce aortic expression levels of several chemokines and cytokines including Tnf.19 Therefore, the effect that we have observed in the livers of Msr1−/−/Cd36−/−-tp mice is likely to be systemic and not restricted to the liver. Similarly, it was described that combined deletion of Cd36 and Msr1, and even deletion of Cd36 alone, resulted in decreased macrophage content, expression levels of serum proinflammatory cytokines, and aortic chemokine expression.18,19,26,27 Moreover, CD36 has been shown to facilitate TLR signaling in response to both oxLDL and diacylglyceride via TLR4/TLR6 and TLR2/6, respectively, and thereby trigger the innate host response.17,27–30 Thus, similar innate immune signaling pathways initiated by SRs may also be activated during steatosis, and these may promote inflammation associated with NASH. Notably, whereas most of the previous reports regarding the role of these SRs in inflammation were performed using the complete knock-out animals in an Apoe−/− background, our current study was performed in Ldlr−/− mice transplanted with bone marrow from mice lacking Msr1 and Cd36. Our approach has the advantage of being independent of the mixed genetic background of the mice, which may have confounded previous studies, and it has also allowed us to specifically examine the contribution of SRs meditated by macrophages on hepatic inflammation.
The decreased fibrosis and expression of fibrogenic-related genes detected in the Msr1−/−/Cd36−/−-tp mice after 3 months of HFC diet were also associated with reduced levels of apoptosis. The effect on apoptosis may be attributed to the dramatic reduction in expression of Tnf, which is known to activate the expression of many proinflammatory cytokines and to induce apoptosis.31 There is considerable evidence contributing to the pathophysiologic concept that excessive apoptosis in the liver acts as a proinflammatory and profibrogenic trigger.32–34 The reduced level of apoptosis could therefore further contribute to the decreased development of fibrosis in the livers of the Msr1−/−/Cd36−/−-tp mice. In addition, Jaeschke35 and Lawson et al36 demonstrated that hepatocyte apoptosis is a potent stimulus for neutrophil infiltration and endotoxin-induced liver injury. These findings are in line with the finding that apoptosis correlates with enhanced myeloid cell infiltration and hepatic fibrosis in our models. Beside the reduced expression levels of inflammatory genes, levels of the anti-inflammatory nuclear receptors Ppar-α and -β were significantly higher in the livers of the Msr1−/−/Cd36−/−-tp mice after 3 months. The recognition that, in addition to their anti-inflammatory response, PPARs are also associated with decreased liver fibrogenesis and tumorigenesis is relevant.37 Thus, the increase in hepatic expression of these nuclear receptors in the Msr1−/−/Cd36−/−-tp mice may further contribute to the improvement of liver pathology in these mice.
Inflammatory Response in Liver Is Attributed to Lipid Oxidation Rather Than to Foam Cell Formation
Whereas our original hypothesis was that the inflammatory response in livers of hyperlipidemic mice would be related to foam cell formation,8 our data demonstrate a link between hepatic inflammation and LDL oxidation, rather than total lipid uptake. KCs lacking both Cd36 and Msr1 were increased in size throughout the duration of the diet but were not significantly different from control macrophages. Although cholesterol content inside KCs was not different between Msr1+/+/Cd36+/+-tp and Msr1−/−/Cd36−/−-tp mice, gene expression analysis of Cd68, Abca1, and Abcg1 in the liver showed already some basal differences. However, it is unlikely that these minor changes in gene expression are associated with a reduced efflux of cholesterol out of KCs via these ATP-binding cassettes.
MSR1 and CD36 have been shown to be critical contributors to modified lipoprotein uptake in macrophages in vitro.14 In addition, it has been shown that a specific CD36-dependent signaling pathway initiated by oxLDL is necessary for foam cell formation in vitro and in vivo.17 However, hyperlipidemic Cd36−/−/Apoe−/− and Msr1−/−/Apoe−/− mice develop abundant foamy macrophages in atherosclerotic plaques, indicating that lipid uptake by intimal macrophages can occur in the absence of CD36 and MSR1.18 In line with these observations, it has also been shown that loss of both CD36 and MSR1 activity reduces atherosclerotic lesion complexity and inflammation in particular, without abrogating foam cell formation in Apoe−/− mice.19
It is important to note that there is still no clear evidence establishing that lipids that generate foam cells in vivo are derived from oxidized lipoproteins. In fact, there is considerable evidence that LDL-derived lipids can enter macrophages via SR-independent pathways.38 Native LDL, which is present in much higher concentrations in plasma compared with modified/oxidized LDL, has been reported to be internalized via macropinocytosis of extracellular fluid.39 In addition, aggregated or enzymatically modified forms of LDL can also be internalized by macrophages. It is therefore possible that, whereas the total amounts of lipids in the hepatic macrophages from the Msr+/+/Cd36+/+-tp and Msr1−/−/Cd36−/−-tp mice are identical, the lipids may be qualitatively different or the mechanism of internalization may trigger more inflammatory pathways.
The reduced inflammatory response in the livers of the mice with targeted deletions of the 2 SRs was associated with diminished cholesterol oxidation and oxidative stress. This effect was also characterized by decreased hepatic MPO in Msr1−/−/Cd36−/−-tp mice compared with Msr1+/+/Cd36+/+-tp mice. MPO is secreted by neutrophils and monocytes that generate many oxidants, which can initiate the oxidation of LDL. Previously, it has been shown that CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species.40 These data suggest the existence of a defensive feedback mechanism to reduce levels of oxidation when CD36 expression is low. In line with the decreased levels of MPO in the liver, we observed significant differences in levels of IgM and IgG autoantibodies against modified LDL in the plasma of both transplanted groups. IgM autoantibodies to MDA-LDL and Cu-OxLDL are increased in Msr1−/−/Cd36−/−-tp mice, suggesting decreased consumption of these IgM, which have been suggested to be natural antibodies to a large part.20 In literature, it has been shown that IgM autoantibodies to MDA-LDL were inversely related with atherosclerosis.21,22 Interestingly, an IgG immune response induced by lipid peroxidation products correlates with hepatic inflammation during alcoholic steatohepatitis and the progression to advanced fibrosis during nonalcoholic steatohepatitis.41,42 Moreover, a number of studies have shown that the levels of IgG autoantibodies are correlated with increased risk for cardiovascular diseases.43,44 Surprisingly, the differences in levels of these autoantibodies between Msr+/+/Cd36+/+-tp and Msr1−/−/Cd36−/−-tp mice were already observed at basal levels. The generation of natural antibodies is known to occur in the complete absence of external antigenic stimulation, and, therefore, it is not surprising that these antibodies are present already before the initiation of the HFC diet. Importantly, the basal differences between recipients of the 2 genotypes suggest that MSR1 and/or CD36 are critically involved in the generation of these antibodies. Thus, our data further add to the breadth of SR functions, in particular, the activation of additional immune responses with analogous specificity. Because this effect is likely to be systemic and not restricted to the liver, our observations regarding lipid oxidation are of general importance and also relevant to the field of atherosclerosis.
In conclusion, our study demonstrates that the SRs MSR1 and CD36 play an important role during early and advanced stages of NASH. Further studies are necessary to investigate the exact contribution of each of these receptors to dissect the molecular mechanisms involved in diet-induced NASH. Our data also establish the hyperlipidemic mice models as a tool to investigate the advanced stages of NASH in a physiologic context and provide a mechanism for the initiation and progression of the disease in these models. Finally, this study demonstrates for the first time an analogy between mechanisms for NASH and atherosclerosis. Thus, the focus on the liver as a crucial driver of inflammation in the metabolic syndrome is expected to provide alternative strategies leading to new therapies for preventing atherosclerosis. This study also provides new evidence for the close and complex link between lipid metabolism and inflammation, as manifested in cardiovascular diseases.
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at doi: 10.1053/j.gastro.2010.02.051.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank Inge van der Made, Pieter Goossens, Monique Vergouwe, and Tim Hendrikx for their excellent technical support; Patrick Lindsey for his statistical advice; and Sander Rensen for providing the MPO antibody and staining protocol.
Funding
Veni: 916.76.070 (2006/00496/MW); Maag Lever Darm Stichting (MLDS) (WO 08-16); Dutch Heart Fondation (NHS) (2002B18); Vidi: 016.066.329.
Abbreviations used in this paper
- ABC
adenosine triphosphate binding cassette
- acLDL
acetylated LDL
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- APOE
apolipoprotein E
- BCA
bicin-choninic acid
- Col1α1
collagen type 1α1
- cDNA
complementary DNA
- Cu-OxLDL
copper oxidized LDL
- FFA
free fatty acid
- HFC
high-fat, high-cholesterol
- Ig
immunoglobulin
- KC
Kupffer cell
- LDL
low-density lipoprotein
- MDA
malondialdehyde
- MMP
matrix metalloprotease
- MPO
myeloperoxidase
- MSR
macrophage scavenger receptor
- NASH
nonalcoholic steatohepatitis
- NF-κB
nuclear factor κ-B
- NIMP
nogo-interacting-mitochondrial protein
- ORO
oil red O staining
- oxLDL
oxidized LDL
- PPAR
peroxisome proliferator activated receptor
- SR
scavenger receptors
- TC
total cholesterol
- TG
triglycerides
- TGF-β
transforming growth factor β
- TIMP
tissue inhibitor of metalloproteinase
- TLR
toll-like receptor
- TNF
tumor necrosis factor
- tp
transplanted
Footnotes
Conflicts of interest
The authors disclose no conflicts.
References
- 1.McCullough AJ. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8:521–533. doi: 10.1016/j.cld.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 2.Parekh S, Anania FA. Abnormal lipid and glucose metabolism in obesity: implications for nonalcoholic fatty liver disease. Gastro-enterology. 2007;132:2191–2207. doi: 10.1053/j.gastro.2007.03.055. [DOI] [PubMed] [Google Scholar]
- 3.Matsuzawa N, Takamura T, Kurita S, et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology. 2007;46:1392–1403. doi: 10.1002/hep.21874. [DOI] [PubMed] [Google Scholar]
- 4.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 6.Ruvolo PP. Intracellular signal transduction pathways activated by ceramide and its metabolites. Pharmacol Res. 2003;47:383–392. doi: 10.1016/s1043-6618(03)00050-1. [DOI] [PubMed] [Google Scholar]
- 7.Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006;45:42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Wouters K, van Gorp PJ, Bieghs V, et al. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology. 2008;48:474–486. doi: 10.1002/hep.22363. [DOI] [PubMed] [Google Scholar]
- 9.Mari M, Caballero F, Colell A, et al. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 10.Vergnes L, Phan J, Strauss M, et al. Cholesterol and cholate components of an atherogenic diet induce distinct stages of hepatic inflammatory gene expression. J Biol Chem. 2003;278:42774–42784. doi: 10.1074/jbc.M306022200. [DOI] [PubMed] [Google Scholar]
- 11.Yoshimatsu M, Terasaki Y, Sakashita N, et al. Induction of macrophage scavenger receptor MARCO in nonalcoholic steatohepatitis indicates possible involvement of endotoxin in its pathogenic process. Int J Exp Pathol. 2004;85:335–343. doi: 10.1111/j.0959-9673.2004.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sano J, Shirakura S, Oda S, et al. Foam cells generated by a combination of hyperglycemia and hyperlipemia in rats. Pathol Int. 2004;54:904–913. doi: 10.1111/j.1440-1827.2004.01778.x. [DOI] [PubMed] [Google Scholar]
- 13.Naito M, Kodama T, Matsumoto A, et al. Tissue distribution, intracellular localization, and in vitro expression of bovine macrophage scavenger receptors. Am J Pathol. 1991;139:1411–1423. [PMC free article] [PubMed] [Google Scholar]
- 14.Kunjathoor VV, Febbraio M, Podrez EA, et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem. 2002;277:49982–49988. doi: 10.1074/jbc.M209649200. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki H, Kurihara Y, Takeya M, et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997;386:292–296. doi: 10.1038/386292a0. [DOI] [PubMed] [Google Scholar]
- 16.Moore KJ, Freeman MW. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol. 2006;26:1702–1711. doi: 10.1161/01.ATV.0000229218.97976.43. [DOI] [PubMed] [Google Scholar]
- 17.Rahaman SO, Lennon DJ, Febbraio M, et al. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006;4:211–221. doi: 10.1016/j.cmet.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore KJ, Kunjathoor VV, Koehn SL, et al. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest. 2005;115:2192–2201. doi: 10.1172/JCI24061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manning-Tobin JJ, Moore KJ, Seimon TA, et al. Loss of SR-A and CD36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 2008;29:19–26. doi: 10.1161/ATVBAHA.108.176644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou MY, Fogelstrand L, Hartvigsen K, et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest. 2009;119:1335–1349. doi: 10.1172/JCI36800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karvonen J, Paivansalo M, Kesaniemi YA, et al. Immunoglobulin M type of autoantibodies to oxidized low-density lipoprotein has an inverse relation to carotid artery atherosclerosis. Circulation. 2003;108:2107–2112. doi: 10.1161/01.CIR.0000092891.55157.A7. [DOI] [PubMed] [Google Scholar]
- 22.Horkko S, Bird DA, Miller E, et al. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J Clin Invest. 1999;103:117–128. doi: 10.1172/JCI4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Platt N, Suzuki H, Kurihara Y, et al. Role for the class A macrophage scavenger receptor in the phagocytosis of apoptotic thymocytes in vitro. Proc Natl Acad Sci U S A. 1996;93:12456–12460. doi: 10.1073/pnas.93.22.12456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. 2001;108:785–791. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cotena A, Gordon S, Platt N. The class A macrophage scavenger receptor attenuates CXC chemokine production and the early infiltration of neutrophils in sterile peritonitis. J Immunol. 2004;173:6427–6432. doi: 10.4049/jimmunol.173.10.6427. [DOI] [PubMed] [Google Scholar]
- 26.Kuchibhotla S, Vanegas D, Kennedy DJ, et al. Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc Res. 2008;78:185–196. doi: 10.1093/cvr/cvm093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stewart CR, Stuart LM, Wilkinson K, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoebe K, Georgel P, Rutschmann S, et al. CD36 is a sensor of diacylglycerides. Nature. 2005;433:523–527. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- 29.Triantafilou M, Gamper FG, Lepper PM, et al. Lipopolysaccharides from atherosclerosis-associated bacteria antagonize TLR4, induce formation of TLR2/1/CD36 complexes in lipid rafts and trigger TLR2-induced inflammatory responses in human vascular endothelial cells. Cell Microbiol. 2007;9:2030–2039. doi: 10.1111/j.1462-5822.2007.00935.x. [DOI] [PubMed] [Google Scholar]
- 30.Stuart LM, Deng J, Silver JM, et al. Response to Staphylococcus aureus requires CD36-mediated phagocytosis triggered by the COOH-terminal cytoplasmic domain. J Cell Biol. 2005;170:477–485. doi: 10.1083/jcb.200501113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rath PC, Aggarwal BB. TNF-induced signaling in apoptosis. J Clin Immunol. 1999;19:350–364. doi: 10.1023/a:1020546615229. [DOI] [PubMed] [Google Scholar]
- 32.Canbay A, Kip SN, Kahraman A, et al. Apoptosis and fibrosis in non-alcoholic fatty liver disease. Turk J Gastroenterol. 2005;16:1–6. [PubMed] [Google Scholar]
- 33.Maher JJ, Scott MK, Saito JM, et al. Adenovirus-mediated expression of cytokine-induced neutrophil chemoattractant in rat liver induces a neutrophilic hepatitis. Hepatology. 1997;25:624–630. doi: 10.1002/hep.510250322. [DOI] [PubMed] [Google Scholar]
- 34.Lauber K, Bohn E, Krober SM, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113:717–730. doi: 10.1016/s0092-8674(03)00422-7. [DOI] [PubMed] [Google Scholar]
- 35.Jaeschke H. Neutrophil-mediated tissue injury in alcoholic hepatitis. Alcohol. 2002;27:23–27. doi: 10.1016/s0741-8329(02)00200-8. [DOI] [PubMed] [Google Scholar]
- 36.Lawson JA, Fisher MA, Simmons CA, et al. Parenchymal cell apoptosis as a signal for sinusoidal sequestration and transendothelial migration of neutrophils in murine models of endotoxin and Fas-antibody-induced liver injury. Hepatology. 1998;28:761–767. doi: 10.1002/hep.510280324. [DOI] [PubMed] [Google Scholar]
- 37.Gervois P, Vu-Dac N, Kleemann R, et al. Negative regulation of human fibrinogen gene expression by peroxisome proliferator-activated receptor alpha agonists via inhibition of CCAAT box/enhancer-binding protein beta. J Biol Chem. 2001;276:33471–33477. doi: 10.1074/jbc.M102839200. [DOI] [PubMed] [Google Scholar]
- 38.Kruth HS, Jones NL, Huang W, et al. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low-density lipoprotein. J Biol Chem. 2005;280:2352–2360. doi: 10.1074/jbc.M407167200. [DOI] [PubMed] [Google Scholar]
- 39.Kruth HS, Huang W, Ishii I, et al. Macrophage foam cell formation with native low-density lipoprotein. J Biol Chem. 2002;277:34573–34580. doi: 10.1074/jbc.M205059200. [DOI] [PubMed] [Google Scholar]
- 40.Podrez EA, Schmitt D, Hoff HF, et al. Myeloperoxidase-generated reactive nitrogen species convert LDL into an atherogenic form in vitro. J Clin Invest. 1999;103:1547–1560. doi: 10.1172/JCI5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Occhino GIA, Hietala J, Sutti S, et al. ESBRA 2007. Abstracts of the 11th Congress of the European Society for Biomedical Research on Alcoholism, 23–26 September, Berlin, Germany. Alcohol Suppl. 2007;42:i1–66. [PubMed] [Google Scholar]
- 42.Albano E, Mottaran E, Vidali M, et al. Immune response towards lipid peroxidation products as a predictor of progression of non-alcoholic fatty liver disease to advanced fibrosis. Gut. 2005;54:987–993. doi: 10.1136/gut.2004.057968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsimikas S, Brilakis ES, Lennon RJ, et al. Relationship of IgG and IgM autoantibodies to oxidized low-density lipoprotein with coronary artery disease and cardiovascular events. J Lipid Res. 2007;48:425–433. doi: 10.1194/jlr.M600361-JLR200. [DOI] [PubMed] [Google Scholar]
- 44.Hulthe J, Wiklund O, Hurt-Camejo E, et al. Antibodies to oxidized LDL in relation to carotid atherosclerosis, cell adhesion molecules, and phospholipase A(2) Arterioscler Thromb Vasc Biol. 2001;21:269–274. doi: 10.1161/01.atv.21.2.269. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.