

Abstract
Tristetraprolin (TTP, Zfp36, Nup475, Tis11) dramatically reduces the stability of target mRNAs by binding to AU-rich elements (AREs) in their 3′UTRs. Through this mechanism, TTP functions as a rheostatic, temporal regulator of gene expression. TTP KO mice exhibit completely penetrant granulocytic hyperplasia. We have shown that the hematopoietic stem-progenitor cell (HSPC) compartment in TTP KO mice is also altered. Although no change was detected in long-term HSC (LT-HSC) frequency or function, as assayed by immunophenotypic markers or limiting dilution transplants, we observed increases in the frequencies and numbers of short-term HSCs (ST-HSCs), multipotent progenitors (MPPs) and granulocyte-monocyte progenitors (GMPs). This pattern is consistent with ‘reactive granulopoiesis,’ in which committed myeloid progenitors and more primitive progenitors cycle more actively to increase production of mature granulocytes in response to infection or adjuvant. We created reverse chimeras by transplanting WT bone marrow (BM) into TTP KO mice and found the ‘reactive granulopoiesis’ phenocopied, indicating a non-HSPC-autonomous mechanism. Correspondingly, we found elevated levels of the granulopoietic TTP targets IL-1β, TNFα and IL-6 in the plasma of TTP KO mice. Consistent with the non-cell autonomous nature of the phenotype, we found elevated levels of IL-1β, TNFα and IL-6 transcripts in the livers of TTP KO mice and no detectable difference in the BMs. These findings demonstrate the importance of TTP in inflammatory homeostasis and highlight the ability of the hematopoietic system to respond to stress without significant numbers of quiescent HSCs entering the cell cycle.
Introduction
‘Steady state’ hematopoiesis produces hundreds of millions of blood cells per day to replace a continuously turning-over pool of myeloid, lymphoid and erythroid cells from a small number of hematopoietic stem cells (HSCs)(1). Upon hematopoietic stress, such as bleeding or infection, expansion of the needed lineages of progenitors is stimulated to respond rapidly and selectively to the specific stress. During infection, ‘steady-state’ neutrophils exit from the circulation into peripheral tissues to combat the invading pathogens. The result is a transient period of peripheral blood neutropenia, which is followed by neutrophilia resulting from a period of ‘reactive’ granulopoiesis in the BM with a concomitant decrease in B lymphopoiesis(2, 3). The exact signaling pathways and cytokines responsible for orchestrating this reactive process are now being elucidated.
It has become clear that reactive granulopoiesis is directed through different pathways from those which regulate steady-state granulopoiesis. For example, the transcription factor Cebp/α is required for steady-state granulopoiesis but dispensible for the reactive response. In contrast, Cebp/β is not necessary for steady-state granulopoiesis but is necessary to mount a reactive response(4). In addition, the cytokines G-CSF, GM-CSF, IL-3 and IL-6, all regulators of steady-state granulopoiesis, were not required for the reactive response following C. albicans challenge(5). Recently, IL-1β was demonstrated to be both necessary for reactive granulopoiesis and sufficient to induce neutrophilia(3, 6). These authors also extended the understanding of reactive granulopoietic regulation to the HSPC compartment, where primitive Lin−Kit+Sca+Flt3− and Lin−Kit+Sca+Flt3+ cells in the bone marrow, known to be enriched for short-term HSCs (ST-HSCs) and multipotent progenitors (MPPs), respectively, sustain reactive granulopoiesis by cycling at rates higher than in steady-state. The entire reactive response was abolished in IL-1R KO mice due to their inability to respond to IL-1β.
AU-rich elements (AREs) are sequences within the 3′ untranslated regions (3′UTRs) of mRNAs that dramatically affect their stability(7). It is predicted that nearly 8% of all transcripts contain AREs(8). Modulation of the half-lives of ARE-containing mRNAs is mediated by ARE binding proteins (ARE-bps) such as TTP and HuR, a destabilizing and a stabilizing ARE-bp, respectively(9). ARE-mediated destabilization via TTP allows for regulation of the strength and duration of responses to various stimuli, for example the production of TNFα in response to LPS(10).
Mice deficient for TTP have an inflammatory phenotype characterized by granulocytic hyperplasia, cachexia, alopecia and decreased B lymphopoiesis(11). We report here that, in the HSPC compartment of TTP KO bone marrow as compared to wild type (WT) control, MPPs and ST-HSCs were more frequent and more actively in cell cycle with no measurable difference in long-term HSCs (LT-HSCs). In addition, we observed lineage skewing towards granulocyte-monocyte progenitors (GMPs), consistent with the reported granulocytic hyperplasia phenotype(11). This pattern is consistent with findings described in reactive granulopoiesis, where early progenitors are stimulated to cycle hyperactively and produce larger numbers of mature granulocytes. The granulocyte hyperplasia and HSPC phenotypes were completely penetrant, arising spontaneously by 4 weeks of age in all TTP KO mice evaluated. The hematopoietic phenotype was non-cell autonomous, in that KO recipients transplanted with WT bone marrow displayed the complete hematopoietic phenotype of granulocyte hyperplasia, while WT recipients of KO bone marrow had normal granulocyte numbers. In the plasma of TTP KO mice, we measured increased protein levels of the granulopoietic TTP targets IL-1β, TNFα and IL-6. The transcript levels for each of these cytokines were elevated in TTP KO livers compared to WT controls; no difference was detected in BM samples. These studies identify the liver as a source tissue for the non-cell autonomous phenotype in TTP KO mice.
Materials and Methods
Mice, limiting dilution transplants and reverse chimeras
TTP heterozygous mice, on the C57Bl/6 background, were provided by Dr. Perry J. Blackshear (National Institutes of Health, National Institute of Environmental Health Sciences, Duke University School of Medicine). TTPheterozygous mice were bred in the Johns Hopkins University School of Medicine animal facility. Offspring genotypes were determined by PCR. All animal studies performed were approved by the Johns Hopkins University Animal Care and Use Committee.
Recipient female 6–8 wk old C57Bl/6.SJL (CD45.1+) mice (Jackson Labs, Bar Harbor, ME) were irradiated with a split dose of 1050cGy from a Cs source gamma irradiator. These mice were then transplanted with a repopulating dose of 250×103 whole bone marrow CD45.1+ cells mixed with dilutions of whole bone marrow from 6–8-wk-old female WT or KO (CD45.2+) mice. Dilutions included 250×103, 100×103, 50×103, 25×103 and 10×103 whole bone marrow cells. Engraftment was evaluated in peripheral blood by flow cytometry. Frequency of engrafting cells was calculated using L-calc software (Stem Cell Technologies, Vancouver, BC).
Reverse chimeras were created by irradiating 6–8-wk-old female WT and KO recipient mice with 950cGy and transplanting 2×106 donor CD45.1+ whole bone marrow cells from 6–8-wk old mice.
Cell isolation and analysis by flow cytometry
Bone marrow cells from 6–8-wk old WT and KO mice were isolated by flushing femurs and tibias with dissection medium (PBS-2.5%FBS+2mM EDTA) or by crushing femurs, tibias, hips and spines in a mortar and pestle with dissection medium. Spleens were harvested and mashed through a 40μm filter (BD Biosciences, San Jose, CA) in staining buffer (PBS-2.5%FBS). After centrifugation, mature red cells were removed with 1× RBC Lysis Buffer (eBioscience, San Diego, CA). Before staining, samples were incubated with Fc Block (2.4G2, BD Biosciences, San Jose, CA).
Mature populations were characterized using PE-Cy7 B220 (RA3-6B2, eBioscience, San Diego, CA), PerCP-Cy5.5 CD3 (145-2C11, BD Biosciences, San Jose, CA), APC CD11b (M1/70, eBioscience, San Diego, CA), APC-Cy7 Gr1 (RB6-8C5, BD Biosciences, San Jose, CA). For chimerism analysis, we used v450 or PE CD45.1 (A20, BD Biosciences, San Jose, CA) and FITC CD45.2 (104, BD Biosciences, San Jose, CA). For analysis of stem and progenitor cells we used biotinylated B220, CD3, Gr1 and Ter119 (BD Biosciences, San Jose, CA), streptavidin-PerCP-Cy5.5 (BD Biosciences, San Jose, CA), APC or v450 Kit (2B8 BD Biosciences, San Jose, CA), PE-Cy7 Sca1 (D7, eBioscience, San Diego, CA), FITC CD34 (RAM34, eBioscience, San Diego, CA), PE Flt3 (A2F10.1 BD Biosciences, San Jose, CA), PE FcRγ (2.4G2, BD Biosciences, San Jose, CA), PE CD150 (TCF-12F12.2, Biolegend, San Diego, CA) and FITC CD48 (HM48-1, BD Biosciences, San Jose, CA). All samples were run on an LSRII flow cytometer (BD Biosciences, San Jose, CA).
Cell Cycle analysis and BrdU staining
Bone marrow was isolated as described, stained with Fc Block and biotinylated Gr1, Ter119, CD3 and B220 (Mouse Lineage Panel, BD Biosciences, San Jose, CA) before lineage depletion using Dynabeads MyOne Streptavidin T1 (Invitrogen, Carlsbad, CA), according to manufacturer’s instructions. Lineage-depleted marrow was stained for Kit, Sca, CD34, Flt3 using the antibodies detailed above and washed with staining buffer. Stained cells were fixed using BD Cytofix (BD Biosciences, San Jose, CA), followed by permeabilization using PBS-0.1%Triton. Cells were stained with APC Ki67 (B56, BD Biosciences, San Jose, CA) for 1 hr at RT, washed with staining buffer and DNA was stained with 7-Aminoactinomycin D (7-AAD) (BD Biosciences, San Jose, CA).
For BrdU incorporation analysis, we used the APC BrdU Flow Kit (BD Biosciences, San Jose, CA) according to the manufacturer’s instructions. Briefly, mice were injected with 100μg BrdU per gram of body weight 16 hours before harvesting bone marrow. Bone marrow was lineage depleted, as above. Cells were stained for cell surface markers then fixed and permeabilized using BD Cytofix followed by BD Cytofix/Cytoperm (BD Biosciences, San Jose, CA). Cells were then treated with DNase and stained for BrdU using APC anti-BrdU. Finally, DNA was stained with 7-AAD.
Enumeration of colony-forming cells (CFC Assays)
Red cell lysed bone marrow or spleen cells (20×103 or 100×103 cells per plate, respectively) were plated in M3434 (Stem Cell Technologies, Vancouver, BC) methylcellulose-containing differentiation media in 35mm dishes in triplicate. Colonies were enumerated on day 7 after plating.
Measurement of cytokine concentrations in mouse plasma
Blood was harvested from 6–12 wk old WT and KO mice by cardiac puncture. Briefly, mice were anesthetized with Avertin (250mg/kg), prepared from 2,2,2 tribromoethanal and 2-methyl-2-butanol (Sigma, St. Louis, MO), by intraperitoneal injection. Blood was drawn using EDTA coated 26G needles. Plasma was removed from blood samples after centrifugation at 10,000 rpm for 10minutes at 4°C. Cytokine concentrations were measured in plasma samples by ELISA using Quantikine kits, according to the manufacturer’s instructions, for IL-1β, TNFα, IL-6, IFNγ, GM-CSF and G-CSF (R&D Systems, Minneapolis, MN).
RNA isolation, cDNA synthesis and real-time PCR reactions
Bone marrow, livers and spleens were harvested from three 6–8 week old WT and KO mice. Bone marrow and spleen samples were harvested as previously described. Livers were harvested and mashed through a 40μm filter (BD Biosciences, San Jose, CA) in PBS 2.5% FBS. The resulting cell suspensions were washed once with PBS and lysed with Qiazol reagent (Qiagen, Valencia, CA). RNA was harvested using the Qiagen miRNeasy kit (Qiagen, Valencia, CA), and cDNA was made using the Invitrogen SSRTIII kit (Invitrogen, Carlsbad, CA), according to the manufacturers instructions. We quantified relative mRNA abundance using Applied Biosystems (Life Technologies, Carlsbad, CA) TaqMan primers for IL-1β (Mm01336189_m1*), IL-6 (Mm00446190_m1*), TNFα (Mm00443258_m1*), G-CSF (Mm00438334_m1*) and IFNγ (Mm01168134_m1*) using GAPDH (Mouse GAPD positive control) as a control. Real-time PCR reactions were run on an Applied Biosystems 7900HT Real-time PCR System using the Applied Biosystems (Life Technologies, Carlsbad, CA) TaqMan Universal PCR Master Mix. Relative abundance was calculated from 3 biological and 2 technical replicates for each organ from WT and KO mice using the ΔΔCt method.
For subset isolation, bone marrow and splenocytes were harvested as described, stained with monoclonal antibodies against T cells, B cells, erythroid progenitors, monocytes and granulocytes, then sorted using a BD FACSAria (BD Biosciences, San Jose, CA). RNA was isolated and cDNA synthesized as described. Real-time PCR for TTP expression was conducted using TTP specific primers (F: TCTCTTCACCAAGGCCATTC, R: GAGAGGAGGTGGTGGGAGTT; IDT, Coralville, IA) and HMBS (F: GTGTTGCACGATCCTGAAACT, R: GTTGCCCATCCTTTATCACTGTA; IDT, Coralville, IA) as a control using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA). Relative expression was determined using the ΔΔCt method with bone marrow mononuclear cells (BMMC) as a reference.
Results
We observed granulocyte hyperplasia in the BM and blood of every adult KO mouse assayed (Fig. 1A and B). In order to fully characterize hematopoiesis in TTP KO mice, we quantified the frequencies and total numbers of mature hematopoietic cells and HSPCs in their BMs and spleens compared to WT littermates. There was no significant difference in BM or spleen cellularity. Overall, the distribution of mature cells in the BMs and spleens were consistent with the reported granulocyte hyperplasia phenotype(11); there was a 2.1-fold increase in the number of Gr1+CD11b+ granulocytes and a 1.6-fold decrease in B220+ B cells in the BMs (Fig. 1B). In spleens, the granulocyte hyperplasia was also evident, but there was only a minor decrease in B cells (Fig. 1C and D). These data are consistent with the description of BM B cell progenitor egress during reactive granulopoiesis(3).
Figure 1. TTP deletion caused a completely penetrant granulocyte hyperplasia in the bone marrow and the spleen.

A Representative FACS plots of mature populations in WT and KO bone marrow mononuclear cells gated on viable cells by forward scatter and side scatter (FSC/SSC). B Frequencies and absolute numbers of each cell population in the bone marrow. C Representative FACS plots of mature populations in WT and KO splenocytes gated on viable cells by FSC/SSC. D Frequencies and absolute numbers of each population in the spleen. Statistical analysis by unpaired t-test on n=3 WT and n=3 KO; * p<0.05, ** p<0.01 and *** p<0.005.
When we examined the lympho-hematopoietic progenitor compartment, we found results consistent with the observed mature cell phenotype. We used established(12, 13) immunophenotypic definitions for common myeloid progenitors (CMP: Lin−Kit+Sca−CD34midFcRgmid), megakaryocyte-erythroid progenitors (MEP: Lin−Kit+Sca−CD34loFcRglo), granulocyte-monocyte progenitors (GMP: Lin−Kit+Sca−CD34hiFcRghi) and common lymphoid progenitors (CLP: Lin−KitmidScamidIL-7Ra+), as well as colony-forming assays to quantify functional progenitors. While the frequencies and numbers of MEPs and CMPs were not different in BMs, the number of GMPs was 1.9-fold higher in TTP KO mice as compared to WT littermates (Fig. 2A and B). In addition, there were more granulocyte colony-forming cells (CFU-G) in the BMs and spleens of TTP KO mice (Fig. 2C and D), consistent with previous characterizations of TTP KO mice(11). We observed no significant change in CLPs (Fig. 2A and B).
Figure 2. Granulocyte hyperplasia was evident in TTP KO myeloid committed progenitors.

A Representative FACS plots of progenitor populations in WT and KO bone marrow mononuclear cells gated on viable cells by FSC/SSC. Sca/Kit plots were gated on viable and Lineage− cells. B Frequencies and absolute numbers of each cell population. CLPs were further gated on IL-7Rα expression. C CFC counts of day 7 colonies from bone marrow and D splenocytes. Statistical analysis by unpaired t-test on n=3 WT and n=3 KO; * p<0.05, ** p<0.01 and *** p<0.005.
An examination of the more primitive HSPC compartment of adult TTP KO vs WT mice revealed, on average, 2.9-fold greater numbers of Lin−Kit+Sca+ (KSLs), a population known to be enriched in HSCs(14) (Fig. 3A and B). The KSL compartment was further sub-divided immnunophenotypically to identify long-term HSCs (LT-HSC: Lin−Kit+Sca+CD34loFlt3−(15) or Lin−Kit+Sca+CD150+CD48−(16)), short-term HSCs (ST-HSC: Lin−Kit+Sca+CD34+Flt3−(15)) and multipotent progenitors (MPP: Lin−Kit+Sca+CD34+Flt3+(17) or Lin−Kit+Sca+CD150−CD48+(18)), using CD34 and Flt3 or SLAM markers based on previous functional characterizations of these populations. In the KSL population, there were increased numbers of immunophenotypic ST-HSCs and MPPs, in TTP KO BMs, using either of the above two cell surface marker definitions of cell subsets (Fig. 3A and B).
Figure 3. TTP deletion resulted in an increase in multipotent progenitor populations but not LT-HSCs.

A Representative FACS plots of primitive HSPC populations in WT and KO bone marrow mononuclear cells. Sca/Kit plots were gated on viable and Lineage− cells. CD34/Flt3 and CD150/CD48 plots were gated on the KSL population. B Frequencies and absolute numbers of each cell population. Statistical analysis by unpaired t-test on n=3 WT and n=3 KO; * p<0.05, ** p<0.01 and *** p<0.005.
Using CD34 and SLAM markers to immunophenotypically identify the population of rare LT-HSCs yielded divergent results. Use of the SLAM markers revealed no difference, while use of CD34/Flt3 markers showed a 3-fold increase in the numbers of LT-HSCs (Fig. 3B). Assessment of the frequency of engrafting HSCs by limiting dilution transplant measured nearly identical frequencies in WT and TTP KO BMs (Fig. 4A), and at each cell dose, the donor chimerism of WT or KO cells was not significantly different between mice transplanted with WT or TTP KO BM. Interestingly, there was no evidence of granulocyte hyperplasia when we analyzed multilineage chimerism in recipients of TTP KO BM (Fig. 4B), suggesting a non-cell autonomous mechanism.
Figure 4. TTP KO multipotent progenitor populations were more actively cycling while LT-HSCs remained quiescent.

A Frequency of engrafting cells as calculated by limiting dilution transplant experiments. Table shows the number of mice that engrafted with WT or KO (CD45.2+) cells at each dose 16 weeks post-transplant. The data are from the combination of two transplant experiments. B Peripheral blood chimerism of mice who received a 1:1 dose of Test:Recipient cells. C Representative FACS plots of cell cycle analysis by icKi67/7-AAD staining. Plots are gated on the primitive population shown. D Frequency of cells in each population in the S-G2-M phase of the cell cycle based on the presence of intracellular Ki67 protein and replicated DNA (icKi67+7-AAD4n). E Representative FACS plots of BrdU incorporation by the primitive cell types shown over a 16hr period in vivo. F Frequencies of BrdU+ cells in each primitive cell population. Statistical analysis by unpaired t-test on n=3 WT and n=3 KO; * p<0.05 and ** p<0.01. Cell cycle and BrdU incorporation experiments were repeated 3 times.
In order to ascertain if the increase in HSPC numbers in TTP KO mice was associated with increased cell cycling, we performed two different flow cytometric cell cycle analyses. By measuring either BrdU uptake or Ki67 expression, we found more MPPs and ST-HSCs actively in the cell cycle in TTP KO BMs (Fig. 4C–F). There was no difference in the cell cycle status of LT-HSCs (Fig. 4C–F).
TTP is known to regulate the half-lives of multiple transcripts encoding intracellular and secreted proteins, such as transcription factors and cytokines. In order to determine whether the hematopoietic phenotypes that we observed in TTP KO mice might be due to factors whose deregulation might be expected to cause a cell-autonomous (e.g. transcription factors) or non-autonomous (e.g. cytokines) effect, we characterized hematopoiesis of WT marrow in a KO environment. Whole BM from WT CD45.1+ donors was transplanted into lethally irradiated WT (WT:WT) or KO (WT:KO) CD45.2+ littermates. At 16 weeks post transplant, 72–85% of BM cells were donor-derived (i.e. CD45.1+; Fig. 5B), with no difference between WT and KO recipients. In addition, there was no significant difference in BM cellularity between recipients of WT or KO BM (Fig. 5A). In the spleens, cells were 87–94% donor with no significant difference in cellularity (Fig. 6A and B). Strikingly, we found that the KO recipients transplanted with WT CD45.1+ cells had granulocytic hyperplasia. In the BMs of WT:KO transplant recipients, the number and frequency of Gr1+CD11b+ cells was significantly increased and the frequency of B220+ cells decreased (Fig. 5C and D). In the spleens of WT:KO transplant recipients, granulocytic hyperplasia was evident with a 3.8-fold increase in the number of Gr1+CD11b+ cells and a 3.5-fold increase in their frequency compared to WT:WT transplant recipient spleens (Fig. 6C and D). There was also a significant increase in the number of CFU-G in the bone marrows and spleens of WT:KO transplanted chimeras (Figs. 5E and 6E).
Figure 5. Reactive granulopoiesis was recapitulated in reverse chimeric bone marrow.
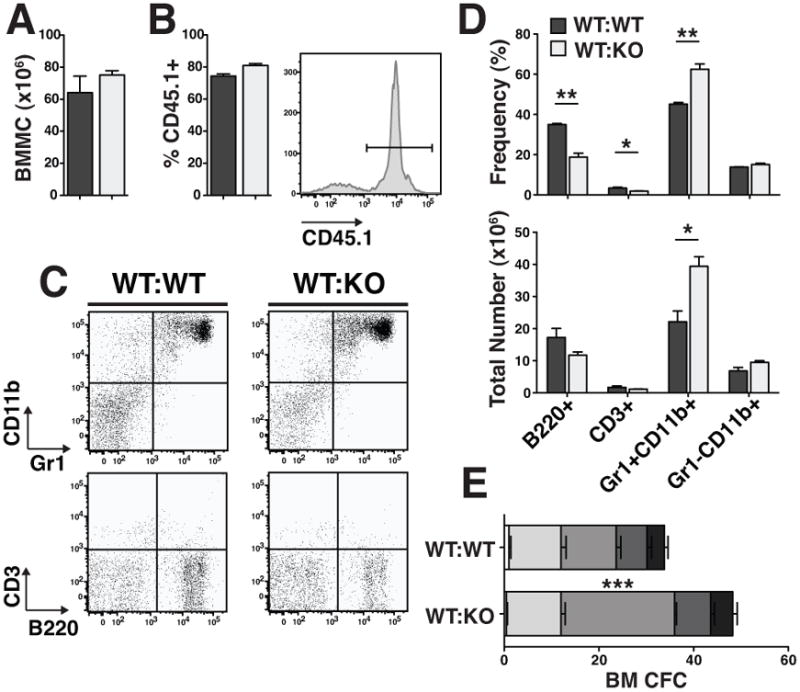
A Total number of bone marrow mononuclear cells in reverse chimeric mice 16 weeks post-transplant. B Chimerism of WT CD45.1+ cells in WT and KO recipient bone marrow. Representative histogram of CD45.1+ cells in the bone marrow of a reverse chimeric mouse. C Representative FACS plots of mature populations gated on viable, CD45.1+ cells. D Frequencies and absolute numbers of cells in each population. E CFC counts of day 7 colonies from whole bone marrow. Statistical analysis by unpaired t-test on n=3 WT:WT and n=4 WT:KO; * p<0.05, ** p<0.01 and *** p<0.005.
Figure 6. Reactive granulopoiesis was recapitulated in reverse chimeric spleens.
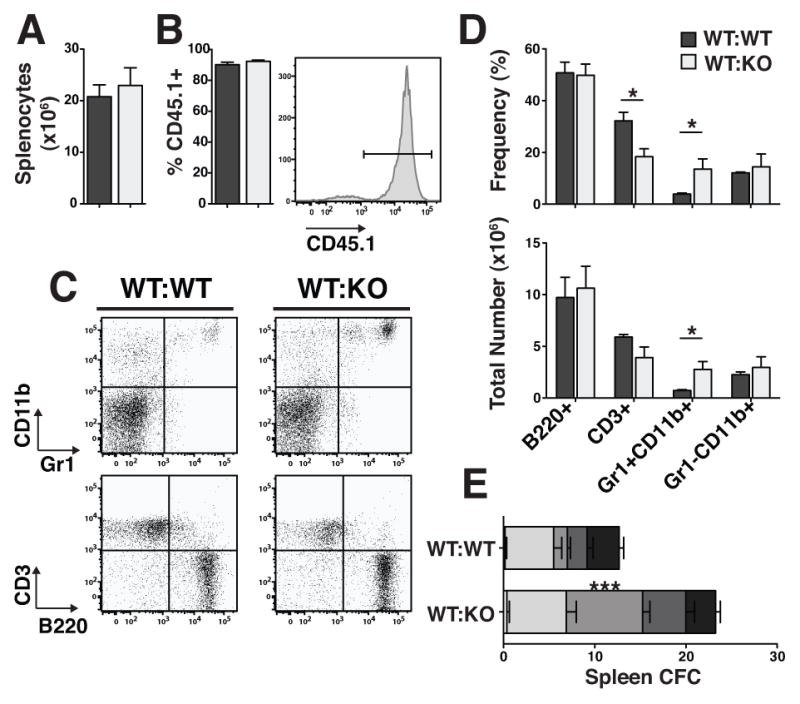
A Total number of splenocytes in reverse chimeric mice 16 weeks post-transplant. B Chimerism of WT CD45.1+ cells in WT and KO recipient spleens. Representative histogram of CD45.1+ cells in the spleens of a reverse chimeric mouse. C Representative FACS plots of mature populations gated on viable, CD45.1+ cells. D Frequencies and absolute numbers of cells in each population. E CFC counts of day 7 colonies from spleens. Statistical analysis by unpaired t-test on n=3 WT:WT and n=4 WT:KO; * p<0.05, ** p<0.01 and *** p<0.005.
In the KSL compartment of BM from WT:KO transplant recipients, we observed the same primitive progenitor expansion phenotype as in KO mice (Fig. 7A and B). In WT:KO transplant recipient marrows, there was a 2.5-fold increase in the number of KSL cells compared to WT:WT transplant chimeras (Fig. 7B). Confirming the immunophenotypic HSPC quantitations in KO mice (Fig. 3A and B), the number of MPPs in WT:KO transplant recipient marrow was increased by 2.7-fold or 2.6-fold using the CD34/Flt3 or SLAM marker phenotypes, respectively (Fig. 7B). Using the KSLCD34lowFlt3− or KSL/SLAM marker conventions to identify a population highly enriched for LT-HSCs in chimeras yielded the same difference that was observed between WT and KO BM (Fig. 7A and B).
Figure 7. Reactive granulopoiesis was recapitulated in the primitive HSPC compartment in reverse chimeric bone marrow.

A Representative FACS plots of primitive HSPC subsets in reverse chimeric bone marrow. Sca/Kit plots are gated on viable, CD45.1+ Lineage− cells. B Frequencies and absolute numbers of primitive HSPC populations. Statistical analysis by unpaired t-test on n=3 WT:WT and n=4 WT:KO; * p<0.05.
Having determined that the hematopoietic phenotype in TTP KO mice is non-cell autonomous, we set out to quantitate the levels of cytokines by ELISA in the plasma of WT and KO littermates. We selected a panel of known granulopoietic cytokines that are also documented TTP targets: IL-1β(19, 20), TNFα(10), IL-6(21, 22), GM-CSF(23) and IFNγ(24) (Fig. 8A). IFNγ and GM-CSF were not detectable in any WT or KO mice assayed. We measured mean IL-1β levels of 16.9pg/mL in KO plasma while IL-1β was undetectable in WT plasma. We also measured elevated levels of plasma IL-6 and TNFa in KOs (20.1 and 16.7pg/mL, respectively) compared to WT (1.0 and 1.5pg/mL, respectively).
Figure 8. Expression of granulopoietic TTP targets was elevated in TTP KO plasma as a result of increased mRNA expression in KO liver.

A Plasma cytokine concentrations as measured by ELISA. Each point represents a plasma sample from a single mouse, horizontal lines represent the mean value. Statistical analysis by two-way ANOVA; * p<0.05 and ** p<0.01. Relative abundance of cytokine mRNAs quantified by real-time PCR in B Bone Marrow, C Spleen and D Liver. Statistical analysis by unpaired t-test on n=3 WT:WT and n=4 WT:KO; * p<0.05, ** p<0.01 and *** p<0.005.
G-CSF is well known to be responsible for maintaining homeostatic levels of granulocytes(25). In addition, G-CSF administration results in a bolus of neutrophil production(4) as well as increased HSC cell cycle entry(26). The mouse G-CSF transcript is not a TTP target, nor does its 3′UTR contain any sequences resembling an ARE. Nevertheless, G-CSF levels were significantly increased in the plasma of KO as compared to WT mice (Fig. 8A; 335.7 and 55.0 pg/mL, respectively).
IL-1β, TNFα and IL-6 have been shown to be overexpressed in the livers of mice transgenic for an active form of NFκB, modeling a syndrome observed in obese animals(27). In addition, TTP is highly expressed in the liver(28). We hypothesized that the livers of TTP KO mice could act as a non-hematopoietic source of these three cytokines, which we observed to be overrepresented in KO plasma (Fig 8A). Therefore, we quantified the relative abundance of the mRNAs encoding these granulopoietic transcripts, including G-CSF, in hematopoietic tissues (BM and spleen) as well as the livers of WT and KO mice. We found no difference in the expression of any of these cytokines in the BM (Fig. 8C). In the spleen, the only difference was a 2.3-fold increase in the abundance of IL-1β mRNA in TTP KOs (Fig. 8D). Consistent with the non-cell autonomous nature of the phenotype, we identified the greatest difference in cytokine expression in the livers of TTP KO mice, where IL-1β, TNFα, IL-6 and G-CSF transcripts were all elevated (Fig. 8E). These data, showing non-hematopoietic expression of the cytokines assayed, correlate well with our reverse chimera results and identify liver as a source organ for the altered cytokine profile in the plasma of TTP KO mice.
Discussion
Deletion of TTP causes a spontaneous, non-cell autonomous granulocyte hyperplasia that we observed in every adult KO mouse assayed; these results are consistent with previously published reports(11). In addition, we extended observations of the TTP deletion phenotype by showing the involvement of uncommitted ST-HSCs and MPPs in sustaining the increased production of granulocytes. Interestingly, LT-HSCs, which remain relatively quiescent at ‘steady state,’ were not measurably affected by this stress. Every aspect of this phenotype that we observed in TTP KO mice was phenocopied by hematopoiesis generated by WT BM transplanted into KO mice, indicating that this phenotype is non-HSPC autonomous. In contrast, KO marrow developing in a chimeric, WT environment, with a 50:50 ratio of WT to KO marrow, did not transplant granulocytic hyperplasia (Fig 4B).
This phenotype strongly resembles reactive granulopoiesis(2, 4, 6), where these mature cells of the immune system respond rapidly to immunogenic insults, such as infection or adjuvant treatment. The hematopoietic system quickly reacts by producing more effector cells, in this case, granulocytes. Granulocytes are the most short-lived cell type in the immune system; a mature granulocyte has a half-life on the order of days before undergoing apoptosis(29, 30). This reactive response, as described(6), is sustained not only by committed granulocytic progenitors but also by the more primitive, uncommitted ST-HSCs and MPPs. The hematopoietic system is capable of mounting similar responses to other stresses, such as stress erythropoiesis in response to blood loss or hemolytic anemia(31), or stress thrombopoiesis in response to platelet consumption or destruction(31, 32). Whether or not HSCs and primitive progenitors play a role in other granulocytic hyperplasias and reactive hematopoietic responses remains to be investigated.
TTP regulates the stability of hundreds of transcripts, encoding a range of molecules from transcription factors like Myc(33) to cytokines like IL-10(34). TTP, an immediate early gene (IEG), is rapidly transcribed following treatment with TPA(35), serum(36) and insulin(37). TTP is rapidly transcribed in the hematopoietic system in response to a variety of stimuli, including LPS(10) treatment and T cell receptor (TCR) stimulation. TTP expression following TCR and CD28 co-stimulation regulates the robustness of the T cell response; TTP KO T cells overproduce IL-2 compared to WT T cells(38).
We propose that constitutive TTP expression moderates the basal expression of inflammatory transcripts. We found that unstimulated leukocytes, lymphocytes and progenitor cells express TTP mRNA (Supplemental Figure 1). TTP is also highly expressed in the livers of WT mice(28). The increased abundance of IL-1β, TNFα and IL-6 mRNA in the livers of TTP KO mice suggests to us that the absence of TTP in certain cell types ‘releases the brake’, allowing increased stability of inflammatory transcripts like IL-1β, TNFα and IL-6. The result is unprovoked expression of multiple inflammatory cytokines and a spontaneous overproduction of granulocytes, an effect that is normally only observed in response to infection or inflammation.
Similar observations have been made in other systems by careful transcriptomic analysis. Studies in embryonic stem cells and B cells have shown that some genes, whose expression is not detectable by microarray, contained permissive chromatin marks (H3K4me3) as well as Pol II at their promoter regions. These genes were shown to be expressed by sensitive RT-PCR assays, indicating that they are expressed, but at low levels(39). It would be interesting to extend these observations to the level of mRNA stability, by examining putative AREs in these transcripts.
Our ‘lack of homeostatic repression’ model is consistent with previous reports in TTP KO mice. The original description of TTP KO mice implicated TNFα mRNA de-repression because KO mice treated with TNF neutralizing antibodies had reduced clinical signs(11). In addition, TTP KO dendritic cells (DCs) have been shown to express increased levels of IL-1β transcript even without stimulation(19). We have extended these results to show that TTP KO mice express higher plasma levels of both these cytokines than their WT littermates. In addition, we have also pinpointed liver as an organ responsible for the production of these cytokines. The liver was an attractive non-hematopoietic organ source given the known expression of TTP in the liver(28) and the observation that liver can be a source of IL-1β, TNFα and IL-6 in a transgenic model of chronic inflammation(27).
The elevated levels of G-CSF in the plasma of TTP KO mice were surprising. G-CSF plays an important role in promoting neutrophil production during homeostasis; G-CSF KO mice have lower neutrophil counts at steady state as compared to WT mice(25). The importance of G-CSF in reactive neutrophil production appears to be context dependent. While G-CSF levels increase rapidly in WT mice challenged with either the fungus C.albicans(5) or the bacterium P. aeruginosa.(40), the neutrophil bolus in response to infection by these two agents in G-CSF KO mice is normal in the case of C. albicans(5) but severely attenuated in response to P. aeruginosa(40).
The cytokines IL-1β, IL-6 and TNFα are each capable of inducing neutrophilia(3, 41–44). Therefore, the spontaneous phenotype we have described in TTP KO mice appears to be the result of a complex cytokine milieu. Plasma concentrations of TNFα, IL-6 and GM-CSF were quantified from the same plasma samples, which allowed us to compare the levels of these cytokines in the same mice (Supplemental Figure 2). While the majority of KO mice displayed elevated levels of both IL-6 and TNFα, two mice, designated KO 1 and KO 2, displayed elevated levels of IL-6 but not TNFα. Conversely, KO 5 displayed elevated TNFα but not IL-6. These data suggest a mechanism where the granulocyte hyperplasia in TTP KO mice is due to the total effects of IL-1β, IL-6 and/or TNFα.
G-CSF is not a known TTP target, nor does its 3′UTR contain any ARE sequences. Therefore, the elevated levels of G-CSF message and protein that we measured in TTP KO mice are most likely not a direct effect of TTP deletion. In the recent description of the requirement of IL-1β in reactive granulopoiesis(6), the data indicated the existence of an intermediate messenger between IL-1β and the response to adjuvant treatments. Since this publication, Dr. Kelsoe’s group has discovered a direct link between IL-1 signaling and G-CSF expression in reactive granulopoiesis (personal communication from their submitted manuscript). Taken together, these results and our new findings strongly suggest that IL-1β, and potentially IL-6 and TNFα, regulate neutrophil production by regulating the expression of G-CSF.
We find evidence of the elegance of the hematopoietic system in that TTP KO environment has no detectable effect on LT-HSCs. Increasing the numbers of short-lived granulocytes is a major burden for the hematopoietic system to accomplish. Still, the earliest cells that respond to the abundance of inflammatory cytokines in the TTP KO mice are the ST-HSCs and MPPs. However, the LT-HSCs, which must remain quiescent in order to ensure that blood production can occur for the animals’ lifetime, are not detectably affected. Recent studies have shown that IFNα(45) and IFNγ(46), both potent inflammatory molecules, cause HSCs to enter the cell cycle. Although we have not tested the levels of IFNα in our system, the IFNα transcript is not a known TTP target and we observed that the levels of IFNγ were undetectable in both WT and KOs.
Overall, these studies further emphasize the importance of RNA binding proteins in regulating the lympho-hematopoietic system. We conclude that TTP is a master regulator of cytokine expression during homeostasis. Without TTP, basal levels of inflammatory cytokines are left unchecked and their increased production stimulates committed and primitive progenitors in the BM to increase the production of granulocytes. Nevertheless, LT-HSCs are insensitive to this stress and remain in their physiologic (WT) relatively quiescent state.
Supplementary Material
Acknowledgments
We would like to thank Dr. Perry J. Blackshear for providing us with TTP+/− mice, and Dr. Ferenc Livak for assistance in the Greenbaum Cancer Center Flow Cytometry Core, University of Maryland School of Medicine.
Footnotes
Grant Support: This research was supported in part by grants from the National Foundation for Cancer Research and the National Institutes of Health (P01CA070970).
References
- 1.Metcalf D. The Molecular Control of Blood Cells. Harvard University Press; Cambridge, MA: 1988. [Google Scholar]
- 2.Ueda Y, Kondo M, Kelsoe G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. The Journal of Experimental Medicine. 2005;201:1771–1780. doi: 10.1084/jem.20041419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ueda Y, Yang K, Foster SJ, Kondo M, Kelsoe G. Inflammation Controls B Lymphopoiesis by Regulating Chemokine CXCL12 Expression. The Journal of Experimental Medicine. 2004;199:47–58. doi: 10.1084/jem.20031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S-i, Imanishi J, Akashi K, Tenen DG. C/EBP[beta] is required for ‘emergency’ granulopoiesis. Nat Immunol. 2006;7:732–739. doi: 10.1038/ni1354. [DOI] [PubMed] [Google Scholar]
- 5.Basu S, Hodgson G, Zhang HH, Katz M, Quilici C, Dunn AR. “Emergency” granulopoiesis in G-CSF-deficient mice in response to Candida albicans infection. Blood. 2000;95:3725–3733. [PubMed] [Google Scholar]
- 6.Ueda Y, Cain DW, Kuraoka M, Kondo M, Kelsoe G. IL-1R type I-dependent hemopoietic stem cell proliferation is necessary for inflammatory granulopoiesis and reactive neutrophilia. J Immunol. 2009;182:6477–6484. doi: 10.4049/jimmunol.0803961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaw G, Kamen R. A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell. 1986;46:659–667. doi: 10.1016/0092-8674(86)90341-7. [DOI] [PubMed] [Google Scholar]
- 8.Bakheet T, Williams BR, Khabar KS. ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Res. 2006;34:D111–114. doi: 10.1093/nar/gkj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Briata P, Ilengo C, Corte G, Moroni C, Rosenfeld MG, Chen CY, Gherzi R. The Wnt/beta-catenin-->Pitx2 pathway controls the turnover of Pitx2 and other unstable mRNAs. Mol Cell. 2003;12:1201–1211. doi: 10.1016/s1097-2765(03)00407-6. [DOI] [PubMed] [Google Scholar]
- 10.Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science. 1998;281:1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 11.Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ. A Pathogenetic Role for TNF[alpha] in the Syndrome of Cachexia, Arthritis, and Autoimmunity Resulting from Tristetraprolin (TTP) Deficiency. Immunity. 1996;4:445–454. doi: 10.1016/s1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- 12.Kondo M, I, Weissman L, Akashi K. Identification of Clonogenic Common Lymphoid Progenitors in Mouse Bone Marrow. Cell. 1997;91:661–672. doi: 10.1016/s0092-8674(00)80453-5. [DOI] [PubMed] [Google Scholar]
- 13.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 14.Osawa M Fau - Nakamura, K., N. Nakamura K Fau - Nishi, N. Nishi N Fau - Takahasi, Y. Takahasi N Fau - Tokuomoto, H. Tokuomoto Y Fau - Inoue, H. Inoue H Fau -Nakauchi, and H. Nakauchi. In vivo self-renewal of c-Kit+ Sca-1+ Lin(low/−) hemopoietic stem cells.
- 15.Yang L, Bryder D, Adolfsson J, Nygren J, Mansson R, Sigvardsson M, Jacobsen SEW. Identification of Lin-Sca1+kit+CD34+Flt3- short-term hematopoietic stem cells capable of rapidly reconstituting and rescuing myeloablated transplant recipients. Blood. 2005;105:2717–2723. doi: 10.1182/blood-2004-06-2159. [DOI] [PubMed] [Google Scholar]
- 16.Kiel MJ, Yilmaz mH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM Family Receptors Distinguish Hematopoietic Stem and Progenitor Cells and Reveal Endothelial Niches for Stem Cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 17.Christensen JL, I, Weissman L. Flk-2 is a marker in hematopoietic stem cell differentiation: A simple method to isolate long-term stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:14541–14546. doi: 10.1073/pnas.261562798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, LiÛ P, MacDonald HR, Trumpp A. Hematopoietic Stem Cells Reversibly Switch from Dormancy to Self-Renewal during Homeostasis and Repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 19.Bros M, Wiechmann N, Besche V, Art J, Pautz A, Grabbe S, Kleinert H, Reske-Kunz AB. The RNA binding protein tristetraprolin influences the activation state of murine dendritic cells. Molecular Immunology. 2010;47:1161–1170. doi: 10.1016/j.molimm.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Chen YL, Huang YL, Lin NY, Chen HC, Chiu WC, Chang CJ. Differential regulation of ARE-mediated TNF[alpha] and IL-1[beta] mRNA stability by lipopolysaccharide in RAW264.7 cells. Biochemical and Biophysical Research Communications. 2006;346:160–168. doi: 10.1016/j.bbrc.2006.05.093. [DOI] [PubMed] [Google Scholar]
- 21.Tudor C, Marchese FP, Hitti E, Aubareda A, Rawlinson L, Gaestel M, Blackshear PJ, Clark AR, Saklatvala J, Dean JL. The p38 MAPK pathway inhibits tristetraprolin-directed decay of interleukin-10 and pro-inflammatory mediator mRNAs in murine macrophages. FEBS Lett. 2009;583:1933–1938. doi: 10.1016/j.febslet.2009.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sauer I, Schaljo B, Vogl C, Gattermeier I, Kolbe T, Muller M, Blackshear PJ, Kovarik P. Interferons limit inflammatory responses by induction of tristetraprolin. Blood. 2006;107:4790–4797. doi: 10.1182/blood-2005-07-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carballo E, Lai WS, Blackshear PJ. Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood. 2000;95:1891–1899. [PubMed] [Google Scholar]
- 24.Ogilvie RL, Sternjohn JR, Rattenbacher B, Vlasova IA, Williams DA, Hau HH, Blackshear PJ, Bohjanen PR. Tristetraprolin mediates interferon-gamma mRNA decay. J Biol Chem. 2009;284:11216–11223. doi: 10.1074/jbc.M901229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, Fowler KJ, Basu S, Zhan YF, Dunn AR. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 1994;84:1737–1746. [PubMed] [Google Scholar]
- 26.Wright DE, Cheshier SH, Wagers AJ, Randall TD, Christensen JL, Weissman IL. Cyclophosphamide/granulocyte colony-stimulating factor causes selective mobilization of bone marrow hematopoietic stem cells into the blood after M phase of the cell cycle. Blood. 2001;97:2278–2285. doi: 10.1182/blood.v97.8.2278. [DOI] [PubMed] [Google Scholar]
- 27.Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu JY, Schneider RJ. Tissue distribution of AU-rich mRNA-binding proteins involved in regulation of mRNA decay. J Biol Chem. 2004;279:12974–12979. doi: 10.1074/jbc.M310433200. [DOI] [PubMed] [Google Scholar]
- 29.Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JAM, Tesselaar K, Koenderman L. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood. 2010;116:625–627. doi: 10.1182/blood-2010-01-259028. [DOI] [PubMed] [Google Scholar]
- 30.Luo HR, Loison F. Constitutive neutrophil apoptosis: mechanisms and regulation. Am J Hematol. 2008;83:288–295. doi: 10.1002/ajh.21078. [DOI] [PubMed] [Google Scholar]
- 31.Perry JM, Harandi OF, Paulson RF. BMP4, SCF, and hypoxia cooperatively regulate the expansion of murine stress erythroid progenitors. Blood. 2007;109:4494–4502. doi: 10.1182/blood-2006-04-016154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wysoczynski M, Kucia M, Ratajczak J, Ratajczak MZ. Cleavage fragments of the third complement component (C3) enhance stromal derived factor-1 (SDF-1)-mediated platelet production during reactive postbleeding thrombocytosis. Leukemia. 2007;21:973–982. doi: 10.1038/sj.leu.2404629. [DOI] [PubMed] [Google Scholar]
- 33.Marderosian M, Sharma A, Funk AP, Vartanian R, Masri J, Jo OD, Gera JF. Tristetraprolin regulates Cyclin D1 and c-Myc mRNA stability in response to rapamycin in an Akt-dependent manner via p38 MAPK signaling. Oncogene. 2006;25:6277–6290. doi: 10.1038/sj.onc.1209645. [DOI] [PubMed] [Google Scholar]
- 34.Stoecklin G, Tenenbaum SA, Mayo T, Chittur SV, George AD, Baroni TE, Blackshear PJ, Anderson P. Genome-wide analysis identifies interleukin-10 mRNA as target of tristetraprolin. J Biol Chem. 2008;283:11689–11699. doi: 10.1074/jbc.M709657200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Varnum BC, Lim RW, Sukhatme VP, Herschman HR. Nucleotide sequence of a cDNA encoding TIS11, a message induced in Swiss 3T3 cells by the tumor promoter tetradecanoyl phorbol acetate. Oncogene. 1989;4:119–120. [PubMed] [Google Scholar]
- 36.DuBois RN, McLane MW, Ryder K, Lau LF, Nathans D. A growth factor-inducible nuclear protein with a novel cysteine/histidine repetitive sequence. J Biol Chem. 1990;265:19185–19191. [PubMed] [Google Scholar]
- 37.Lai WS, Stumpo DJ, Blackshear PJ. Rapid insulin-stimulated accumulation of an mRNA encoding a proline-rich protein. J Biol Chem. 1990;265:16556–16563. [PubMed] [Google Scholar]
- 38.Ogilvie RL, Abelson M, Hau HH, Vlasova I, Blackshear PJ, Bohjanen PR. Tristetraprolin down-regulates IL-2 gene expression through AU-rich element-mediated mRNA decay. J Immunol. 2005;174:953–961. doi: 10.4049/jimmunol.174.2.953. [DOI] [PubMed] [Google Scholar]
- 39.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A Chromatin Landmark and Transcription Initiation at Most Promoters in Human Cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hogue ADLA, Ferkol TW, Link DC. Regulation of systemic and local neutrophil responses by G-CSF during pulmonary Pseudomonas aeruginosa infection. Blood. 2007;109:3235–3243. doi: 10.1182/blood-2005-01-015081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ulich TR, del Castillo J, Guo K, Souza L. The hematologic effects of chronic administration of the monokines tumor necrosis factor, interleukin-1, and granulocyte-colony stimulating factor on bone marrow and circulation. Am J Pathol. 1989;134:149–159. [PMC free article] [PubMed] [Google Scholar]
- 42.Ulich TR, del Castillo J, Guo KZ. In vivo hematologic effects of recombinant interleukin-6 on hematopoiesis and circulating numbers of RBCs and WBCs. Blood. 1989;73:108–110. [PubMed] [Google Scholar]
- 43.Ulich TR, del Castillo J, Yin S. Tumor necrosis factor exerts dose-dependent effects on erythropoiesis and myelopoiesis in vivo. Exp Hematol. 1990;18:311–315. [PubMed] [Google Scholar]
- 44.Ulich TR, Guo KZ, Remick D, del Castillo J, Yin SM. Endotoxin-induced cytokine gene expression in vivo. III. IL-6 mRNA and serum protein expression and the in vivo hematologic effects of IL-6. J Immunol. 1991;146:2316–2323. [PubMed] [Google Scholar]
- 45.Essers MAG, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A. IFN[agr] activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 46.Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465:793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.