

Abstract
The interaction between a chemical and a cell may strongly depend on whether this cell is normal or pathological. Side effects of anticancer drugs may sometimes overcome their benefit action, so it is important to investigate their effect in both the target and normal cells. Capecitabine (Xeloda, CAP), a prodrug of 5-fluorouracil, is mainly used in colon cancer, but little is known about its action in head and neck cancer. We compared the cyto- and genotoxicity of CAP in head and neck HTB-43 cells and normal human lymphocytes by comet assay and flow cytometry. CAP at concentration up to 50 μM significantly decreased the viability of the cancer cells, whereas it did not affect normal lymphocytes. The drug did not interact with isolated plasmid DNA, but it damaged DNA in both cancer and normal cells. However, the extent of the damage in the former was much higher than in the latter. CAP induced apoptosis in the cancer cells, but not in normal lymphocytes. Pre-treatment of the cells with the nitrone spin traps α-(4-pyridil-1-oxide)-N-tert-butylnitrone and N-tert-butyl-α-phenylnitrone decreased the extent of CAP induced DNA damage, suggesting that free radicals may be involved in the formation of DNA lesions induced by CAP. The drug evoked an increase in the G0/G1 cell population accompanied by a decrease in the S cell population. CAP may evoke a pronounced cyto- and genotoxic effects in head and neck cancer cells, whereas it may or may not induce such effects in normal cells to far lesser extent.
Keywords: Capecitabine, Head and neck cancer, DNA damage, DNA repair, Apoptosis, Cell cycle
Introduction
Chemotherapy in head and neck cancer is a multi-stages process, in which 5-fluorouracil (5-FU), a fluorinated analog of uracil, plays a pivotal role [1, 2]. This drug displays activity in various regimes of therapy of squamous cell carcinoma of the head and neck (HNSCC) and its combination with cisplatin and radiation improves the efficacy of the treatment. After entering the cell, 5-FU is metabolized in a rather complex way, including at least four pathways, one of which gives rise to 5-FUTP [3]. Because the atom of fluorine is similar in size to hydrogen in UTP, RNA polymerases include 5-FU in all classes of RNA. 5-FU may also produce 5-FdUMP, which is an inhibitor of thymidylate synthase (TS), involved in the synthesis of DNA. TS inhibiting results in blocking of formation of dTMP and, therefore, decreasing the availability of dTTP for DNA replication and repair. Cytotoxic effect of 5-FU can also be mediated by its incorporation into DNA and changes in some membrane function in cancer cells treated with the drug [4]. The use of 5-FU has several limitations. The activity of the drug is limited by its rapid degradation by the cytosolic enzyme dihydropyrimidine dehydrogenase, the first enzyme in the catabolic route of 5-FU [3]. Because this enzyme displays a considerable variation between tissues within one subject and among different subjects, the bioavailability of 5-FU can be unpredictable [5]. Although 5-FU proved to be effective in many chemotherapy regimes in several cancer types, it is not free from adverse side effects, including fever, febrile neutropenia, anorexia, nausea, vomiting, diarrhea, stomatitis, alopecia, fatigue, hand-foot syndrome [6].
To overcome some limitations in therapy with 5-FU, its oral prodrugs have been synthesized. Doxifluridine (5′-deoxy-5-fluoridine, 5′-DFUR) is a prodrug of 5-FU and its carbamate derivative is capecitabine (Xeloda, N4-pentyloxycarbonyl-5′-deoxy-5-fluorocytidine, CAP, Fig. 1) that is absorbed through intestine in the prodrug form and it is metabolized to 5-FU in three activation stages. The final stage require thymidine phosphorylase, an enzyme present in tumor cells in much higher concentration than in normal cells, which results in almost threefold higher concentration of 5-FU in malignant compared with non-malignant tissues [7]. CAP was used with success in the treatment of colorectal cancers [8]. Recently, an anti-angiogenic potential of CAP in human colorectal cancer xenograft was reported [9].
Fig. 1.
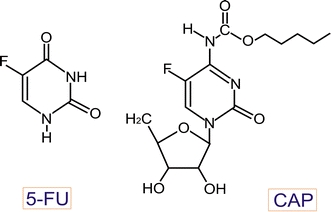
Structure of 5-fluorouracil (5-FU) and CAP
Because major chemotherapeutic treatment in head and neck cancer is based on 5-FU and CAP may improve the anticancer action of 5-FU, it was rational to consider it in head and neck cancer therapy [10]. The drug has been used in HNSCC chemotherapy singly or in a combination with other drugs: cisplatin, oxaliplatin, vorinostat, a histone deacetylase inhibitor, docetaxel, paclitaxel and several other agents [11–16]. It was successfully applied in the therapy of nasopharyngeal cancer as a single agent and its monotherapy is postulated to be considered in patients with recurrent/metastatic disease [17].
The results of several studies indicate that CAP can induce several side effects, among them hematological syndromes—neutropenia, anemia and thrombocytopenia seem to be most serious [18]. Besides hematological disturbances several other effects, including diarrhea, hand-foot syndrome, hypotension, abdominal pain, and asthenia have been reported [19]. Therefore, in the light of the research performed so far, blood toxicity seems to be a major side effect of CAP. Several other effects not-related to blood may also be associated with anticancer therapy with CAP. However, little is know about the interaction of the drug with DNA of normal cells, but this issue should be addressed, because genotoxic effect of an anticancer drug in normal cells may lead to genomic instability, mutations and induction of secondary malignances, which can be considered as a major side effect of chemotherapy.
In the present work we investigated cyto- and genotoxic action of CAP in HNSCC HTB-43 cells and human lymphocytes obtained from healthy individuals. We measured cell viability, DNA damage and repair, cell cycle and apoptosis in these cells exposed to CAP.
Materials and methods
Chemicals
Capecitabine was a gift of Roche Pharmaceuticals (Basel, Switzerland). QIAamp DNA Blood Mini Kit for isolation of high-molecular-weight DNA was obtained from QIAGEN (Chatsworth, CA, USA). Plasmid DNA purification kit was provided by EURx (Gdansk, Poland). Cell viability, apoptosis and cell cycle kits were purchased in BD Biosciences (San Jose, CA, USA). Gradisol, low melting point (LMP) and normal melting point (NMP) agarose, phosphate buffered saline (PBS), 4′, 6-diamidino-2-phenylindole (DAPI), dimethyl sulfoxide (DMSO), fetal bovine serum (FBS), MTT, lectin, penicillin, streptomycin, α-(4-pyridil-1-oxide)-N-tert-butylnitrone (POBN), N-tert-butyl-α-phenylnitrone (PBN), Bradford reagent were purchased from Sigma (St. Louis, MO, USA). ApoAlert Caspase Colorimetric Assay Kit was bought from Clontech Laboratories Inc (Palo Alto, CA, USA). All other chemicals were of the highest commercial grade available.
Cells
Human lymphocytes were isolated from peripheral blood obtained from healthy, sex- and age matched donors by centrifugation in a density gradient of Gradisol L (15 min, 280×g, 4°C). The cell viability was measured by trypan blue exclusion staining and was found to be about 99%. Human larynx cancer cell line HTB-43 was purchased from the ATCC (Rockville, MD, USA). The cells were grown in EMEM medium with 10% FBS, 100 IU/ml penicillin and 100 g/ml streptomycin in a 5% CO2 atmosphere at 37°C for 3–4 days. Just before use, they were trypsinized and re-suspended in EMEM to a final concentration of 1–2 × 105 cells/ml.
Escherichia coli cells, strain DH5α cells with pUC19 plasmid, were grown in a LB broth at 37°C overnight.
Cell treatment
CPA at final concentrations from 0.5 to 500 μM was added to the cells in EMEM from its 50 mM ethanolic solution. The final concentration of ethanol in the samples did not exceed 0.3%, which did not influence processes under study (results not shown). The control cells received only the growth medium. To examine DNA damage and cell viability, the cells were incubated with the drug for 1 h at 37°C. Each experiment included a positive control, which was hydrogen peroxide at 20 μM for 15 min on ice. H2O2 produced pronounced DNA damage, which resulted in tail DNA of 30–40%.
Cell viability
The BD Cell Viability Kit was used. The kit contains thiazole orange (TO) solution to stain all cells and propidium iodide (PI) to stain dead cells. Aliquots of 2.0 μl of TO and 1.0 μl of PI were added to 1 ml of cell suspension containing 5 × 105 cells, producing the final staining concentrations of 84 nM and 4.3 μM, respectively. The mixture was gently shaken and incubated for 5 min in room temperature to allow both dyes to enter the cells. The samples were acquired on a LSRII (Becton Dickinson, San Jose, USA) flow cytometer equipped with 488 nm laser excitation and BD FACSDiva software v 4.1.2. 5 × 104 cells were analyzed in each experiment and each experiment was repeated in triplicate. In this and all next experiments the samples were coded prior to measurements, so the investigator did not know the exposure mode for any particular sample.
Plasmid relaxation assay
pUC19 plasmids were isolated from DH5α Escherichia coli cells with Genematrix Plasmid Miniprep DNA Purification Kit (EURx, Gdansk, Poland) according to the manufacturer instruction. Plasmids were exposed to UV irradiation at 35 J/m2 (positive control) to check the migration of its multimeric forms (supercoiled, nicked circular and linear). UV irradiation induced strand breaks of DNA and caused the relaxation of supercoiled plasmid—one break is enough to relax one molecule of the plasmid. Structural differences between supercoiled, nicked circular and linear forms of the plasmid accounted for their different electrophoretic mobility. Plasmid samples at 250 ng/μl were subjected to 1% agarose gel electrophoresis carried out in Tris–Borate–EDTA (TBE) buffer. The gel was stained with ethidium bromide (0.5 mg/ml) and the plasmid DNA was visualized under ultraviolet light (302 nm), scanned by a CCD camera and densitometry analysis was performed with the GeneTools by Syngene (Cambridge, UK) software. UV irradiation was performed at 4°C with UVC-6-12 lamp (NeoLab, Heidelberg, Germany) emitting UV light at 254 nm at dose rate of 0.12 J m−2 s−1. The ability of CAP to damage DNA was quantified by calculating the ratio of open circular DNA to the total amount of DNA (R). The values for supercoiled DNA were multiplied by 1.66 to correct for the decreased intercalating ability of ethidium bromide [20].
Comet assay
The comet assay was performed under alkaline conditions essentially according to the procedure of Singh et al. with modifications as described previously [21–23]. A freshly prepared suspension of cells in 0.75% LMP agarose dissolved in PBS was spread onto microscope slides precoated with 0.5% NMP agarose. The cells were then lysed for 1 h at 4°C in a buffer consisting of 2.5 M NaCl, 100 mM EDTA, 1% Triton X-100, 10 mM Tris, pH 10. After lysis, the slides were placed in an electrophoresis units, the DNA was allowed to unwind for 40 min in the electrophoretic solution consisting of 300 mM NaOH, 1 mM EDTA, pH > 13. Electrophoresis was conducted at 4°C (the temperature of the running buffer did not exceeded 12°C) for 20 min at an electric field strength of 0.73 V/cm (29 mA). The slides were then neutralized with 0.4 M Tris, pH 7.5, stained with 2 μg/ml of DAPI and covered with cover slips. To prevent additional DNA damage, all the steps described above were conducted under dimmed light or in the dark.
In the neutral version of the comet assay, electrophoresis was run in a buffer consisting of 100 mM Tris and 300 mM sodium acetate at pH adjusted to 9.0 by glacial acetic acid [24]. Electrophoresis was conducted for 60 min, after a 20 min equilibrium period, at an electric field strength of 0.41 V/cm (50 mA) at 4°C.
The slides were examined at 200× magnification in an Eclipse fluorescence microscope (Nikon, Tokyo, Japan) attached to a COHU 4910 video camera (Cohu, Inc., San Diego, CA) equipped with a UV filter block consisting an excitation filter (359 nm) and barrier filter (461 nm) and connected to a personal computer-based image analysis system, Lucia-Comet v. 4.51 (Laboratory Imaging, Praha, Czech Republic). Fifty images were randomly selected from each sample and the comet tail DNA was measured. Two parallel tests with aliquots of the same sample of cells were performed for a total of 100 cells. Each experiment was repeated three times. Percentage of DNA in the tail (% tail DNA) was analyzed. It is positively correlated with the level of DNA breakage or/and alkali labile sites in the cell and is negatively correlated with the level of DNA crosslinks [25]. For neutral version, this quantity correlates positively with DNA double strand breaks. The mean value of the % tail DNA in a particular sample was taken as an index of DNA damage in this sample.
DNA repair enzyme treatment
To assess the role of oxidative modifications to the DNA bases in the genotoxicity of CAP, we employed two DNA repair enzymes, endonuclease III (Endo III) and formamidopyrimidine-DNA glycosylase (Fpg). After incubation with CAP and cell lysis the slides from the comet assay were washed 3× in the enzyme buffer containing 40 mM HEPES–KOH, 0.1 M KCl, 0.5 mM EDTA, 0.2 mg/ml bovine serum albumin, pH 8.0 and drained. The agarose on slides was covered with 30 μl of the enzyme buffer either with or without enzyme at 1 μg/ml, sealed with a cover glass and incubated for 30 min at 37°C [24]. The slides were processed as described in “Comet assay”. To check the ability of both enzymes to recognize the DNA oxidative damage, we exposed the lymphocytes to 10 μM hydrogen peroxide for 10 min on ice (positive control). Since enzymatic buffer induced DNA damage the values of the % DNA in tail in the buffer only were subtracted from the value for the enzyme with buffer.
Spin trap treatment
In spin trapping experiments, the incubation with CAP was carried out in the presence of POBN or PBN at a final concentration of 100 μM. Each spin trap was added to the cells 15 min prior to the CAP incubation. The cells were then processed as described previously. Each experiment included a positive control, which was hydrogen peroxide at 10 μM applied for 10 min on ice.
Apoptosis
The BD Annexin V-FITC Apoptosis Detection Kit I was used to measure apoptosis. The kit contains Annexin V conjugated to the fluorochrome FITC that has affinity to the phosphatidylserine, which is transferred through cell membrane in the earlier stages of apoptosis. PI was used to distinguish early apoptotic cells from cells undergo late apoptosis or necrosis. Cells that are viable are Annexin V-FITC and PI negative, cells that are in early apoptosis are Annexin-FITC positive and PI negative, cells that are in late apoptosis are both Annexin-FITC and PI positive, cells already dead are only PI positive. After 6 h of incubation with UDMA cells were washed in cold PBS and resuspended in 1× binding buffer at 106 cells/ml. Five microliter of Annexin V-FITC and 5 μl of PI were added to an aliquot of 100 μl (105 cells) of cells suspension, gently mixed by pipetting and incubated for 30 min at room temperature in the dark. Next, 400 μl of 1× binding buffer was added to each tube and samples were analyzed by flow cytometry. Each experiment had a negative, positive and unstained control samples. About 10,000 events were counted per sample. The apoptosis ratio was calculated as a percent of apoptotic cells in a sample.
Cell cycle
In the cell cycle experiment, we replaced lymphocytes with highly proliferating human cancer cells. The Cycle TEST PLUS DNA Reagent Kit was used to determine the DNA index (DI) and cell cycle phase distributions. Nuclei were isolated, stained with PI and afterward analyzed on the LSRII flow cytometer according to the manufacturer instruction. The DI was calculated by dividing the mean of the relative content of the exposed G0/G1 population by the mean of the control G0/G1 population. Results were analyzed by Flow Jo software (v. 7.2.4).
DNA repair
To examine DNA repair, the cells after a 1 h pre-treatment with 10 or 50 μM CAP at 37°C were washed and re-suspended in a fresh, CAP-free RPMI 1640 medium preheated to 37°C. Aliquots of the suspension were taken immediately and 60 and 120 min later. Placing the samples in an ice bath stopped the repair incubation. Cells exposed to 10 μM H2O2 served as a positive control, whereas lymphocytes with neither CAP nor H2O2 treatment were negative controls.
Data analysis
The values in this study were expressed as mean ± SEM from three experiments, i.e. the data from three experiments were pooled and the statistical parameters were calculated. Data obtained from cell viability were expressed as mean ± SD. Mann–Whitney U test was used to determine differences between samples with the abnormal distribution (Kolmogorov–Smirnov test). The differences between samples with the normal distribution were evaluated by applying the Student’s t test.
Results
Cell viability
Capecitabine at concentrations up to 500 μM did not affect the viability of normal lymphocytes (Fig. 2, open symbols). The drug evoked a dose-depended decrease in the viability of HTB-43 cancer cells (Fig. 2, closed symbols). At the highest CAP concentration, 500 μM, almost all cancer cells were dead, whereas the viability of normal lymphocytes was about 88%.
Fig. 2.
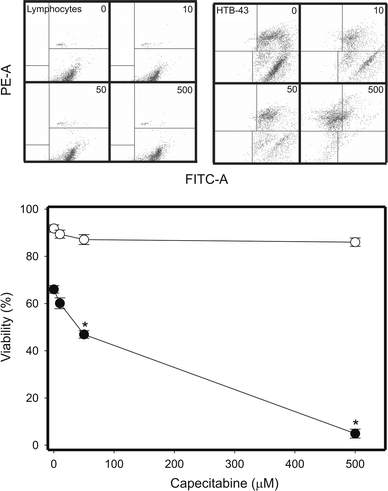
Viability of head and neck squamous cell carcinoma HTB-43 cells (closed symbols) and normal human lymphocytes (open symbols) exposed to CAP for 1 h at 37°C and measured by flow cytometry with thiazole orange and PI. Displayed is the mean of three experiments of 5 × 104 measurements each, error bars denote standard deviation, *P < 0.001. The contour diagrams above the plot show results for one representative experiment out of three for each CAP concentration
DNA damage in vitro
Capecitabine did not introduce DNA breaks to isolated DNA, as assessed by the plasmid relaxation assay, in which the ratio of the amount of open circular form of plasmid DNA to the total amount of DNA was calculated (Table 1, Fig. 3). The plasmid used in our experiment was sensitive to DNA-breaking agents, as checked by UV irradiation.
Table 1.
Ratio of open circular DNA to the total amount of DNA (R) of isolated pUC19 plasmid exposed to capecitabine
| Capecitabine (μM) | R |
|---|---|
| 0 (negative control) | 0.084 |
| 50 | 0.097 |
| 500 | 0.101 |
| UV (positive control) | 0.458* |
The values for supercoiled DNA were multiplied by 1.66 to correct for the decreased intercalating ability of ethidium bromide
* P < 0.001 as compared with negative control
Fig. 3.
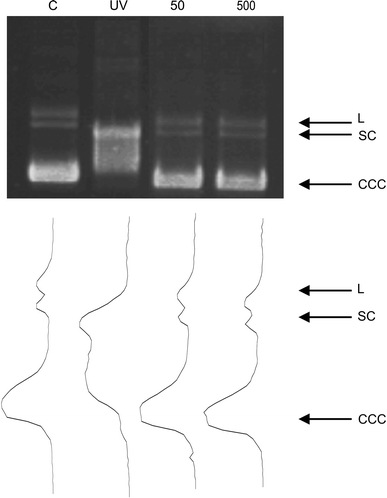
DNA damage in isolated pUC19 plasmid incubated with CAP. The picture shows three forms of the plasmid: linear (L), open circular (OC) and supercoiled (SC) exposed to CAP at indicated concentrations or 254 nm UV at dose rate of 0.12 J m−2 s−1 (positive control). The samples were electrophoresed on a 1% agarose gel, stained with ethidium bromide and visualized in UV light. Optical density of each lane is plotted above it
DNA damage in HTB-43 cells and lymphocytes
The dependence of DNA damage in human lymphocytes in the alkaline version of the comet assay on CAP concentration is displayed in Fig. 4. Single and double DNA strand breaks as well as alkali labile sites can be detected in this version of the technique. CAP evoked a dose-dependent increase in the percentage of DNA in the percentage of DNA in comet tail of HTB-43 cells and normal lymphocytes, but this increase was significantly (P < 0.05) higher in the former than in the latter. The drug did not affect the comet tail DNA in the neutral version of the comet assay (data not shown), so we concluded that it had no ability to induce this kind of DNA damage.
Fig. 4.
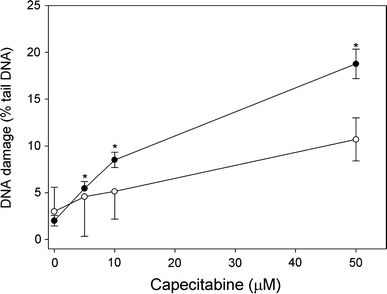
DNA damage in head and neck squamous cell carcinoma HTB-43 cells (closed symbols) and normal human lymphocytes (open symbols) exposed to CAP for 1 h at 37°C. DNA damage was measured as percentage in the tail DNA in comets of the alkaline version of the comet assay. The mean value for one hundred cells analyzed in each treatment in three independent experiments is displayed, error bars represent SEM, *P < 0.05 as compared with lymphocytes at the same concentration
DNA repair
We analyzed the kinetics of DNA repair in HTB-43 cancer cells and normal human lymphocytes by measuring the extent of DNA damage in the cells exposed to CAP at 10 or 50 μM immediately after the exposure as well as 60 and 120 min thereafter (Fig. 5). In all cases, the comet tail DNA of the control cells was constant. The cells exposed to 10 μM hydrogen peroxide (positive control) were able to recover within 45 min (results not shown). Both kinds of the cells recovered completely from DNA damage after 120 min.
Fig. 5.
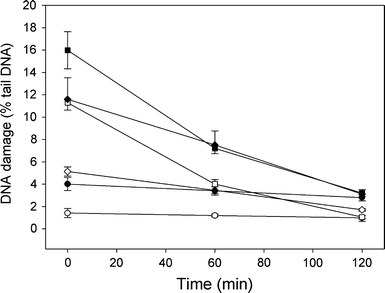
Time-course of DNA repair in HNSCC HTB-43 cells (closed symbols) and normal human lymphocytes (open symbols) exposed to CAP for 1 h at 37°C at 10 (diamonds) or 50 μM (squares). DNA damage was measured as percentage in the tail DNA in comets of the alkaline version of the comet assay. The control HTB-43 cells (closed circles) and lymphocytes (open circles) were not exposed to CAP. After the exposure, the cells were washed and incubated in a cap-free medium at 37°C. The number of cells analyzed in each time-interval was 100. The results are mean of three independent experiments. Error bars denote SEM, for both HTB-43 and lymphocytes the extent of DNA damage at 60 and 120 min was significantly (P < 0.001) lower than at 0 min
Detection of DNA damage by DNA repair enzymes
Table 2 presents the mean percentage tail DNA of human lymphocytes and HTB-43 cells exposed for 1 h at 37°C to CAP, lysed and post-treated with Endo III or Fpg, as compared with the cells without treatment with any enzyme. Both kinds of cells exposed to CAP and treated with Endo III and Fpg showed greater percentage tail DNA than those untreated with any enzyme. The difference increased with the increasing concentration of CAP. We observed also a significant difference in the percentage tail DNA of the cells post-treated and untreated with Endo III in the absence of CAP, which arisen likely from the endogenous oxidative damage to the DNA bases as well as from the oxidative damage to DNA introduced during isolation and subsequent processing of the cells.
Table 2.
DNA damage measured as percentage of DNA in tail comet in human lymphocytes and head and neck cancer HTB-43 cells exposed for 1 h at 37°C to capecitabine with subsequent treatment with Endo III or Fpg at 1 μg/ml
| Capecitabine (μM) | %Tail DNA | |||
|---|---|---|---|---|
| Lymphocytes | HTB-43 | |||
| Endo III | Fpg | Endo III | Fpg | |
| 0 | 3.2 ± 0.6 | 6.3 ± 0.8 | 4.1 ± 0.6 | 7.2 ± 0.9 |
| 5 | 7.4 ± 0.9*** | 12.9 ± 1.4*** | 10.8 ± 1.3*** | 18.3 ± 2.3*** |
| 10 | 13.2 ± 1.4*** | 19.4 ± 2.2*** | 16.4 ± 1.7*** | 22.1 ± 2.6*** |
| 50 | 16.9 ± 2.1*** | 23.7 ± 2.5*** | 24.6 ± 3.0*** | 28.3 ± 3.1*** |
| 10 μM H2O2 (positive control) | 12.4 ± 1.4*** | 16.3 ± 1.7*** | 14.2 ± 1.6*** | 19.7 ± 2.1*** |
The values given in the table are normalized to the values for cells treated with enzyme buffer only
Mean ± SE; *** P < 0.001 as compared with appropriate controls not exposed to the drug
Spin trapping
The mean percentage tail DNA of human lymphocytes and HTB-43 cells exposed to CAP at 5 and 10 μM after pre-treatment with 100 μM of the nitrone spin traps PBN and POBN is presented in Fig. 6. Both traps significantly (P < 0.001 for either CAP concentration) decreased the extent of DNA damage evoked by CAP in both kinds of cells.
Fig. 6.
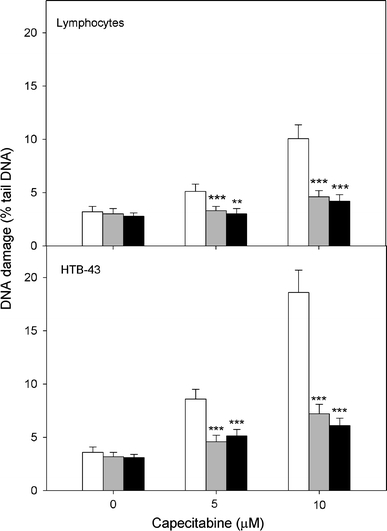
DNA damage measured as mean comet tail DNA of normal human lymphocytes and head and neck HTB-43 cancer cells incubated for 15 h at 37°C with CAP in the absence and in the presence of POBN (black bars) or PBN (grey bars) at 100 M compared with appropriate controls (empty bars). The number of cells scored in each treatment was 100. The figure shows mean results from three independent experiments. Error bars denote SEM; **P < 0.01, ***P < 0.001
Apoptosis
Capecitabine induced apoptosis in HTB-43 cells in a dose-dependent manner (Fig. 7). At highest drug concentration, 500 μM, almost all cells underwent apoptosis and even at 50 μM the fraction of apoptotic cells exceeded 80%. CAP did not induced apoptosis in normal lymphocytes. Untreated lymphocytes were primarily Annexin V-FITC and PI negative, indicating that they were viable and not undergoing apoptosis, but this was not the case for the HTB-43 cells, which significant (about 30%) fraction went apoptosis. After incubation with CAP there were basically two populations of cancer cells: Annexin V-FITC positive and PI negative (undergoing apoptosis) and Annexin V-FITC and PI positive, indicating that they were in the end stage of apoptosis or already dead.
Fig. 7.
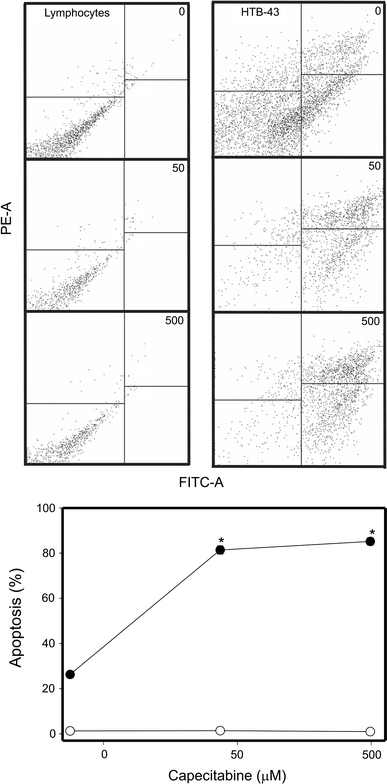
Apoptosis of HNSCC HTB-43 cells (closed symbols) and normal human lymphocytes (open symbols) exposed to CAP for 1 h at 37°C at 10 (diamonds) or 50 μM (squares). Apoptosis was assessed by flow cytometry with Annexin V-FITC/PI. Displayed is the mean of three experiments of 5 × 104 measurements each, error bars denote standard deviation. The contour diagrams above the plot show one representative experiment out of three for each CAP concentration. The lower left quadrant of each diagrams show the viable cells, which exclude PI and are negative for Annexin V-FITC binding. The upper right quadrants contain the non-viable, necrotic cells, positive for Annexin V-FITC binding and for PI uptake. The lower left quadrants represent the apoptotic cells, Annexin V-FITC positive and PI negative, demonstrating cytoplasmic membrane integrity. The apoptosis was expressed as a ratio of the number of early and late apoptotic cells to the number of cells with no measurable apoptosis
Cell cycle
In order to determine whether CAP had an influence on the progression of cell cycle, HTB-43 cells were exposed to CAP at 50 and 500 μM and to 5 M nocodazole as a positive control (Fig. 8). CAP evoked a slow and mild increase in the G0/G1 cell population accompanied by a faster decrease in the S cell population. The G2/M population was unaffected by CAP.
Fig. 8.
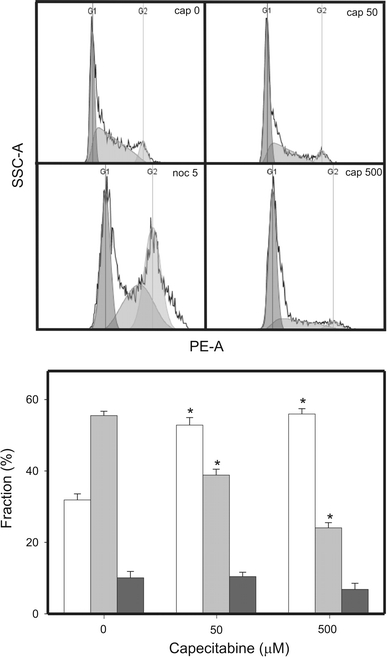
Cell cycle analysis in HTB-43 cells exposed to CAP. Fraction of cells in G0/G1 (white bars), S (light grey bars) and G2/M (dark grey bars) stage of cell cycle after treatment with CAP was presented along with histograms for each CAP concentration. Nocodazole at 5 μM was used as a positive control. Data are expressed as means of at least three independent experiments, error bars denote SD, *P < 0.001 as compared with unexposed control
Discussion
The interaction between a chemical and a cell may depend on the state of the cell, which, in turn, may be determined by the state of an organism, from which this cell was derived. This feature is very important in chemotherapy, when the effect induced by a drug in target cancer cells should be much more pronounced than in non-target normal cells. Therefore, it is important to identify mechanisms determining differential response to a chemical in various kinds of cells, including normal and cancer ones.
Capecitabine is an anticancer drug applied in metastatic colorectal cancer. The drug is also approved for use in metastatic breast cancer patients who are resistant to anthracycline- and paclitaxel-based regimens or whom further anthracycline treatment is not recommended. CAP can be also used in combination with docetaxel, when anthracycline-based chemotherapy is not efficient. It is also reported to bring benefits in patients with prostate, pancreatic, renal cell, and ovarian cancers. There are reports on potential usefulness of CAP in head and neck cancer therapy. Three aspects of anticancer drug should be taken into account, when its usefulness in therapy of a particular cancer is assessed. First, it should affectively kill cancer cells or inhibit their proliferation. Second, it should have tolerable side effects. Third, target cells should not acquire resistance to it. In the present work we explored the first two issues of this dogma in the context of cyto- and genotoxicity of CAP in cancer and normal cells. We showed that CAP effectively killed HTB-43 head and neck cancer cells, whereas it did not affect the viability of normal human lymphocytes. Moreover, CAP induced DNA damage in HTB-43 cells, in contrary to normal lymphocytes. Therefore, our results support the idea of keeping on investigating on usefulness of CAP in head and neck therapy.
The main general mode of the anti proliferative action of CAP is interfering with DNA synthesis in highly proliferating cancer cells. We showed that the drug was not able to interact with isolated DNA, which suggests that such interaction, if any, requires chemical transformation of CAP or would be mediated by the presence of other biological macromolecules, present in the cell. It was partly confirmed by the results of our next experiment, in which we showed that CAP could induce damage to DNA in head and neck cancer cells. This means that a cellular environment is needed to activate CAP for its DNA-damaging action. However, this cellular environment can be attributed to cancer cells, since no DNA damage was observed in normal cells. This is in line with the idea of the prodrug and general mechanism of CAP action, in which it requires metabolic activation by thymidine phosphorylase, present in tumor cells in much higher amount than in normal cells.
In our research CAP effectively induced alkali labile sites and/or DNA strand breaks in HTB-43 cells, as evaluated by the alkaline comet assay. However, we excluded the induction of DNA double strand breaks from the mechanism underlying genotoxicity of CAP, since the drug did not caused any increase in the percentage of DNA in tail of comet in the neutral version of the comet assay.
In general Fpg recognized more DNA damage than Endo III, which may suggest that CAP preferentially interacts with the DNA purines, but this conclusion is not very important due to rather moderate differences between the extent of DNA damage recognized by these two enzymes and moderate specificity of both enzymes. But the results obtained in this experiment directly show the presence of oxidative damage to DNA after the action of CAP.
The immediate question arises from the results obtained on DNA-damaging potential of CAP in cancer cells: whether it manifests a new mechanism of the action of CAP in cancer cells or it is a consequence of its anti-metabolite action. If we look at basic mechanisms of CAP action, at least two of them may bring DNA damage as a consequence. First, the inhibition of TS activity by CAP causes a decrease in the amount of dTTT in replication and repair, which may lead to decreased process of DNA polymerases and eventually stop the synthesis of DNA. Such incomplete synthesis would result in DNA structures with single-stranded overhangs, recognized as damaged DNA. Second, the effect of CAP may be intermediated by incorporation of 5-FU in DNA. Such modified base is recognized preferentially by MBD4, a DNA glycosylase of base excision repair pathway [26]. This enzyme harbours an apurinic/apyrimidinic lyase activity in addition to its DNA glycosylase activity. Therefore, MBD4 may create DNA single strand breaks in sites of 5-FU incorporation. Normally, these breaks are sealed by DNA polymerase α, but 5-FU may block TS activity and the polymerase may encounter difficulties in putting dTTT in the site previously occupied by 5-FU. This may result in the persistence of DNA strand breaks, which can be detected in the comet assay. Therefore, we suggest that the DNA-damaging effect can be attributed to CAP, as a consequence of its multi-pathway action. However, it should be noted, that there are reports suggesting that uracil incorporation into DNA did not predict genotoxicity of 5-FU followed from the inhibition of TS [27]. It was reported, that uracil excision initiated by a uracil glycosylase did not adequately explain the toxicity caused by TS inhibition, which confirms an earlier studies indicating, that the expression of uracil DNA glycosylase did not affect cellular sensitivity to TS inhibition [28]. These reports did not indicate that the DNA-damaging effect of CAP is not important for the toxicity of the drug, but they rather suggest that the action of a uracil glycosylase itself may not result in an immediate cellular toxicity. We share this conclusion and add that a cross-talk between mechanisms of different pathways of CAP toxicity, including DNA-damaging effect, may contribute to overall toxicity of the drug in cancer cells.
We showed also that the drug induced apoptosis in cancer cells. This effect can be rather related to the anti-metabolic action of CAP, than to its ability to induce DNA damage, since in the light of the lack of double strand break breaks, the drug would have induced enormous amount of single strand breaks and alkali labile sites and this is rather unlikely, that it would do so only in cancer cells. It was also suggested that genotoxic stress resulting from the inhibition of TS might activate programmed cell death pathways [29]. The observed changes in the cell cycle progression are generally in line with results of other results showing arrest of cells in early S phase after treatment with TS inhibitors and resuming replication progression during recovery from the treatment. Our results suggest that this effect can be rather associated with anti-metabolite than the DNA-damaging action of CAP, since we did not observe changes in the G2/M population.
Conclusions
Our results suggest that CAP may exert a pronounced cyto- and genotoxic effect in head and neck cancer cells, while affecting normal lymphocytes to a much lesser degree. This may be useful in planning chemotherapeutic strategy in head and neck cancer.
Acknowledgments
This work was supported by the grants number NN403 2955 33 from Ministry of Science and Higher Education and 505/376 from University of Lodz. The authors thank Ms. Monika Kicinska for helping them in preparing the manuscript and Dr. Marek Garbaczonek of Roche Poland for providing them with capecitabine. Presented in part at Society for Free Radicals Research Europe Meeting, Rome, Italy, August 26–29, 2009.
Conflict of interest
None
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Colevas AD, Amrein PC, Gomolin H, et al. A phase II study of combined oral uracil and ftorafur with leucovorin for patients with squamous cell carcinoma of the head and neck. Cancer. 2001;92:326–331. doi: 10.1002/1097-0142(20010715)92:2<326::AID-CNCR1326>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 2.Bazarbashi S, Abdelsalam M, Amin T, et al. Phase II trial of neo adjuvant cisplatin, 5-fluorouracil and interferon-alpha in operable squamous cell carcinoma of the esophagus. Chemotherapy. 2008;54:315–322. doi: 10.1159/000151353. [DOI] [PubMed] [Google Scholar]
- 3.Malet-Martino M, Martino R. Clinical studies of three oral pro drugs of 5-fluorouracil (capecitabine, UFT, S-1): a review. Oncologist. 2002;7:288–323. doi: 10.1634/theoncologist.7-4-288. [DOI] [PubMed] [Google Scholar]
- 4.Grem JL. 5-fluorouracil: forty-plus and still ticking. A review of its preclinical and clinical development. Investig New Drugs. 2000;18:31–299. doi: 10.1023/a:1006416410198. [DOI] [PubMed] [Google Scholar]
- 5.Diasio RB, Johnson MR. Dihydropyrimidine dehydrogenase: its role in 5-fluorouracil clinical toxicity and tumor resistance. Clin Cancer Res. 1999;5:2672–2673. [PubMed] [Google Scholar]
- 6.Fuse N, Doi T, Ohtsu A, et al. Safety of irinotecan and infusional fluorouracil/leucovorin (FOLFIRI) in Japan: a retrospective review of 48 patients with metastatic colorectal cancer. Int J Clin Oncol. 2008;13:144–149. doi: 10.1007/s10147-007-0737-z. [DOI] [PubMed] [Google Scholar]
- 7.Miwa M, Ura M, Nishida M, et al. Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur J Cancer. 1998;34:1274–1281. doi: 10.1016/S0959-8049(98)00058-6. [DOI] [PubMed] [Google Scholar]
- 8.Comella P. A review of the role of capecitabine in the treatment of colorectal cancer. Ther Clin Risk Manag. 2007;3:421–431. [PMC free article] [PubMed] [Google Scholar]
- 9.Ooyama A, Oka T, Zhao HY, et al. Anti-angiogenic effect of 5-fluorouracil-based drugs against human colon cancer xenograft. Cancer Lett. 2008;267:26–36. doi: 10.1016/j.canlet.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Walko CM, Lindley C. Capecitabine: a review. Clin Ther. 2005;27:23–44. doi: 10.1016/j.clinthera.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Mackean M, Planting A, Twelves C, et al. Phase I and pharmacologic study of intermittent twice-daily oral therapy with capecitabine in patients with advanced and/or metastatic cancer. J Clin Oncol. 1998;16:2977–2985. doi: 10.1200/JCO.1998.16.9.2977. [DOI] [PubMed] [Google Scholar]
- 12.Pivot X, Chamorey E, Guardiola E, et al. Phase I and pharmacokinetic study of the association of capecitabine-cisplatin in head and neck cancer patients. Ann Oncol. 2003;14:1578–1586. doi: 10.1093/annonc/mdg410. [DOI] [PubMed] [Google Scholar]
- 13.Diaz-Rubio E, Evans TR, Tabernero J, et al. Capecitabine (Xeloda) in combination with oxaliplatin: a phase I dose-escalation study in patients with advanced or metastatic solid tumors. Ann Oncol. 2002;13:558–565. doi: 10.1093/annonc/mdf065. [DOI] [PubMed] [Google Scholar]
- 14.Tang p, Oza A, Townsley C, et al. Phase I study of vorinostat (VOR) in combination with capecitabine (CAP) in patients (pts) with advanced solid tumors. J Clin Oncol. 2007;25:3576. doi: 10.1200/JCO.2006.08.1935. [DOI] [PubMed] [Google Scholar]
- 15.Pronk LC, Vasey P, Sparreboom A, et al. A phase I and pharmacokinetic study of the combination of capecitabine and docetaxel in patients with advanced solid tumours. Br J Cancer. 2000;83:22–29. doi: 10.1054/bjoc.2000.1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villalona-Calero MA, Weiss GR, Burris HA, et al. Phase I and pharmacokinetic study of the oral fluoropyrimidine capecitabine in combination with paclitaxel in patients with advanced solid malignancies. J Clin Oncol. 1999;17:1915–1925. doi: 10.1200/JCO.1999.17.6.1915. [DOI] [PubMed] [Google Scholar]
- 17.Chua D, Wei WI, Sham JS, et al. Capecitabine monotherapy for recurrent and metastatic nasopharyngeal cancer. Jpn J Clin Oncol. 2008;38:244–249. doi: 10.1093/jjco/hyn022. [DOI] [PubMed] [Google Scholar]
- 18.Alexandre J, Kahatt C, Bertheault-Cvitkovic F, et al. A phase I and pharmacokinetic study of irofulven and capecitabine administered every 2 weeks in patients with advanced solid tumors. Invest New Drugs. 2007;25:453–462. doi: 10.1007/s10637-007-9071-6. [DOI] [PubMed] [Google Scholar]
- 19.Becerra BC, Verma UN, Tran HT, et al. Phase I dose escalation study with irinotecan, capecitabine, epirubicin, and granulocyte colony-stimulating factor support for patients with solid malignancies. Am J Clin Oncol. 2008;31:219–225. doi: 10.1097/COC.0b013e31815a438f. [DOI] [PubMed] [Google Scholar]
- 20.Kainthla R, Zewail-Foote M. Oxidative DNA damage following photoexcitation of daunomycin: direct role of oxygen. J Photochem Photobiol A. 2008;198:200–204. doi: 10.1016/j.jphotochem.2008.03.009. [DOI] [Google Scholar]
- 21.Singh NP, McCoy T, Tice RR, et al. A simple technique for quantification of low levels of DNA damage in individual cells. Exp Cell Res. 1998;175:184–192. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 22.Klaude M, Eriksson S, Nygren J, et al. The comet assay: mechanisms and technical considerations. Mutat Res. 1996;12:89–96. doi: 10.1016/0921-8777(95)00063-1. [DOI] [PubMed] [Google Scholar]
- 23.Blasiak J, Gloc E, Drzewoski J, et al. Free radicals scavengers can differentially modulate the genotoxicity of amsacrine in normal and cancer cells. Mutat Res. 2003;535:25–34. doi: 10.1016/s1383-5718(02)00289-9. [DOI] [PubMed] [Google Scholar]
- 24.Singh NP, Stephens RE. Microgel electrophoresis: sensitivity, mechanisms and DNA electro stretching. Mutat Res. 1997;383:167–175. doi: 10.1016/s0921-8777(96)00056-0. [DOI] [PubMed] [Google Scholar]
- 25.Tice RR, Agurell E, Anderson D, et al. Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen. 2000;35:206–221. doi: 10.1002/(SICI)1098-2280(2000)35:3<206::AID-EM8>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 26.Petronzelli F, Riccio A, Markham GD, et al. Investigation of the substrate spectrum of the human mismatch-specific DNA N-glycosylase MED1 (MBD4): fundamental role of the catalytic domain. J Cell Physiol. 2000;185:473–480. doi: 10.1002/1097-4652(200012)185:3<473::AID-JCP19>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 27.Luo Y, Walla M, Wyatt MD. Uracil incorporation into genomic DNA does not predict toxicity caused by chemotherapeutic inhibition of thymidylate synthase. DNA Repair. 2008;7:162–169. doi: 10.1016/j.dnarep.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Welsh SJ, Hobbs S, Aherne GW. Expression of uracil DNA glycosylase (UDG) does not affect cellular sensitivity to thymidylate synthase (TS) inhibition. Eur J Cancer. 2003;39:378–387. doi: 10.1016/S0959-8049(02)00610-X. [DOI] [PubMed] [Google Scholar]
- 29.Rustum YM. Thymidylate synthase: a critical target in cancer therapy? Front Biosci. 2004;9:2467–2473. doi: 10.2741/1408. [DOI] [PubMed] [Google Scholar]