

Abstract
Background
Mutations in the DNMT3A, TET2, IDH1, and IDH2 genes carry prognostic significance and occur frequently in adult AML. Leukemic mutations in all four genes have recently been implicated in aberrant DNA methylation, a hallmark of neoplasia. We previously reported that IDH1 mutations were absent whereas TET2 mutations were present in 6% of pediatric AML patients; in the present study we determined the prevalence of DNMT3A and IDH2 mutations in pediatric AML.
Methods
We screened for DNMT3A and IDH2 mutations by direct sequencing of diagnostic specimens from 180 children treated on the Children’s Oncology Group clinical trial AAML03P1. Clinical characteristics, the presence of other leukemic mutations, and survival outcome was determined for mutation-positive patients.
Results
No disease-associated DNMT3A mutations were detected. IDH2 mutations were detected in 4/180 patients (2.2%), affecting codons R140 (n=3) and R172 (n=1). Two patients with IDH2 mutations harbored t(8;21), one patient harbored an MLL translocation, and one patient had a concomitant NPM1 mutation. FLT3, CEBPA, and WT1 mutations did not occur together with IDH2 mutations in our study.
Conclusion
DNMT3A and IDH2 mutations are uncommon in pediatric AML. The low prevalence of methylation-associated mutations in our study highlights the differences in the pathogenesis of pediatric vs. adult AML, at the genetic as well as potentially the epigenetic level. The age-specific characteristics of AML underscore the importance of studying the molecular biology of both childhood and adult forms of this leukemia in parallel, as the development of novel therapeutics should account for these biologic differences.
Keywords: AML, pediatric AML, DNMT3A, IDH2, Methylation
INTRODUCTION
The methylation of DNA cytosine bases is an epigenetic regulatory phenomenon critical to many physiologic processes. Aberrant DNA methylation, including both global hypomethylation as well as hypermethylation-induced silencing at the promoter sites of specific tumor-suppressor genes, is a hallmark of cancer.1 Unique methylation signatures have been associated with specific cytogenetic subtypes of acute myeloid leukemia (AML), particularly MLL rearrangements.2 Additionally, other distinct methylation signatures in AML have been described which do not correspond to any particular cytogenetic alteration.3 A molecular lesion such as a somatic gene mutation may prove to be the unifying characteristic for some leukemic methylation phenotypes.
The human DNA methyltransferase genes (DNMT1, DNMT3A, and DNMT3B) encode enzymes which catalyze the addition of a methyl group to the 5-position of cytosine, generating 5 methylcytosine (5mC). By this mechanism, the DNA methyltransferases mediate the downregulation of target genes via the methylation of upstream CpG islands. Somatic mutations which putatively alter enzyme function have now been described in leukemia in one of these genes: large-scale targeted resequencing efforts of adult leukemia genomes led to the initial identification of a missense mutation at the Arginine 882 residue of DNMT3A.4 Ley5 detected an additional DNMT3A mutation upon massively-parallel sequencing of an adult normal karyotype AML genome. These investigators went on to screen a cohort of 282 adult AML patients and reported the presence of DNMT3A mutations in 62 patients (22%). FLT3, NPM1, and IDH1 mutations were enriched in the DNMT3A-mutated cohort, while favorable-risk cytogenetic abnormalities were absent. Notably, DNMT3A mutations were independently and significantly associated with decreased survival in this adult AML study.
The oxidation of 5mC to 5-hydroxymethylcytosine (5hmC) is catalyzed by the TET family of proteins and appears to be an important post-methylation modification. The TET genes may be involved in turning off the DNA methylation process, as the presence of 5hmC prevents the re-methylation of cytosine by DNA methyltransferases during cell division, and 5hmC functions as an intermediary which may lead to passive de-methylation.6 Hypomorphic mutations in the TET2 gene are present in a variety of myeloid disorders, occurring at high frequencies in adult myeloproliferative diseases;7 we previously reported TET2 mutations in 6% of pediatric AML patients.8 The oxidative function of TET2 is dependent on the substrate alpha-ketoglutarate. This metabolite is produced in the citric acid cycle by the oxidative decarboxylation of isocitrate by the isocitrate dehydrogenase enzymes. Mutations in the isocitrate dehydrogenase genes IDH1 and IDH2 themselves are now a well-described phenomenon in adult AML. Neomorphic mutations altering arginine residues (R132 in IDH1, and R140 and R172 in IDH2) in exon 4 of these genes together affect over 15% of unselected adult AML patients and are also associated with poor prognosis.9 These mutations result in the production of an alternative metabolite, 2-hydroxyglutarate (2HG), rather than alpha-ketoglutarate. IDH1 and IDH2 mutations are associated with global DNA hypermethylation, impairment of TET2 function, and a specific hypermethylation phenotype.10 However, we previously reported that IDH1 mutations were absent in pediatric AML.11 Given the novel finding of DNMT3A mutations in a significant proportion of adult AML patients, and the disparity in prevalence of leukemia-associated genes in adult vs. childhood forms of the disease, we sought to determine the previously unknown prevalence of DNMT3A and IDH2 mutations in 180 children enrolled on the Children’s Oncology Group (COG) trial AAML03P1.
METHODS
Patient Samples
COG-AAML03P1 enrolled 340 patients (age 1 month to 21 years) with newly diagnosed de novo AML. Patients with acute promyelocytic leukemia (M3 AML) were excluded, as were children with Down syndrome, prior myelodysplastic syndrome, or inherited bone marrow failure syndromes. This pilot study evaluated the efficacy and safety of a 5-cycle modified MRC-like chemotherapy regimen including gemtuzumab ozogamicin (GO) in selected cycles, as previously described.12 Genomic DNA extracted from diagnostic marrow specimens was available from 180 patients. In accordance with the principles of the Declaration of Helsinki, consent was obtained from all study participants or their parents. Institutional review board approval was obtained from the Fred Hutchinson Cancer Research Center prior to mutation analysis, and this study was approved by the Myeloid Disease Biology Committee of the COG.
Molecular Genotyping
Genomic DNA was extracted from diagnostic marrow specimens with the AllPrep DNA/RNA Mini Kit using the QIAcube automated system (Qiagen, Valencia, CA). PCR amplification of the entire coding region of DNMT3A was performed using 23 overlapping primer pairs (Supplemental Table I). The reaction mixture was carried out in a volume of 25 microliters and consisted of Failsafe PCR 2x Premix Buffer (Epicentre Biotechonologies, Madison WI), 0.5 microliters of 10 mM deoxyribonucloetide triphosphates, 0.5 microliters of 50 mM MgCl2, 1.25U Platinum Taq DNA Polymerase (Invitrogen Corporation, Carlsbad, CA), 5pmol of each primer, and 10 ng of genomic DNA. Thermocycler conditions were as follows: 94°C for 5 minutes, followed by 45 cycles at 94°C for 30 seconds, 60°C for 45 seconds, and 72° for 60 seconds, with a final extension step at 72°C for 7 minutes. High-throughput direct sequencing was performed and sequences were run on an ABI 3730xl DNA analyzer (Applied Biosystems, Foster City, CA) as previously described.13
Exon 4 of IDH2, containing the mutational hotspots R140 and R172, was sequenced via the following PCR reaction: Failsafe PCR 2x Premix Buffer K, Platinum Taq DNA polymerase, 5pmol of each primer (IDH2F: 5′-GCTGCAGTGGGACCACTAT-3′ and IDH2R: 5′-GTGCCCAGGTCAGTGGAT-3′), and 10 ng of genomic DNA. Thermocycler conditions were 94°C for 5 minutes, 35 cycles at 94°C for 30 seconds, 60°C for 30 seconds, and 72° for 30 seconds, with a final extension step at 72°C for 7 minutes. Direct sequencing was carried out in both directions using the Big Dye Terminator v3.1 Cycle Sequencing Reaction (Applied Biosystems) and run on an ABI 3730xL DNA analyzer.
Statistical Methods
The Chi-squared test was used to test the significance of observed differences in proportions; Fisher’s exact test was used when sample sizes were small. The Mann-Whitney test was used to determine the significance between differences in medians.
RESULTS
Patient Population
Cryopreserved diagnostic specimens were available from 180 (53%) of the 340 eligible pediatric patients enrolled on COG-AAML03P1. Demographics, laboratory features, and clinical characteristics of patients for whom specimens were and were not available were compared (Table I), to determine whether patients included in this analysis were representative of patients enrolled on the study at large. Patients with available samples had higher median diagnostic white blood cell (WBC) count (p<0.001). The cohort of patients with samples available for this study contained a higher proportion of FAB M4 patients (p=0.012) and a lower number of FAB M5 patients (p=0.027). All cases of the rare translocation t(6;9) occurred in the cohort of patients with available samples. There were no significant differences in age, gender, race, or median diagnostic blast percentage between the two groups (Supplemental Table II). Outcome measures also did not differ significantly between patients with and without available specimens.
Table I.
Laboratory Characteristics, FAB Classification, and Cytogenetics of Study Patients
| Patients Tested (n=180) | Patients Not Tested (n=160) | p-value | |||
|---|---|---|---|---|---|
| N | % | N | % | ||
| WBC (x103/μL) - median (range) | 34.1 | (1.4 – 495) | 11.6 | (0.7 – 324) | <0.001 |
| BM Blasts % - median (range) | 70 | (0 – 100) | 67 | (0 – 99) | 0.457 |
| FAB Classification | |||||
| M0 | 2 | 1% | 6 | 4% | 0.151 |
| M1 | 19 | 12% | 15 | 11% | 0.775 |
| M2 | 47 | 30% | 30 | 22% | 0.127 |
| M4 | 49 | 31% | 25 | 18% | 0.012 |
| M5 | 26 | 17% | 37 | 27% | 0.027 |
| M6 | 2 | 1% | 5 | 4% | 0.256 |
| M7 | 12 | 8% | 18 | 13% | 0.115 |
| De novo (NOS) | 23 | 24 | |||
| Cytogenetics | |||||
| Normal | 37 | 22% | 33 | 22% | 0.903 |
| t(8;21) | 20 | 12% | 16 | 11% | 0.670 |
| Inv(16) | 27 | 16% | 15 | 10% | 0.093 |
| Abnormal 11 | 33 | 20% | 34 | 23% | 0.585 |
| t(6;9)(p23;q34) | 7 | 4% | 0 | 0% | 0.015 |
| Monosomy 7 | 3 | 2% | 7 | 5% | 0.203 |
| Del7q | 3 | 2% | 2 | 1% | 1.000 |
| -5/5q- | 2 | 1% | 1 | 1% | 1.000 |
| +8 | 14 | 8% | 13 | 9% | 0.968 |
| Other | 19 | 12% | 30 | 20% | 0.040 |
| Missing/Unknown | 15 | 9 |
DNMT3A Mutations
In the adult study, DNMT3A mutations occurred throughout the gene, but clustered in the C-terminal methyltransferase domain, with over half of the reported mutations being missense alterations affecting codon R882.5 We sequenced the entire coding region (23 exons) of DNMT3A. Within the 180 pediatric AML patients in our study, no DNMT3A mutations were detected (Figure 1).
Fig. 1.
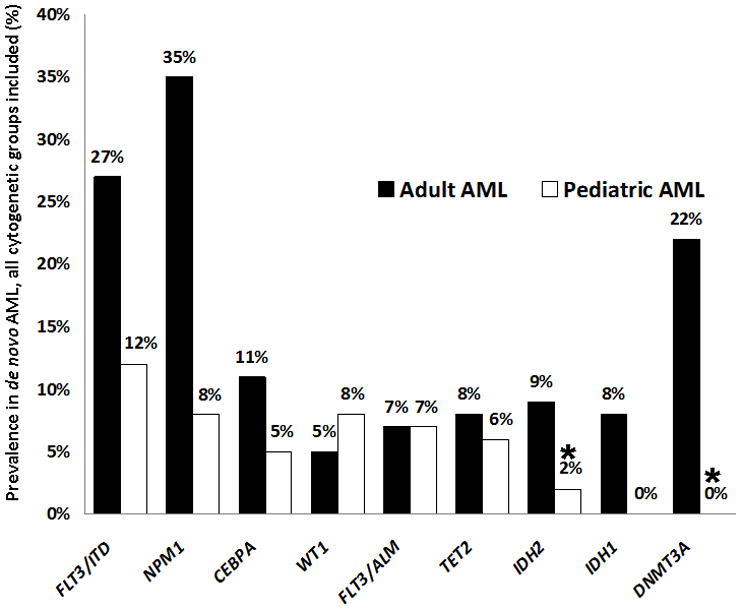
Prevalence of Leukemia-Associated Mutations in Pediatric vs. Adult AML. Prevalence of mutations in pediatric patients treated on COG AML trials is compared to corresponding reported mutation prevalence in adult AML. Results from the present study are indicated with an asterisk. WT126,27 and FLT3/ALM21,23 mutations occurred at similar frequencies between the two cohorts, while FLT3/ITD,21,23 NPM1,22,24 and CEBPA12,25 mutations occurred more commonly in adult AML. Although TET28,16 mutations were nearly as prevalent in childhood as compared to adult AML, functional mutations of the other methylation-associated genes–IDH111 or IDH2 (combined mutation prevalence of 17% in adult AML9) and DNMT3A (22% in adult AML5)–were notably less common (rare or absent) in pediatric patients.
IDH2 Mutations
2HG-producing neomorphic mutations occur in exon 4 of the isocitrate dehydrogenase genes. In IDH2, R172 missense mutations have been reported in both adult AML and adult gliomas, while R140 mutations are exclusive to AML.14 We detected IDH2 mutations in 4 of 180 patients (2.2%, Figure 1); 3 mutations were R140 alterations (R140Q, n=2; R140W, n=1) and 1 was a R172K alteration (Table II). All mutations were heterozygous. Three of the four patients who harbored IDH2 mutations are classified as favorable-risk in current COG classification schemes, two by virtue of t(8;21) and one due to the presence of an NPM1 mutation. Both t(8;21) patients had additional cytogenetic abnormalities as well: −Y and del 9q in the first patient, and +4 in the second patient. A third IDH2-mutated patient harbored an 11q23 rearrangement due to a t(11;19)(q23p13.3) translocation. None of the patients with IDH2 mutations had concomitant FLT3/ITD, WT1, CEBPA, IDH1, or TET2 mutations.
Table II.
Characteristics and Clinical Profile of Patients with IDH2 Mutations
| Patient # | Mutation | WBC X 10^9/L | FAB | Cyto | NPM | FLT3 | CEBPA | WT1 |
|---|---|---|---|---|---|---|---|---|
| 1 | R140Q | 12.5 | M2 | t(8;21); −Y; del(9)(q11q22) | Neg | Neg | Neg | Neg |
| 2 | R140Q | 8.5 | M4 | t(8;21); +4 | Neg | Neg | Neg | Neg |
| 3 | R140W | 90.7 | AML not further classified | t(11;19)(q23;p13.3) | Neg | Neg | Neg | Neg |
| 4 | R172K | 5 | M1 | unknown | Pos | Neg | Neg | Neg |
Outcome parameters were examined for patients with IDH2 mutations as numbered in Table II. Patient 2 withdrew from the clinical study after enrolling and providing diagnostic specimens, and Patient 4 was lost to follow-up. Patients 1 and 3 both achieved CR; Patient 1 died of treatment-related mortality following hematopoietic stem cell transplantation, while Patient 3 is alive in continuous remission at 5 years from diagnosis.
DISCUSSION
Aberrant DNA methylation patterns have long been associated with neoplasia. An emerging class of newly described mutations in adult AML affecting the TET2, IDH1, IDH2, and DNMT3A genes are now being reported to occur in association with epigenetic consequences related to alterations of the DNA cytosine-5 methylation pathway (Figure 2).5,7,15 We reported in a prior study that TET2 mutations were detected in 6% of pediatric AML patients,8 similar to the 7.6% prevalence of these mutations in adult AML.16 We also previously found that IDH1 mutations were absent in a cohort of 257 pediatric AML patients treated on COG-AAML03P1.11 In the present study of 180 children with available specimens from the COG-AAML03P1 trial, we found that DNMT3A mutations are also absent, while IDH2 mutations are present in 4 patients (2.2%). Two of the IDH2-mutated patients in our study harbored t(8;21). In contrast, isocitrate dehydrogenase mutations rarely occur in patients with favorable cytogenetic abnormalities in adult AML.10 Three of the 4 IDH2-mutated patients are classified as favorable-risk based on current COG risk-stratification schemas (on the basis of t(8;21) in two patients and an NPM1 mutation in the third). In adult AML, isocitrate dehydrogenase mutations are of poor prognostic significance, particularly in the NPM1-mutated/FLT3-wild-type subgroup.9 The low number of patients with IDH2 mutations on our study precludes meaningful outcome analysis, and molecular genotyping of IDH2 is unlikely to be useful in upfront risk-stratification due to the low prevalence of isocitrate dehydrogenase mutations in pediatric AML.
Fig. 2.
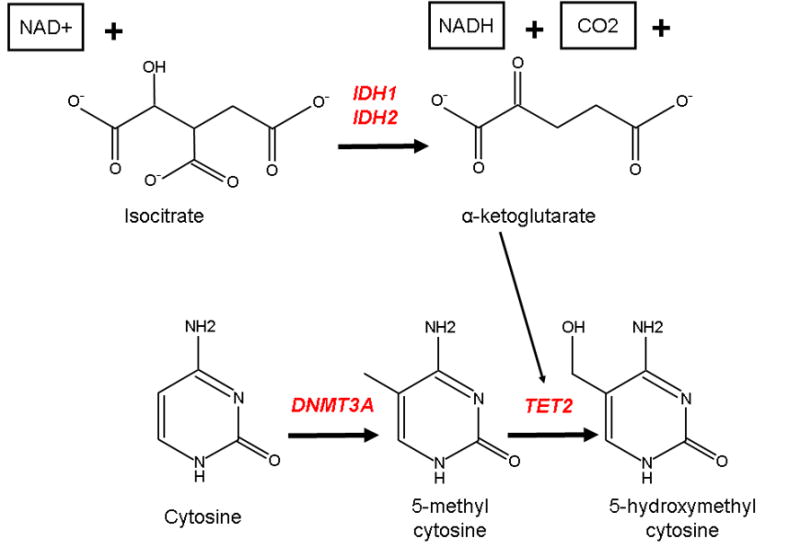
Role of the leukemia-associated genes DNMT3A, TET2, IDH1, and IDH2 in DNA methylation DNMT3A encodes a DNA methyltransferase gene responsible for methylation at the 5 position of cytosine, resulting in 5-methylcytosine (5mc). TET2 catalyzes the conversion of 5mc to 5-hydroxymethylcytosine (5hmc), a reaction which may ultimately result in the demethylation of DNA. The TET2 enzyme requires the substrate alpha-ketoglutarate, which is the product of the oxidative decarboxylation of isocitrate by IDH1 or IDH2. Function-altering mutations of all four genes have been implicated in AML and may be involved in the deregulation of DNA methylation/demethylation.
Myeloid leukemogenesis is a multi-step process. In the classic model of molecular-genetic cooperativity,17 an initial “class II” event, such as a core-binding factor translocation or a CEBPA mutation, interrupts terminal differentiation. A second, “class I” molecular event which provides a proliferative signal, such as a FLT3/ITD or a c-Kit mutation, is then required for leukemic transformation. The recent description of mutations in the enzyme-encoding genes TET2, DNMT3A, IDH1, and IDH2 in AML adds an additional layer of complexity to this model of leukemia pathogenesis in a subset of patients. All four of these genes have now been demonstrated to play a functional role in cytosine methylation pathways; disordered DNA methylation may be the neoplastic mechanism common to these mutations. A vital epigenetic function of DNA methylation/demethylation is the regulation of gene expression at various stages of development. Recent work suggests that the TET family of proteins may play a role in the maintenance of stemness through the regulation of the balance between differentiation vs. self-renewal.18 Disruption of these criticial functions likely plays a role in the development of leukemia. However, it is notable that somatic mutations in this group of genes arise in a substantially smaller proportion of pediatric AML cases as compared to adult AML.
The mechanisms of abnormal methylation in most cases of AML are not well understood. There are likely other significant mechanisms of epigenetic dysregulation in childhood AML patients which have yet to be well-described. For instance, certain cytogenetic abnormalities, particularly 11q23 rearrangements,2 result in specific methylation abnormalities; the MLL gene at 11q23 encodes a histone methyltransferase with putative epigenetic function, and in our study 1 patient with an IDH2 mutation had a concomitant 11q23 translocation. The MLL gene is also fused to the TET1 gene in the rare recurrent t(10;11) translocation, providing yet another possible mechanism of dysregulated methylation.6 Further, overexpression of all three DNA methyltransferase genes (DNMT1, DNMT3A, and DNMT3B) has also been documented in AML.19 COG investigators are currently conducting studies of global methylation signatures in pediatric AML.
Molecular genotyping studies in the past decade have greatly enriched our understanding of leukemogenesis in both pediatric and adult leukemias, and lead to the identification of several prognostically relevant gene alterations.20 Currently, the presence of FLT3 internal tandem duplications (high-risk),21 as well as mutations in either NPM122 or CEBPA12 in the absence of FLT3/ITD (favorable-risk), are used in COG risk-stratification schemas. The prevalence of most AML-associated mutations, including FLT3/ITD,21,23 NPM1,22,24 CEBPA,12,25 and WT1,26,27 differs between pediatric and adult patient cohorts (Figure 1). Further, the prognostic significance of gene mutations may be dramatically different between pediatric and adult AML, as we have reported in the case of the WT1 gene,26 and the c-Kit gene in core-binding factor AML.28 In contrast to TET2, which is mutated at a similar rate in adult and pediatric AML, disparities in mutation frequency are particularly apparent in the remaining methylation-associated genes: DNMT3A and IDH1 mutations are absent in pediatric AML patients in our studies, while IDH2 mutations occur at a low rate. This underscores the distinct biology of pediatric vs. adult AML. The application of powerful next-generation sequencing technology to childhood and adult forms of leukemia in parallel will facilitate our understanding of the similarities and differences in molecular pathogenesis between these two categories of AML. Future studies should focus on the differences in age-specific genetic and epigenetic mechanisms of myeloid leukemogenesis, as these differences may have significant ramifications for the development of targeted therapy.
Supplementary Material
Acknowledgments
We are grateful to the patients and families who consented to the use of biologic specimens in these trials. We also thank the COG AML Reference Laboratory for providing diagnostic specimens, and Dr. Vani Shanker for scientific editing. This work was supported by the National Institutes of Health grants K12 HD043376 (P.A.H.), T32 CA009351 (M.A.K.), R21 CA10262 (S.M.), R01 CA114563 (S.M.), and COG Chair’s Grant U10 CA98543, as well as the Mary Claire Satterly Foundation (P.A.H.).
Footnotes
CONFLICT OF INTEREST
The authors declare no relevant conflicts of interest.
References
- 1.Esteller M. Epigenetics in Cancer. N Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez S, Suela J, Valencia A, et al. DNA methylation profiles and their relationship with cytogenetic status in adult acute myeloid leukemia. PLoS One. 2010;5:e12197. doi: 10.1371/journal.pone.0012197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Figueroa ME, Lugthart S, Li Y, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17:13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamashita Y, Yuan J, Suetake I, et al. Array-based genomic resequencing of human leukemia. Oncogene. 2010;29:3723–31. doi: 10.1038/onc.2010.117. [DOI] [PubMed] [Google Scholar]
- 5.Ley TJ, Ding L, Walter MJ, et al. DNMT3A Mutations in Acute Myeloid Leukemia. N Engl J Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dahl C, Gronbaek K, Guldberg P. Advances in DNA methylation: 5-hydroxymethylcytosine revisited. Clin Chim Acta. 2011 doi: 10.1016/j.cca.2011.02.013. Prepublished online Feb 12. [DOI] [PubMed] [Google Scholar]
- 7.Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 8.Kutny MA, Alonzo TA, Gerbing RB, et al. TET2 SNP rs2454206 (I1762V) Correlates with Improved Survival in Pediatric Acute Myelogenous Leukemia, a Report From the Children’s Oncology Group. Blood. 2010;116:949. [Abstract] [Google Scholar]
- 9.Paschka P, Schlenk RF, Gaidzik VI, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28(22):3636–3643. doi: 10.1200/JCO.2010.28.3762. [DOI] [PubMed] [Google Scholar]
- 10.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ho PA, Alonzo TA, Kopecky KJ, et al. Molecular alterations of the IDH1 gene in AML: a Children’s Oncology Group and Southwest Oncology Group study. Leukemia. 2010;24(5):909–913. doi: 10.1038/leu.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho PA, Alonzo TA, Gerbing RB, et al. Prevalence and prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia (AML): A report from the Children’s Oncology Group. Blood. 2009;113(26):6558–6566. doi: 10.1182/blood-2008-10-184747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geraghty DE, Daza R, Williams LM, et al. Genetics of the immune response: identifying immune variation within the MHC and throughout the genome. Immunol Rev. 2002;190:69–85. doi: 10.1034/j.1600-065x.2002.19006.x. [DOI] [PubMed] [Google Scholar]
- 14.Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst. 2010;102(13):932–941. doi: 10.1093/jnci/djq187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ko M, Huang Y, Jankowska AM, et al. Impaired hydroxylation of 5-methylctyosine in myeloid cancers with mutant TET2. Nature. 2010;468(7325):839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaidzik VI, Schlenk RF, Paschka P, et al. TET2 Mutations in Acute Myeloid Leukemia (AML): Results on 783 Patients Treated within the AML HD98A Study of the AML Study Group (AMLSG) Blood. 2010;116:97. [Abstract] [Google Scholar]
- 17.Ito S, D’Alessio AC, Taranova OV, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010 Aug 26;466(7310):1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Renneville A, Roumier C, Biggio V, et al. Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia. 2008;22(5):915–931. doi: 10.1038/leu.2008.19. [DOI] [PubMed] [Google Scholar]
- 19.Mizuno S, Chijiwa T, Okamura T, et al. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood. 2001;97(5):1172–1179. doi: 10.1182/blood.v97.5.1172. [DOI] [PubMed] [Google Scholar]
- 20.Gaidzik V, Dohner K. Prognostic implications of gene mutations in acute myeloid leukemia with normal cytogenetics. Semin Oncol. 2008;35(4):346–355. doi: 10.1053/j.seminoncol.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Meshinchi S, Alonzo TA, Stirewalt DL, et al. Clinical implications of FLT3 mutations in pediatric AML. Blood. 2006;108:3654–3661. doi: 10.1182/blood-2006-03-009233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown P, McIntyre E, Rau R, et al. The incidence and clinical significance of nucleophosmin mutations in childhood AML. Blood. 2007;110(3):979–985. doi: 10.1182/blood-2007-02-076604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meshinchi S, Appelbaum FR. Structural and functional alterations of FLT3 in acute myeloid leukemia. Clin Cancer Res. 2009;15(13):4263–4269. doi: 10.1158/1078-0432.CCR-08-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Falini B, Mecucci C, Tiacci E, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352(3):254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 25.Preudhomme C, Sagot C, Boissel N, et al. Favorable prognostic significance of CEBPA mutations in patients with de novo acute myeloid leukemia: a study from the Acute Leukemia French Association (ALFA) Blood. 2002;100(8):2717–2723. doi: 10.1182/blood-2002-03-0990. [DOI] [PubMed] [Google Scholar]
- 26.Ho PA, Zeng R, Alonzo TA, et al. Prevalence and prognostic implications of WT1 mutations in pediatric acute myeloid leukemia (AML): a report from the Children’s Oncology Group. Blood. 2010;116(5):702–10. doi: 10.1182/blood-2010-02-268953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Renneville A, Boissel N, Zurawski V, et al. Wilms tumor 1 gene mutations are associated with a higher risk of recurrence in young adults with acute myeloid leukemia: a study from the Acute Leukemia French Association. Cancer. 2009;115(16):3719–3727. doi: 10.1002/cncr.24442. [DOI] [PubMed] [Google Scholar]
- 28.Pollard JA, Alonzo TA, Gerbing RB, et al. Prevalence and prognostic significance of KIT mutations in pediatric patients with core binding factor AML enrolled on serial pediatric cooperative trials for de novo AML. Blood. 2010;115(12):2372–2379. doi: 10.1182/blood-2009-09-241075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.